Dinuclear and mononuclear manganese(IV)-radical complexes and their catalytic catecholase activity†
Soumen Mukherjeea, Thomas Weyhermüllera, Eberhard Bothea, Karl Wieghardta and Phalguni Chaudhuri*b
aMax-Planck-Institut für Bioanorganische Chemie, Stiftstrasse 34-36, D-45470 Mülheim an der Ruhr, Germany
bMax-Planck-Institut für Bioanorganische Chemie, Stiftstrasse 34-36, D-45470 Mülheim an der Ruhr, Germany. E-mail: Chaudh@mpi-muelheim-mpg.de
First published on 20th October 2004
Abstract
Seven o-aminophenol ligands based on aniline and m-phenylenediamine, which act as mono- and di-nucleating non-innocent ligands, respectively, together with their seven Mn(IV)-complexes, 1–7 are described. One of them, 1, is dinuclear and 2–7 are mononuclear, in which the ligands are coordinated in their oxidized o-iminobenzosemiquinone radical forms. The crystal structures of 1 and 3 were determined by X-ray diffraction and the electronic structures were established by various physical methods including EPR and variable-temperature (2–290 K) susceptibility measurements. Electrochemical measurements (CV and SQW) indicate primarily ligand-centered redox processes. The Mn(IV)-radical complexes, 1–7, catalyze the oxidation of 3,5-di-tert-butylcatechol with molecular oxygen as the sole oxidant to afford 3,5-di-tert-butylquinone quantitatively under mild conditions to mimic the function of the copper-containing enzyme catechol oxidase. An “on–off” mechanism of the radicals without redox participation of the metal center is proposed for the catalytic oxidation process. Complex 1 is found to be a good catalyst for oxidative C–C coupling of hindered phenols to diphenoquinones.
Introduction
Widespread occurrence of tyrosine radicals in metalloproteins1 involved in oxygen-dependent enzymatic radical catalysis has sparked and fuelled the interest of bioinorganic chemists in using phenol-containing ligands2 to synthesize model compounds that contain coordinated phenolate and its one-electron oxidized phenoxyl radical. Additionally, in the field of molecular magnetism3 redox-active paramagnetic ligands are gaining increasing attention for the generation of molecular high-spin materials based on the hybridization of organic–inorganic molecules in which paramagnetic metal ions are coordinated to organic open-shell radical ligands.4 This surge of interest has been also due partly to the relevance of such molecules to biological electron-transfer processes.The ubiquitous enzymes catechol oxidases,1b,5 found in bacteria, fungi and plants, belong to the class of type 3 copper proteins and are also known as o-diphenol oxidases or polyphenol oxidases. In contrast to tyrosinase, also a type 3 dicopper protein, catechol oxidases catalyze exclusively the two-electron oxidation of o-diphenols (i.e. catechols) to the corresponding o-quinones (called catecholase activity) coupled with the reduction of dioxygen to water. The resulting highly reactive quinones autopolymerize to form brown polyphenolic catechol melanins, a process that is thought to be utilized by the plants for their defense against the possible damage caused by pathogens or insects. This enzyme contains an EPR-silent, i.e. a strongly antiferromagnetically coupled, dicopper(II) center in the resting oxidized state. Three dimensional X-ray crystal structural analysis of catechol oxidase from sweet potato in the resting dicupric Cu(II)Cu(II) state, in the reduced Cu(I)Cu(I) form and in complex with the inhibitor phenylthiourea, have been reported.5c,6 In the deoxy or reduced state the two copper ions are in the +I oxidation state at a distance of 4.4 Å. In the met oxidized form each copper center is coordinated to three histidine residues and a bridging probably hydroxide ligand, thus resulting in a coordination number of four for each cupric ion with a Cu(II)⋯Cu(II) distance of 2.9 Å. Consequently, the search for complexes capable of mimicking the function of catechol oxidase is primarily involved with dicopper(II) complexes,7 although complexes of other transition metal ions, particularly Mn,8 are also known to exhibit catechol oxidase activity.
Recently we have described two 2-aminophenol ligands, based on aniline9 and m-phenylenediamine,10 which act as mononucleating (H2L2) and dinucleating (H4L1) non-innocent ligands, respectively.

These ligands can coordinate not only in their different deprotonated forms but also in their oxidized o-imonobenzosemiquinonate radical forms. The different forms which have been characterized unambiguously are depicted in Scheme 1.
 | ||
| Scheme 1 | ||
These two ligands react with different metal ions in the presence of a base and air to yield radical complexes, in which in some cases as many as six iminosemiquinone radicals are present. As a preliminary communication10 we have also reported that the Mn(IV)-complex of the dinucleating ligand H4L1, Mn2IV(L1A˙)2(L1˙˙), containing four radicals, can catalyze the oxidation of catechol with molecular oxygen as the sole oxidant to afford quinone quantitatively under mild conditions to mimic the function of the copper-containing enzyme catechol oxidase. Interestingly, the corresponding dicopper(II) complex, CuII2(L1˙˙)2, containing four radicals exhibits much less catechol oxidase activity than that of the corresponding dimanganese(IV) complex. In this work we extend our previous study to similar substituted aniline-containing ligands together with the catecholase activity of manganese(IV) complexes containing the ligands L1–L7 in detail (Scheme 2).
 | ||
| Scheme 2 | ||
Moreover, we describe here some preliminary results of typical radical mediated reaction, viz. oxidative coupling of 2,6-di-tert-butylphenol catalytically by the dimanganese(IV) complex MnIV2(L1˙A)2(L1˙˙) in the presence of air.
Results and discussion
The ligands H4L1 and H2L2–H2L7 are readily available11 in good yields from the reaction of 3,5-di-tert-butylcatechol and m-phenylenediamine (2 ∶ 1) (for H4L1) or differently substituted aniline (1 ∶ 1) for H2L2–H2L7 in n-heptane in the presence of air and the base triethylamine.In the IR the ligands show characteristic strong peaks at 3392–3370, 3350–3330 and 2960–2860 cm−1 attributable to ν(OH), ν(NH) and ν(CH) of the tert-butyl groups, respectively. Selected IR peaks are listed in the Experimental section. The ligands were characterized also by different other spectroscopic techniques viz.1H NMR, MS and melting points. They exhibit “normal” behaviour (see Experimental section) and do not warrant any special discussion.
Schemes 1 and 2 show the ligands in different oxidation states observed and the complexes prepared with their labels, respectively. When a reaction mixture of the ligand either H4L1 or H2L2–H2L7, [Bu4N][OCH3] and manganese(III) acetate [MnIII3(μ3-O)(CH3COO)6]CH3COO, in methanol was heated to reflux in the presence of air, deep brown microcrystalline crystals of complexes 1–7 were obtained.
The most salient features observed for all complexes in the IR are the absence of the frequencies attributable to ν(NH) and ν(OH) stretching. A medium intense band at ∼1570 cm−1 assigned to ν(C–N) and a sharp peak at ∼1440 cm−1 due to ν(C-O) appear for all complexes. Details of the IR peaks are given in the Experimental section.
Molecule-ion peaks for complexes were observed in EI-MS. For example, m/z = 1646 was obtained for complex 1, showing clearly the composition to be [Mn2L13], thus indicating that all of the ligands cannot be present in their “simple” innocent form. Similarly, the metal ∶ ligand ratio of 1 ∶ 3 was ascertained from the MS-data for other complexes. Mass spectrometric data for all complexes are summarized in the Experimental section.
The electronic spectrum of 1 in dichloromethane solution shown in Fig. 1 exhibits above 450 nm several maxima at 550 (ε
= 1![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 880), 648 (ε
= 11500), 807 nm (ε
= 10700 M−1 cm−1) and a shoulder at 953 nm (ε
∼ 8430 M−1 cm−1) which are assigned to intraligand π–π* transitions of the iminosemiquinone ligands. In contrast, the spectra of 3–7 resemble closely that reported for the Mn(IV)-compound122 with the ligand H2L2.
880), 648 (ε
= 11500), 807 nm (ε
= 10700 M−1 cm−1) and a shoulder at 953 nm (ε
∼ 8430 M−1 cm−1) which are assigned to intraligand π–π* transitions of the iminosemiquinone ligands. In contrast, the spectra of 3–7 resemble closely that reported for the Mn(IV)-compound122 with the ligand H2L2.
![Electronic spectra of complex 1, [MnIV2(L1˙A)2(L1˙˙)], and its two electrochemically oxidized forms in CH2Cl2.](/image/article/2004/DT/b410842f/b410842f-f1.gif) | ||
| Fig. 1 Electronic spectra of complex 1, [MnIV2(L1˙A)2(L1˙˙)], and its two electrochemically oxidized forms in CH2Cl2. | ||
Description of the structures
The crystal structures of complexes 1 and 3 have been determined by single-crystal X-ray crystallography at 100(2) K. Fig. 2 shows the structure of a neutral molecule in crystals of 1. Selected bond lengths and angles for complex 1 are summarized in Table 1.
|
|
| |||
|---|---|---|---|---|---|
| Mn(1)–O(1) | 1.884(3) | Mn(1)–O(41) | 1.933(3) | Mn(1)–O(81) | 1.967(3) |
| Mn(2)–O(20) | 1.921(3) | Mn(2)–O(60) | 1.999(3) | Mn(2)–O(100) | 1.905(3) |
| Mn(1)–N(7) | 1.936(3) | Mn(1)–N(47) | 1.959(3) | Mn(1)–N(87) | 1.972(3) |
| Mn(2)–N(14) | 1.975(3) | Mn(2)–N(54) | 1.988(3) | Mn(2)–N(94) | 1.933(3) |
| C(1)–O(1) | 1.323(5) | C(41)–O(41) | 1.310(5) | C(81)–O(81) | 1.297(4) |
| C(6)–N(7) | 1.381(5) | C(46)–N(47) | 1.360(5) | C(86)–N(87) | 1.342(5) |
| C(8)–N(7) | 1.427(5) | C(48)–N(47) | 1.438(5) | C(88)–N(87) | 1.425(5) |
| C(1)–C(2) | 1.415(5) | C(41)–C(42) | 1.414(6) | C(81)–C(82) | 1.428(6) |
| C(2)–C(3) | 1.395(6) | C(42)–C(43) | 1.383(5) | C(82)–C(83) | 1.368(5) |
| C(3)–C(4) | 1.410(6) | C(43)–C(44) | 1.418(6) | C(83)–C(84) | 1.434(5) |
| C(4)–C(5) | 1.385(6) | C(44)–C(45) | 1.369(6) | C(84)–C(85) | 1.369(5) |
| C(5)–C(6) | 1.397(6) | C(45)–C(46) | 1.414(6) | C(85)–C(86) | 1.420(5) |
| C(6)–C(1) | 1.423(6) | C(46)–C(41) | 1.427(6) | C(86)–C(81) | 1.427(5) |
| C(20)–O(20) | 1.318(5) | C(60)–O(60) | 1.294(4) | C(100)–O(100) | 1.330(5) |
| C(15)–N(14) | 1.359(5) | C(55)–N(54) | 1.343(5) | C(95)–N(94) | 1.383(5) |
| C(12)–N(14) | 1.436(5) | C(52)–N(54) | 1.440(5) | C(92)–N(94) | 1.414(5) |
| C(20)–C(19) | 1.410(6) | C(60)–C(59) | 1.421(6) | C(100)–C(99) | 1.417(5) |
| C(19)–C(18) | 1.382(6) | C(59)–C(58) | 1.367(6) | C(99)–C(98) | 1.381(6) |
| C(18)–C(17) | 1.427(6) | C(58)–C(57) | 1.429(5) | C(98)–C(97) | 1.407(6) |
| C(16)–C(17) | 1.368(6) | C(57)–C(56) | 1.355(5) | C(97)–C(96) | 1.377(6) |
| C(16)–C(15) | 1.406(6) | C(56)–C(55) | 1.415(5) | C(96)–C(95) | 1.405(6) |
| C(15)–C(20) | 1.426(6) | C(55)–C(60) | 1.445(5) | C(95)–C(100) | 1.410(5) |
| Mn(1)⋯Mn(2) | 6.762(2) | ||||


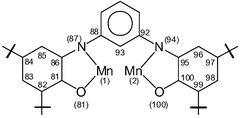
 | ||
| Fig. 2 Molecular structure of the neutral complex 1. | ||
Complex 1 crystallizes in the monoclinic space group P21/n (Z = 4), the same as that for the corresponding [FeIII2(L1˙˙)3],10 thus rendering them isomorphous. It is misleading to infer from this fact that complex 1 and [FeIII2(L1˙˙)3] are isostructural, and in particular, that they contain the same charge distributions among the metal ions and ligands, i.e. three bis(o-iminosemiquinonato(−) radicals) and two trivalent metal ions. That manganese centers, Mn(1) and Mn(2), cannot be ascribed to an oxidation state of +III is clearly borne out by the facts that (i) none of the manganese centers show any Jahn–Teller effect, as is expected for a d4 system, and (ii) the observed Mn–N bond distances, av. 1.961 Å are too short in comparison to the corresponding bond lengths reported for Mn(III)-complexes.13 As will be shown below, each manganese center must be ascribed to a +IV (d3) oxidation level.
The charge distributions of three ligands, acting each of them as expected (as a bis(O,N-bidentate)) bridging ligand with a m-phenylene group as a spacer, are different (see Table 1). The ligand containing the donor atoms O(41), N(47) on one side of the m-phenylene spacer and O(60), N(54) on the other side exhibits typical characteristics of the charge distribution for two o-iminobenzosemiquinonate(1−) π-radical ligands, [L1˙˙]2−: two short C–N and C–O bond distances, approaching double bonds, together with the loss of aromaticity at the phenyl rings containing the tert-butyl substituents. The second ligand with the donor atoms O(1)/N(7) and O(20)/N(14) has a mixed-valence character [L1˙A]3−, one part with an iminobenzoquinonate(1−) radical and the other part displaying the characteristic features of an O,N-coordinated aromatic amidophenolate dianion with C(1)–O(1) 1.323 Å, C(6)–N(7) 1.381 Å and six equidistant C–C at 1.405 (±0.020) Å of the phenyl ring containing the tert-butyl substituents; both nitrogen atoms are sp2 hybridized. Thus the ligand contributes three negative charges towards electroneutrality. The third ligand with O(81), N(87), O(100), N(84) exhibits features which are very similar to the second one just described; one part consists of the amidophenolate(2−) ring and the other, the iminobenzosemiquinone(1−) form. Thus complex 1 consists of four iminobenzosemiquinone(1−) π-radicals, and two O,N-coordinated aromatic amidophenolatedianion(2−). Hence each manganese ion must be in +4 oxidation state and is coordinated to (i) two phenolate oxygens and two imine-nitrogens from two o-iminobenzosemiquinonate(1−) radicals, and (ii) one phenolate oxygen and one amide-nitrogen from the amidophenolate dianion(2−), thus rendering the complex to be neutral. Summarily, the crystal structure of 1 and its physical properties (loc. cit.) are in excellent agreement with a dimanganese(IV)-formulation with four radicals.
Dark brown crystals of complex 3 afforded by diethyl ether–methanol solution was subjected to single-crystal X-ray diffraction study at 100(2) K. Selected bond lengths and angles are summarized in Table 2. The neutral molecule in crystals of 3, shown in Fig. 3, contains two O,N-coordinated o-iminobenzosemiquinonate(1−) π radical ligands, as is clearly borne out by the observation that (i) both nitrogens, N(7) and N(67), are sp2 hybridized and not protonated, (ii) the six-membered ring of the iminobenzosemiquinonate part displays the typical quinoid distortions, comprising a short, a long, and another short C–C bond followed by three long ones, (iii) the C–O and C–N bond lengths are short, approaching double bonds. The third ligand, in contrast, is N-deprotonated; the corresponding nitrogen N(37) is sp2 hybridized (the sum of three bond angles C36–N37–C38 118.2(2), C35–N37–Mn(1) 113.0(1) and C38–N37–Mn(1) 126.8(1) is 358.0°), thus providing an o-amidophenolate dianion(2−) ligand. The six C–C bonds of the amidophenolate ring are equidistant at 1.40 ± 0.015 Å and C(31)–O(31) and C(36)–N(37) distances at 1.330(2) and 1.384(2) Å, respectively are long. Thus, the central Mn(1) must be ascribed to +IV (d3) oxidation level. The observed Mn–O and Mn–N bond distances, av. 1.90 (±0.035) and 1.94 (±0.025) Å, respectively, support this view; they are shorter than the comparable reported values for Mn(III)-complexes13 and similar to those for Mn(IV)-complexes.2j Additionally, the pseudo-octahedral O3N3Mn polyhedron in complex 3 does not show any Jahn–Teller distortion. The crystal structure of complex 3 is in excellent agreement with the charge distribution of [MnIV(L3A)(L3˙)2] and is identical as expected with that for the Mn(IV)-complex with the ligand H2L2, complex 2, reported earlier by us.12
|
|
| |
|---|---|---|---|
| Mn(1)–O(31) | 1.8676(13) | O(31)–Mn(1)–O(1) | 178.12(6) |
| Mn(1)–O(1) | 1.9050(13) | O(31)–Mn(1)–N(37) | 83.45(6) |
| Mn(1)–N(37) | 1.9189(16) | O(1)–Mn(1)–N(37) | 96.27(6) |
| Mn(1)–O(61) | 1.9394(13) | O(31)–Mn(1)–O(61) | 89.43(6) |
| Mn(1)–N(7) | 1.9522(16) | O(1)–Mn(1)–O(61) | 90.87(6) |
| Mn(1)–N(67) | 1.9664(17) | N(37)–Mn(1)–O(61) | 172.84(6) |
| C(1)–O(1) | 1.303(2) | ||
| C(1)–C(6) | 1.428(3) | ||
| C(1)–C(2) | 1.428(3) | ||
| C(2)–C(3) | 1.375(3) | ||
| C(3)–C(4) | 1.431(3) | ||
| C(4)–C(5) | 1.368(3) | ||
| C(5)–C(6) | 1.418(3) | ||
| C(6)–N(7) | 1.348(2) | ||
| N(7)–C(8) | 1.425(2) | ||
| O(31)–C(31) | 1.330(2) | O(31)–Mn(1)–N(7) | 96.61(6) |
| C(31)–C(32) | 1.412(3) | O(1)–Mn(1)–N(7) | 81.56(6) |
| C(31)–C(36) | 1.413(3) | N(37)–Mn(1)–N(7) | 96.08(7) |
| C(32)–C(33) | 1.390(3) | O(61)–Mn(1)–N(7) | 85.44(6) |
| C(33)–C(34) | 1.411(3) | O(31)–Mn(1)–N(67) | 96.44(6) |
| C(34)–C(35) | 1.385(3) | O(1)–Mn(1)–N(67) | 85.44(6) |
| C(35)–C(36) | 1.404(3) | N(37)–Mn(1)–N(67) | 98.95(7) |
| C(36)–N(37) | 1.384(2) | O(61)–Mn(1)–N(67) | 81.07(6) |
| N(37)–C(38) | 1.429(2) | N(7)–Mn(1)–N(67) | 161.09(7) |
| C(6)–N(7)–C(8) | 120.90(16) | ||
| C(6)–N(7)–Mn(1) | 113.47(13) | ||
| C(8)–N(7)–Mn(1) | 124.52(12) | ||
| C(1)–O(1)–Mn(1) | 115.18(12) | ||
| C(61)–O(61) | 1.297(2) | C(31)–O(31)–Mn(1) | 115.33(12) |
| C(61)–C(62) | 1.426(3) | C(36)–N(37)–C(38) | 118.18(16) |
| C(61)–C(66) | 1.441(3) | C(36)–N(37)–Mn(1) | 113.00(12) |
| C(62)–C(63) | 1.379(3) | C(38)–N(37)–Mn(1) | 126.78(12) |
| C(63)–C(64) | 1.424(3) | C(61)–O(61)–Mn(1) | 114.57(12) |
| C(64)–C(65) | 1.373(3) | C(66)–N(67)–C(68) | 121.10(17) |
| C(65)–C(66) | 1.414(3) | C(66)–N(67)–Mn(1) | 113.12(12) |
| C(66)–N(67) | 1.353(2) | C(68)–N(67)–Mn(1) | 124.12(13) |
| N(67)–C(68) | 1.422(2) | ||

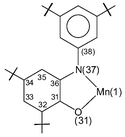
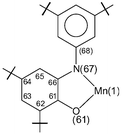
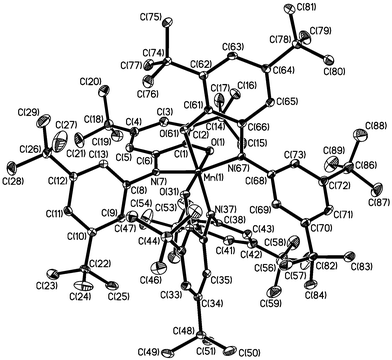 | ||
| Fig. 3 Perspective view of the neutral molecule in 3. | ||
It is obvious that the structures of other complexes, 4–7, are identical to those of 2 and 3 and hence, we have refrained from determining the crystal structures of them.
Magnetic susceptibility and EPR data
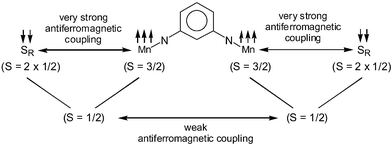
In order to establish the electronic ground states for complexes 1–7, variable–temperature (2–290 K) magnetic susceptibility measurements on powdered samples of 1 and 3 as a representative of the mononuclear complexes 2–7 were performed by using a SQUID magnetometer in an external magnetic field of 1.0 T.Fig. 4 shows the temperature dependence of the magnetic moment, μeff, and of the susceptibility χM for compound 1. The μeff-value of 2.72 μB (χMT = 0.924 cm3 mol−1 K) at 290 K does not change drastically till 30 K to reach a value of 2.44 μB (χMT = 0.744 cm3 mol−1 K). These values are very close to 2.45 μB, expected for two uncoupled S = 1/2 states and indicate clearly strong antiferromagnetic interaction operative between the paramagnetic Mn(IV) (S = 3/2) and the radical (S = 1/2) centers. On each side of the m-phenylene-spacer there are two radicals and one Mn(IV) center, which are strongly antiferromagnetically coupled yielding thus two S = 1/2 residual fictive spins for the whole molecule. This qualitative picture is schematically shown in Scheme 3.
 | ||
| Fig. 4 Plots of μeff and χMvs. T for complex 1. The solid lines represent the simulations using the parameters given in the text. | ||
 | ||
| Scheme 3 | ||
We have performed susceptibility measurements by collecting 24 data points at an interval of 1 K in the temperature range of 2.0–25.0 K and 27 data points in the range 30.0–290.0 K, which are shown as a plot of χMvs. T, in which a maximum at 5 K with μeff
= 1.58 μB
(χMT
= 0.31 cm3 mol−1 K) is observed. This is in accord with the qualitative picture (Scheme 3) and represents the weak antiferromagnetic coupling yielding an St
= 0 ground state resulting from the two fictive S
= 1/2 states. The susceptibilities of 1 were calculated by using the spin-Hamiltonian:
Ĥ
=
−2J![[S with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0053_20d1.gif) 1·2 1·2 |
Fig. 5 shows the temperature dependence of the effective magnetic moment μeff for complex 3. On lowering the temperature μeff decreases only from 1.80 at 290 K to 1.70 μB at 15 K. This behaviour indicates a ground state of St
= 1/2 for 3. As the overall magnetic behaviour is dominated by much stronger antiferromagnetic interactions between ligand radicals and metal-based d-electrons, the decrease of μeff on lowering the temperature could well be fitted by a single coupling constant J
=
J12
=
J23
[coupling between the radical centers (S
= 1/2) and Mn(IV) center (S
= 3/2)] with J13
[coupling between the radical centers (S
= 1/2)] set to zero. Using the Hamiltonian:
| H = 2J(S1·S2 + S2S3) − 2J13(S1·S3) |
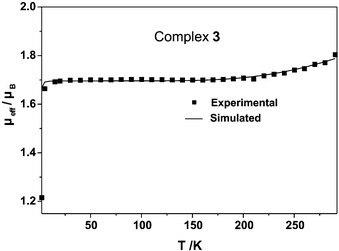 | ||
| Fig. 5 Temperature dependence of the magnetic moments μeff of complex 3. The solid line represents the best fit of the experimental data using the parameters given in the text. | ||
The manganese-based St = 1/2 ground state for 3 has also been established by X-band EPR spectroscopy. Fig. 6 shows the EPR spectrum of 3 in CH2Cl2 solution at 298 K. The spectrum displays an isotropic signal at giso = 2.009 with hyperfine coupling to the 55Mn nucleus (I = 5/2) of Aiso = 106.6 G, and in addition, superhyperfine coupling to three 14N (I = 1) donor atoms and two protons (I = 1/2) was clearly detected, A(14N) = 4.29 G, A(1H) = 5.15 G, A(1H) = 2.83 G. This spectrum resembles closely to that reported for [MnIV(L2˙A)(L2˙)2].12
 | ||
| Fig. 6 X-Band EPR spectrum of 3 in CH2Cl2 at 298 K. Conditions: frequency 9.44 GHz; power 10 mW; modulation amplitude 8 G. | ||
The EPR-spectrum of 1 at 10 K is described later under the Catalytic catecholase activity section.
Electrochemistry and spectroelectrochemistry
Cyclic voltammograms (CV) and square wave voltammograms (SQW) of complex 1 and the analogous CuII2(L1˙˙)2, FeIII2(L1˙˙)3 and CoIII2(L1˙˙)3, whose structures have been reported earlier,10 and which have been included here to support the assignment of the Mn complexes, have been recorded in CH2Cl2 solutions containing 0.1 M TBAPF6 as supporting electrolyte at a glassy carbon working electrode and using a Ag/AgNO3 reference electrode. Ferrocene was added as an internal standard after completion of a set of experiments, and potentials are referenced vs. the ferrocenium/ferrocene couple (Fc+/Fc). Coulometric experiments were performed at appropriate potentials (at −25 °C) to determine the number of electrons/molecule transferred in the redox process under investigation. During coulometry electronic spectra were recorded in order to obtain information about the mechanism of the redox process. The CV and SQW voltammograms recorded revealed that in each complex a number of oxidations and reductions are discernible, the redox potentials of which are compiled in Table 3. Figs. 7 and 8 show CV and SQW voltammograms for the MnIV2 (1) and FeIII2-complexes, respectively.| Complex | E1/22(ox) | E1/21(ox) | E1/21(red) | E1/22(red) | |||
|---|---|---|---|---|---|---|---|
| irr Redox process is irreversible on the time scale of voltammetric experiments (e.g. in CV at scan rates up to 0.8 V/s); peak potentials are given. a Two electron processes. | |||||||
| CuII2(L1˙˙)2 | +0.44a | −0.120 | −0.210 | −0.960 | −1.13 | −1.56 | |
| CoIII2(L1˙˙)3 | +0.247 | +0.174 | −0.277 | −0.431 | −1.24irr | ||
| FeIII2(L1˙˙)3 | +0.481 | +0.382 | −0.108 | −0.288 | −1.114 | −1.278 | −1.557irr |
| 1 MnIV2(L1˙A)2(L1˙˙) | +0.575 | +0.101irr | −0.120 | −0.341 | −1.019 | −1.234 | −1.632irr |
 | ||
| Fig. 7 Cyclic voltammograms (bold) and square-wave voltammograms (dotted) of 1 in CH2Cl2 at a scan rate of 50 mV s−1 and at a frequency of 20 Hz, respectively. | ||
 | ||
| Fig. 8 (a) Cyclic voltammograms (bold) and square-wave voltammograms (dotted) of FeIII2(L1˙˙)3 in CH2Cl2; (b) electronic spectral changes during the coulometric two-electron oxidation of FeIII2(L1˙˙)3. | ||
It is seen from column E1/21(ox) of the table that the dimeric complexes, can be twice reversibly oxidized at two low potentials of close vicinity (E1/21(ox); range −0.12 to −0.43 V vs. Fc+/Fc). A second set of pairwise, reversible oxidations occur with the iron and cobalt complexes at somewhat higher potentials (E1/22(ox); range +0.2 to +0.5 V), with the same pattern as those at E1/21(ox). Also, at negative potentials (E1/21(red); at −0.96 to −1.3 V) similar reversible reduction couples occur with copper, iron and manganese (complex 1) complexes.
Coulometric experiments with the oxidations show that a charge of two Faradays/mol is transferred for each couple, i.e. each single peak corresponds to an one electron transfer process. We can assign all these couples firmly to ligand-centered oxidations of two radicals to the corresponding quinoid forms of the ligand as (i) the redox potentials as well as the electronic spectra (vide infra), of the oxidized species are similar for all complexes irrespective of the nature of the central metal ion, and (ii) with the respective analogous monomeric complexes it was demonstrated earlier11d that their oxidations are ligand-centered, and they occur at potentials which are very similar to those of the present dimeric forms.
The close vicinity of the redox potentials for each couple shows that there is only little interaction between the oxidation sites, i.e. the two radicals, which conforms with the magnetic measurements. Therefore we assign the two oxidation sites of a couple to two radicals at the same ligand, i.e. each is coordinated to two metal ions.
With the dicopper(II) compound, a single broad CV wave is observed in the range of E1/22(ox), with a peak separation between oxidation and re-reduction peaks which became increasingly higher with higher scan rates. This behaviour is typical for sluggish heterogeneous electron transfer kinetics, and under these conditions two steps cannot be distinguished. During coulometry, the oxidized product formed at E1/22(ox) was found to be not fully stable, but about two electrons per complex molecule passed. Therefore we assign the same mechanism to the oxidation processes for the dimeric complexes. With complex 1 (MnIV2), the oxidative wave in the second range is only very poorly developed and irreversible, but in view of the similarity of the structure of 1, and the potential at which the oxidation takes place, it might occur at the same place.
Reductions, E1/21(red), take place at around −1 V, again pairwise with Cu(II), Fe(III) and Mn(IV). The interpretation is analogous to the oxidations: reduction of two radicals of the same ligand to its dianionic form. As to the number of electrons, with the reductions we only can compare their peak heights in the voltammograms with the peak heights for oxidation, because none of the products of E1/21(red) (and a forteriori of E1/22(red)) was sufficiently stable to allow the collection of reliable coulometric data. The reductions at E1/22(red) are irreversible in the time scale of the voltammetric experiments (with exception of a single reversible wave at −1.56 V with dicopper(II)) and do not warrant further discussion.
The UV/VIS spectral changes recorded (Fig. 1 for complex 1 and Fig. 8 for the analogous FeIII2(L1˙˙)3) during coulometry showed very similar features with all oxidations of all complexes: the appearance of an broad absorption band extending from 400–600 nm with a maximum at 500 nm. With CoIII2- and FeIII2-complexes, which could be four times reversibly oxidized, the spectral changes maintain almost isosbestic behaviour over all four oxidations. This confirms that the underlying oxidation process is the same for all oxidations, namely formation of quinoid structures. It was shown earlier11d that generation of such structures is accompanied by the formation of a new absorption band in the above wavelength range.
CV and SQW experiments were also carried out with the monomeric, two radical containing MnIV complexes 2–7, which differ only in the meta-substituents at the aniline moieties. In analogy to the dimeric complexes we expect now two ligand-centered oxidations and two ligand-centered reductions for oxidation and reduction of the two radicals, respectively. The results listed in Table 4 show that this is indeed the case. It is seen that the nature of the substituents causes a shift of the reversible redox potentials in the expected manner: electron-withdrawing groups facilitate the reductions and impede the oxidations and vice versa. The total shifts introduced by the substituents in the meta positions amount to 0.56 V for the reversible reductions and 0.49 V for the reversible oxidations, which correspond to alterations of the free energy by 54 and 47 kJ mol−1, respectively. This difference is not reflected in the rate constant ksub for the catecholase activity described later; there is no correlation visible between the rate constants and the electronic properties of the substituents (or the reduction potential). Therefore we conclude that the rate limiting step in the reaction sequence of the catalysis is not the electron transfer.
| Complex | Substituent | E1/22(ox)a | E1/21(ox)b | E1/21(red)b | E1/22(red)b |
|---|---|---|---|---|---|
| a The redox processes described by E1/22(ox) and E1/22(red) are all irreversible on the time scale of the voltammetric experiments and peak potentials at a scan rate of 0.2 V s−1 are given.b The values of E1/21(ox) and E1/21(red) refer to reversible processes, with the exception of -H, (complex 2) where already the first reduction is irreversible. | |||||
| 2 | H | +0.46 | +0.41 | −1.05 | −1.35 |
| 3 | C(CH3)3 | +0.52 | −0.48 | −1.24 | −1.84 |
| 4 | CF3 | n.d. | +0.01 | −0.68 | −1.12 |
| 5 | CH3 | +0.50 | −0.48 | −1.11 | −1.38 |
| 6 | Cl | +0.75 | −0.13 | −0.76 | −1.56 |
| 7 | OCH3 | +0.48 | −0.40 | −1.03 | −1.35 |
Catalytic catecholase activity
We have examined the catalytic activity of complexes 1–7 for oxidation of 3,5-di-tert-butylcatechol (3,5-DTBC) by O2 to o-quinone(3,5-DTBQ) in air-saturated dichloromethane according to the reaction: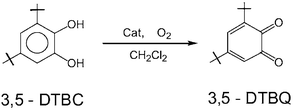
The reactivity studies were performed in dichloromethane solution because of the good solubility of the complexes as well as of the substrate and of its product. No base was added to the solutions to avoid the autooxidation of the substrate by air in presence of base. The above catalytic oxidation does not proceed in the absence of the catalysts, the manganese(IV) complexes 1–7.
Prior to a detailed kinetic study we evaluated quantitatively the catalytic activity of the complexes. For this purpose, 5 × 10−6 mol of a complex (catalyst) in 25 ml of dichloromethane was treated with 50 equivalents of 3,5-DTBC and stirred in air at ambient temperature. The products were analyzed by liquid chromatography (LC). The retention time in LC for 3,5-DTBC and 3,5-DTBQ was found to be 8.0 and 10.5 min, respectively (column: Luna-5-phenylhexyl, eluent: methanol–water (3 ∶ 1), rate 0.8 ml min−1) as observed for commercially available pure compound. It was found that for all the oxidative reactions involving 3,5-DTBC as the substrate, 3,5-DTBQ was the only oxidized product.
The dinuclear copper(II) complex of the ligand H4L1, CuII2(L1˙˙)2, described earlier by us,10 was used also as a catalyst. Stirring in air a dichloromethane solution of CuII2(L1˙˙)2 and 3,5-DTBC in the ratio of 1 ∶ 50 yielded at the end of 24 h only ∼16% 3,5-DTBQ and other peaks in LC, which are assigned to the decomposition products of the catalyst CuII2(L1˙˙)2 by LC-MS coupling measurements. Thus the turnover number turned out to be only eight indicating that the dicopper complex is not a good catalyst for such 2e-oxidation of 3,5-DTBC to 3,5-DTBQ. This result might be indicative of non-involvement of any radical mechanism and is in line with the presently accepted mechanism for the dicopper enzyme catechol oxidase.5
When complex 1, MnIV2(L1˙A)2(L1˙˙), and 3,5-DTBC in the ratio 1 ∶ 50 was dissolved in CH2Cl2 in the presence of air, the dark brown color changes slowly to yellow and then finally to red. Analysis of the resulting solution after 24 h shows a 100% conversion of 3,5-DTBC to 3,5-DTBQ. On increasing the amount of 3,5-DTBC the turnover number (TON) increases and the maximum turnover reached after 24 h is 500, indicating that complex 1 is a good catalyst for the two-electron oxidation of 3,5-DTBC to 3,5-DTBQ and thus mimicking the function of the dicopper-protein catechol oxidase.
Reaction kinetics were performed by observing the change in absorbance at 408 nm, which is characteristic of 3,5-DTBQ in CH2Cl2. In a typical reaction the catalyst was dissolved in CH2Cl2 and the substrate, 3,5-DTBC was added, and the resulting solution was stirred in presence of air; the resultant spectral change as a function of time was measured. For calculation of rate constants, initial rate method was used and the velocity of the reaction was obtained from the slope of the tangent to the absorbance vs. time curve. This procedure was used for all kinetic measurements.
The kinetics of the oxidative reaction at a constant catalyst concentration ([complex 1] = 2 × 10−7 mol per 10 ml) in dichloromethane (10 ml) were investigated with variation of the substrate concentration, (2–20) × 10−6 mol per 10 ml. As an example, the electronic spectra for [cat]/[subs] = 1/30 shows an increase in the intensity of the peak at 408 nm with time and are depicted in Fig. 9(a). The difference in absorbance ΔA at 408 nm, was plotted against time to obtain the velocity (ro) for that particular catalyst to substrate concentration ratio (Fig. 9(b)). From variation of the velocity as a function of substrate concentration, the rate of the reaction with respect to substrate was found to be first order with ksub = 9.13 × 103 mol−1 min−1. Similar procedures with varying catalyst concentrations keeping a constant substrate concentration (5 × 10−6 mol per 10 ml) yielded different velocities (r’o), thus leading to the result for the order of the reaction with respect to the catalyst.
 | ||
| Fig. 9 (a) Electronic spectral change of a solution of 1 (2 × 10−7 mol) and 3,5-DTBC (6 × 10−6 mol) in CH2Cl2 (10 ml); (b) a plot of the difference in absorbance (ΔA) vs time to evaluate the velocity of the catalysis. | ||
A first-order dependence with kcat
= 2.26 × 105 mol−1 min−1 was obtained, resulting in an overall rate-law:
| Rate = k[catalyst][substrate] |
Similar kinetic experiments were performed with other Mn(IV)-radical complexes, 2–7, and the same rate-law was observed. The kinetic data together with the relevant reduction potentials Ered are summarized in Table 5.
| Complex | Substituent R | ERed/V (vs. Fc+/Fc) | 10−4ksubs/mol−1 min−1 | Maximum TONa/24 h | Catalytic activitya/min−1 |
|---|---|---|---|---|---|
| a Turnover number = [number of mol of product]/[number of mol of catalyst].b Activity expressed as μmol substrate oxidized per mg catalyst. | |||||
| 1 MnIV2(L1˙A)2(L1˙˙) | — | −1.02, 1.23 | 0.9 | 500 | 0.210 |
| 2 MnIV(L2A)(L2˙)2 | H | −1.05, −1.35 | 1.5 | 48 | 0.355 |
| 3 MnIV(L3A)(L3˙)2 | C(CH3)3 | −1.24, −1.84 | 2.8 | 169 | 0.920 |
| 4 MnIV(L4A)(L4˙)2 | CF3 | −0.68, −1.12 | 4 | 48 | 0.247 |
| 5 MnIV(L5A)(L5˙)2 | CH3 | −1.11, −1.38 | 4.6 | 84 | 0.569 |
| 6 MnIV(L6A)(L6˙)2 | Cl | −0.76, −1.56 | 5.3 | 45 | 0.272 |
| 7 MnIV(L7A)(L7˙)2 | OCH3 | −1.03, −1.35 | 9.1 | 40 | 0.260 |
To throw more light into this two-electron oxidation process, we performed additional experiments to demonstrate the following salient points: (i) the radicals coordinated to the manganese(IV) centers are participating in the conversion process; (ii) oxygen of air is essential for the catalysis, and (iii) the stoichiometry and fate of oxygen involved.
One equivalent of the dinuclear complex 1 and 10 equivalents of the substrate 3,5-DTBC in CH2Cl2 were allowed to react for 1 h under argon in a glove box.
An aliquot of this reaction solution was divided into two parts, (i) to determine by LC the amount of 3,5-DTBQ formed in absence of air, and (ii) to measure an EPR spectrum for comparison with that of 1 in CH2Cl2 at 10 K.
From the quantitative LC-study, also under argon, formation of only 2 equivalents of 3,5-DTBQ could be observed, indicating that four radical electrons of 1 (see EPR) are involved.
The X-band EPR spectra at 10 K of 1 in CH2Cl2 and of the frozen solution after the reaction with 3,5-DTBC in glove box are shown in Fig. 10 (ESI†). The spectrum of only 1 consists of a broad signal centered at g ≈ 2.0 and a weak 11-line hyperfine signal at g ≈ 4.05. The assignment of the spectrum is certainly not straightforward and needs further study. A more complicated EPR spectrum was observed when 1 was reacted with two equivalents of DTBC under argon. The spectrum shows broad peaks between g ≈ 20 and g ≈ 4 along with hyperfine signals centered at g ∼ 2.0 presumably due to the formation of two S = 3/2 states at each manganese center of the dimer.
The remaining reaction solution containing 1 from the glove box was subjected to a quantitative LC-study after long exposure to air to ascertain that 10 equivalents of 3,5-DTBQ have been formed, indicating the essentiality of aerial oxygen for the catalytic process.
The results of the same type of experiments with complex 2, a mononuclear Mn(IV) (S = 3/2) with two radicals, are the following: (i) one equivalent of 3,5-DTBQ is formed in glove box in absence of aerial oxygen, i.e. involvement of only two radical electrons; (ii) the EPR spectrum of the solution containing 2 and 3,5-DTBC from the glove-box exhibits peak centered at g ∼ 3.15 and a low intensity peak at g ∼ 2.0, which could be assigned to an S = 3/2 system due to the Mn(IV)-center, (iii) long exposure to air of the reaction solution results in quantitative oxidation of added 3,5-DTBC to 3,5-DTBQ unambiguously showing the necessity of aerial oxygen for the catalysis.
Oxygen-uptake measurements
As complex 3, characterized by most of the methods available to us, exhibits highest catalytic activity (Table 5), we choose 3 for oxygen uptake measurements which were performed at −25 °C and at −10 °C. These low-temperatures were selected considering the high vapor pressure of the solvent dichloromethane and to reduce, as much as possible, decomposition of hydrogen peroxide, which might be expected as a product of the 2e-oxidation of 3,5-DTBC to 3,5-DTBQ by oxygen.At −25 °C, one equivalent of oxygen was needed to convert all (0.5 mmol) 3,5-DTBC to 3,5-DTBQ; the reaction was followed by using a gas-burette method for 320 min till saturation of O2-uptake occurred. The presence of hydrogen peroxide15 in the catalytic solution was confirmed both by detection of titanyl(IV)–peroxo complex and by liberation of iodine from an acidic potassium iodide solution. Therefore, O2-uptake measurements at −25 °C results in the following stoichiometry for the reaction:

At −25 °C, catalase-activity of 3 is very slow and H2O2 formed did not undergo appreciable decomposition to H2O and O2. However, at −10 °C, 0.5 equivalent of oxygen was required in order to convert all 3,5-DTBC (0.5 mmol) to 3,5-DTBQ; saturation of O2-uptake occurred after 152 min reaction-time. At −10 °C formed hydrogen peroxide decomposed to H2O and O2, and the latter was also taken up by the substrate to reduce the total amount of O2 consumed. At −10 °C and hence at room temperature the stoichiometry changes to:
| 2(3,5-DTBC) + 2O2
→ 2(3,5-DTBQ) + 2H2O2 |
| 2H2O2→ 2H2O + O2 |
| 2(3,5-DTBC) + O2
→ 2(3,5-DTBQ) + 2H2O |
Mechanism
It is tempting to propose a mechanism based on the electrochemical data, O2-uptake measurements and the experiments described earlier. Additionally, the EPR data show that the intermediate could be the two-electron reduced form, i.e. involvement of both radicals in the oxidation, of the Mn(IV)-complex. With incoming oxygen of air the reduced ligands get re-oxidized to the radical form of the complex which carries out the next turnover.The proton transfer followed by the electron transfer as implied in Scheme 4 may occur also as hydrogen atom transfer. However, the proposed “on–off” mechanism of the radicals without redox-participation of the metal center seems to be very common for the catalysis involving such metal-radical complexes.16
 | ||
| Scheme 4 | ||
The results of the catalytic oxidations are presented in Table 5. Despite the fact that no clear correlation between the redox potentials of the catalysts, the Mn(IV)-complexes, and the velocity of the catecholase activity was revealed for 1–7, a rough correlation is envisagable between the catalytic activity and the substituents on the aniline ring. Leaving aside complex 1 because of its dinuclear nature, the catalytic activities of the complexes range from a high 0.920 μmol substrate (mg catalyst min)−1 for the di-tert-butyl substituted aniline ring, complex 3, to a low of 0.247 μmol substrate (mg catalyst min)−1 for the ditrifluoromethyl-substituted ligand, complex 4. It is surprising that the value of 0.210 is so low for the dinuclear complex 1, considering that it contains two additional radicals. However, as a general rule-of-thumb it can be concluded that electron-withdrawing substituents reduce the activity, whereas electron-pushing groups make the catalyst more active by increasing the electron density on the aniline-nitrogen, which presumably thus positively influences the hydrogen bond-formation with the substrate, as has been proposed in the pre-equilibrium step prior to oxidation (Scheme 4). Apparently, the polarization effect resulting from the effect on the resonance interaction of the highly polarizing meta-substituents, R, with the aniline ring is operative.17 Hydrogen-bond formation between the catalyst and the substrate seems to be the driving force for the equilibrium shown in Scheme 4.
However, it is clear from Table 5 that there is a complex interplay between the structural and electronic properties (as evidenced by their electrochemical properties) of the manganese(IV) complexes in determining the hydrogen bond-formation in the equilibrium described in Scheme 4, essential for the oxidative process. For this series of complexes, it appears that the redox properties are secondary in importance to the hydrogen bond-formation in determining the catalytic properties of the complexes.
Oxidative C-C coupling of hindered phenols to diphenoquinones: conversion of 2,6-di-tert-butylphenol to 3,3′-5,5′-tetra-tert-butyldiphenoquinone
As the oxidative carbon–carbon coupling of hindered phenols leading to diphenoquinones is radical-mediated reactions, we have studied complex 1 containing radicals for oxidative studies with 2,6-di-tert-butylphenol. When 2,6-di-tert-butylphenol (DTBP) (2 mmol) is added to complex 1 (0.01 mmol) in dichloromethane–methanol (1 ∶ 1) solvent mix (50 ml) the dark brown colour slowly turns to red in presence of air. It was found that after 48 h 100% of the phenol was converted to 3,3′-5,5′-tetra-tert-butyldiphenolquinone (TTBD), thus resulting in a turnover number of 200.
For the reaction a maximum turnover number of 1284/48 h was observed. The course of the reaction was followed by optical spectroscopy, as TTBD shows a characteristic peak in the electronic spectrum at 425 nm. Such changes in electronic spectra with different time intervals were monitored by removing the catalyst from the reaction solution using a column of Amberlyst-resin and are depicted in Fig. 11 (ESI†). The resultant plot of absorbance vs. time indicates a complicated nature of the kinetics and warrants a more detailed study.
Concluding remarks
To conclude, the following points of this study deserve particular attention:As an obvious progression of our earlier reports9–12 dinuclear transition metal–iminosemiquinone complexes containing as high as six radicals can be rationally synthesized. A dimanganese(IV) complex containing four radicals has been structurally and spectroscopically characterized. On each side of the m-phenylene spacer in 1 there are two radicals (S = 1/2) and one Mn(IV) center (SMn = 3/2), which are strong antiferromagnetically coupled yielding thus two S = 1/2 residual fictive spins even at room temperature for the whole molecule. Antiferromagnetic exchange interaction between these two fictive residual spins is weak with a singlet–triplet gap of ∼7 cm−1. The strong antiferromagnetic coupling between the radical ligands and the Mn(IV) ions can be ascribed to the large covalency of the Mn–N bonds in 1.
A series of disubstituted 2-anilino-4,6-di-tert-butylphenol ligands has been prepared to synthesize six new radical-containing Mn(IV) compounds. As expected, these Mn(IV) compounds, 3–7, exhibit strong antiferromagnetic coupling yielding an St = 1/2 ground state, like that for 2.12
Electrochemical measurements indicate redox processes which are mainly ligand-centered. Thus for each complex a number of oxidations and reductions, resulting in quinoid and phenolate forms of the ligands, respectively, are discernible.
The investigation emphasizes that the Mn(IV)–radical complexes 1–7 can catalyze the oxidation of catechol with molecular oxygen as the sole oxidant to afford quinone in excellent yield under mild conditions to mimic the function of the copper-containing enzyme catechol oxidase.

Although the nature of the substituents on the aniline moiety of the ligand causes a shift of the reversible redox potentials in the expected manner, i.e. electron-withdrawing groups facilitate the reductions and impede the oxidations and vice versa, this change in potentials is not reflected in the rate constant for the catecholase activity, thus indicating that the rate-determining step in the reaction sequence of the catalysis (Scheme 4) probably is not the electron transfer.
An “on–off” mechanism of the radicals without redox-participation of the metal center manganese has been proposed for the catalytic process, in which hydrogen-bond formation between the catalyst and the substrate catechol seems to be the driving force for the oxidation. It appears that the redox properties are secondary in importance to the hydrogen-bond formation in determining the catalytic properties of the complexes.
Complex 1 has been found to be a good catalyst for oxidative C–C coupling of hindered phenols to diphenoquinones. Thus, 2,6-di-tert-butylphenol (DTBP) was oxidized by aerial oxygen quantitatively to 3,3′-5,5′-tetra-tert-butyl-4,4′-diphenoquinone (TTBD) by using complex 1 as a catalyst.
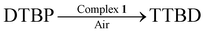
Thus, as is expected, a radical-mediated reaction is catalyzed by 1.
Experimental
Materials and physical measurements
Commercial grade chemicals were used for the synthetic purposes and solvents were distilled and dried before use. Fourier transform IR spectroscopy on KBr pellets was performed on a Perkin-Elmer 2000 FT-IR instrument. Solution electronic spectra were measured on a Perkin-Elmer Lambda 19 spectrophotometer. Mass spectra were recorded either in the EI or ESI (in CH2Cl2) mode with a FinniganMAT 95 or 8200 spectrometer. Magnetic susceptibilities of the polycrystalline samples were recorded on a SQUID magnetometer (MPMS, Quantum Design) in the temperature range 2–290 K with an applied field of 1 T. Diamagnetic contributions were estimated for each compound by using Pascal's constants. X-Band EPR spectra were recorded on a Bruker ESP 300E spectrometer equipped with a helium flow cryostat (Oxford Instruments ESR 910), an NMR field probe (Bruker 035M), and a microwave frequency counter HP5352B. Spin-Hamiltonian simulations of the EPR spectra were performed by iteration of the (an)isotropic g values, hyperfine coupling constants, and line widths. GC of the organic products during catalysis were performed either on HP 5890 II or HP 6890 instruments using RTX-1701 15 m S-41 columns. GC-MS was performed using the above columns coupled with a HP 5973 mass spectrometer with mass selective detector. NMR spectra were measured using Bruker ARX 250 (250 and 63 MHz for 1H and 13C NMR, respectively).Cyclic voltammograms, square-wave voltammograms, and coulometric experiments were performed using an EG&G potentiostat/galvanostat. Simulations of the cyclic voltammograms were obtained using the program DigiSim 3.0 (Bioanalytical Systems, Inc., West Lafayette, IN).
LC analysis of the reaction solutions during catalysis were performed on HPLC instrumentation using a Gilson M305 pump, and the Diode-Array-Detector (DAD) SPDM 10 AV (Shimadzu Corporation). MeOH and water in the ratio 3 ∶ 1 with flow velocity 0.8 ml min−1 was used as eluent through a Luna-5 phenylhexyl column.
Catalytic oxidation of 3,5-di-tert-butylcatechol (3,5-DTBC)
In dichloromethane (25 ml), the Mn(IV) complexes 1–7, were dissolved and 3,5-DTBC was added and the solution was stirred in room temperature for 24 h. The solution was then subjected to liquid chromatographic studies.For kinetic measurements, 1 ml from the reaction solution was diluted to 10 ml with dichloromethane. The change in the concentration of the product, 3,5-di-tert-butyl-o-benzoquinone, was monitored by electronic spectroscopy.
Catalytic oxidation of 2,6-di-tert-butylphenol
In a dichloromethane/methanol solvent mixture (1 ∶ 1; 50 ml) complex 1 was dissolved. To it 2,6-di-tert-butylphenol was added and the solution was stirred for 48 h. The solution was then filtered and the filtrate was evaporated to dryness. 5 ml of methanol were added to dissolve the excess 2,6-di-tert-butylphenol and the methanolic solution was subjected to GC studies with the presence of hexadecane (C16) as standard.For measuring the progress of the reaction, 50 μl from the aliquot was passed through an Amberlyst cationic ion-exchanger and washed with 10 ml (2 × 5) dichloromethane. The change in the concentration of product 3,3’-5,5’-tetra-tert-butyldiphenoquinone, was monitored by optical spectroscopy.
X-Ray crystallographic data collection and refinement of the structures
Dark brown single crystals of 1 and 3 were coated with perfluoropolyether, picked up with glass fibers, and mounted on a Nonius Kappa-CCD diffractometer equipped with a cryogenic nitrogen cold stream at 100 K. Graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å) was used. Intensity data were corrected for Lorentz and polarization effects. The intensity data were not corrected for absorption due to a low absorption coefficient. The SHELXTL software package18 was used for solution, refinement, and artwork of the structure, and neutral atom scattering factors of the program were used. All structures were solved and refined by direct methods and difference Fourier techniques. Non-hydrogen atoms were refined anisotropically, and hydrogen atoms were placed at calculated positions and refined as riding atoms with isotropic displacement parameters. A dichloromethane solvent molecule in 1 was found to be not fully occupied. The C and Cl atoms were anisotropically refined with a given occupation factor of 0.5. Details of the data collection and structure refinements are summarized in Table 6.| 1 | 3 | |
|---|---|---|
| Empirical formula | C102H132Mn2N6O6·0.5CH2Cl2 | C84H123MnN3O3 |
| Mr | 1690.48 | 1277.79 |
| T/K | 100(2) | 100(2) |
| λ/Å | 0.71073 | 0.71073 |
| Crystal system | Monoclinic | Monoclinic |
| Space group | P21/n | P21/c |
| Unit cell dimensions | a = 25.0716(9) Å | a = 16.6726(4) Å |
| b = 15.7152(6) Å | b = 14.5993(4) Å | |
| c = 26.2842(12) Å | c = 33.4362(12) Å | |
| β = 105.43(1)° | β = 97.20(1)° | |
| V/Å3, Z | 9982.8(7), 4 | 8074.5(4), 4 |
| Dc/Mg m−3 | 1.125 | 1.051 |
| μ/mm−1 | 0.332 | 0.209 |
| F(000) | 3620 | 2788 |
| Crystal size/mm | 0.25 × 0.20 × 0.16 | 0.22 × 0.14 × 0.14 |
| θ range for data collection/° | 2.06 to 22.50 | 2.96 to 27.50 |
| Index ranges, hkl | −27 to 27, −16 to 17, −29 to 23 | −21 to 21, −18 to 18, −43 to 43 |
| Reflections collected | 56918 | 35212 |
| Independent reflections (Rint) | 13020 (0.0922) | 18437 (0.0410) |
| Data/restraints/param. | 12926/0/1072 | 18437/0/856 |
| Goodness-of-fit on F2 | 1.027 | 1.062 |
| Final R indices [I >2σ(I)] | R1 = 0.0592, wR2 = 0.1236 | R1 = 0.0590, wR2 = 0.1285 |
| R indices (all data) | R1 = 0.1005, wR2 = 0.1444 | R1 = 0.0830, wR2 = 0.1394 |
| Largest diff. peak, hole/e A−3 | 0.822, −0.586 | 1.247, −0.570 |
CCDC reference numbers 199330 for 1 and 244678 for 3.
See http://www.rsc.org/suppdata/dt/b4/b410842f/ for crystallographic data in CIF or other electronic format.
Preparation of the ligands H4L1, H2L2–H2L7
The ligands were prepared according to a very similar procedure described in ref. 11.The ligand H2L2, 2-anilino-4,6-di-tert-butylphenol, has been described earlier.11d
H2L3–H2L7, 2-(3,5-disubstituted anilino)-4,6-di-tert-butylphenol
As the 3,5-disubstituted anilino-ligands were prepared by a similar protocol, a representative method is only described. A solution containing 3,5-di-tert-butylcatechol (4.44 g, 20 mmol), the disubstituted aniline (20 mmol) and triethylamine (0.2 ml) in n-heptane (40 ml) was stirred at ambient temperature for 24 h to yield a brown suspension. The brown solid was collected by filtration and washed with cold n-heptane and air-dried. The compounds were recrystallized from n-hexane yielding colorless crystalline materials. The yields were in the range 60–80% based on the starting catechol.Mp, IR, 1H NMR, 13C NMR, EI-MS and microanalysis data for H2LCH3, H2LCl, H2LOCH3 are given as ESI.†
Preparation of complexes
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 1470s ν(C–O), and other peaks 1520m, 1360s, 1268s, 1021m, 703w. UV-VIS (CH2Cl2): λ/nm (ε/M−1 cm−1): 550 (11880), 648 (11500), 807 (10700).
N), 1470s ν(C–O), and other peaks 1520m, 1360s, 1268s, 1021m, 703w. UV-VIS (CH2Cl2): λ/nm (ε/M−1 cm−1): 550 (11880), 648 (11500), 807 (10700).For complexes 4–7 the analytical data, ESI-MS (positive) and the experimental magnetic moments (2–290 K) together with their simulations are given as ESI.†
Acknowledgements
Financial support from the German Research Council (DFG) is gratefully acknowledged (Project: Priority Program Ch111/2-2). Skilful technical assistance of Mrs H. Schucht, Mrs R. Wagner, Mrs U. Westhoff, Mr A. Göbels and Mr U. Pieper is thankfully acknowledged.References
- (a) J. Stubbe and W. A. van der Donk, Chem. Rev., 1998, 98, 705 CrossRef CAS; (b) R. H. Holm and E. I. Solomon, Guest Editors, Chem. Rev., 1996, 96(7) Search PubMed; (c) G. T. Babcock, M. Espe, C. Hoganson, N. Lydakis-Simantiris, J. McCracken, W. Shi, S. Styring, C. Tommas and K. Warncke, Acta Chem. Scand., 1997, 51, 533 CAS; (d) V. Mahadevan, R. J. M. Klein Gebbink and T. D. P. Stack, Curr. Opin. Chem. Biol., 2000, 4, 228 CrossRef CAS; (e) M. M. Fontecave and J. L. Pierre, Bull. Soc. Chim. Fr., 1996, 133, 653 CAS; (f) P. Chaudhuri and K. Wieghardt, Prog. Inorg. Chem., 2001, 50, 151 CAS; (g) J. W. Whittaker, Chem. Rev., 2003, 103, 2347 CrossRef CAS; (h) J. Stubbe, Chem. Commun., 2003, 2511 RSC.
- (a) Selected examples: J. A. Halfen, B. A. Jazdzewski, S. Mahapatra, L. M. Berreau, E. C. Wilkinson, L. Que, Jr. and W. B. Tolman, J. Am. Chem. Soc., 1997, 119, 8217 Search PubMed; (b) Y. Wang and T. D. P. Stack, J. Am. Chem. Soc., 1996, 118, 1309 CrossRef CAS; (c) D. Zurita, I. Gautier-Luneau, S. Menage, J. L. Pierre and E. Saint-Aman, J. Biol. Inorg. Chem., 1997, 2, 46 CrossRef CAS; (d) E. Bill, J. Müller, T. Weyher-Müller and K. Wieghardt, Inorg. Chem., 1999, 38, 5795 CrossRef CAS; (e) M. A. Halcrow, L. M. Lindy Chia, X. Liu, E. J. L. McInnes, L. J. Yellowlees, F. E. Mabbs, I. J. Scowen, M. McPartlin and J. E. Davies, J. Chem. Soc., Dalton Trans., 1999, 1753 RSC; (f) S. Itoh, S. Takayama, R. Arakawa, A. Furuta, M. Kumatsu, A. Ishida, S. Takamuku and S. Fukuzumi, Inorg. Chem., 1997, 36, 1407 CrossRef CAS; (g) Y. Shimazaki, S. Huth, A. Odani and Y. Yamauchi, Angew. Chem., Int. Ed., 2000, 112, 1666 CrossRef CAS; (h) M. Vaidyanathan and M. Palaniandavar, Proc. Indian Acad. Sci. (Chem. Sci.), 2000, 112, 223 Search PubMed; (i) C. N. Verani, E. Bothe, D. Burdinski, T. Weyhermüller, U. Flörke and P. Chaudhuri, Eur. J. Inorg. Chem., 2001, 2161 CrossRef CAS; (j) T. Weyhermüller, T. K. Paine, E. Bothe, E. Bill and P. Chaudhuri, Inorg. Chim. Acta, 2002, 337, 344 CrossRef CAS.
- (a) O. Kahn, Molecular Magnetism, VCH, New York, 1993 Search PubMed; (b) Molecular Magnetism: New Magnetic Materials, ed. K. Itoh, M. Kinoshita, Gordon & Breach, Amsterdam, The Netherlands, p. 2000 Search PubMed; (c) H. Iwamura, K. Inoue and T. Hayamizu, Pure Appl. Chem., 1996, 68, 24; (d) A. Racja, Chem. Rev., 1994, 94, 871 CrossRef CAS; (e) Guest ed. K. R. Dunbar:, J. Solid State Chem., 2001, 159 Search PubMed; (f) Special issue on Molecular Magnets, Electrochem. Soc. Interface, 2002, 11 Search PubMed (no. 3).
- (a) A. Caneschi, D. Gatteschi and P. Rey, Prog. Inorg. Chem., 1992, 39, 331; (b) C. G. Pierpont and C. W. Lange, Prog. Inorg. Chem., 1994, 41, 331 CAS; (c) W. Kaim, Dalton Trans., 2003, 761 RSC.
- (a) E. I. Solomon, U. M. Sundaram and T. E. Machonkin, Chem. Rev., 1996, 96, 2563 CrossRef CAS; (b) J. R. Walker and P. H. Ferrar, Biotechnol. Genet. Eng. Rev., 1998, 15, 457 Search PubMed; (c) C. Gerdemann, C. Eicken and B. Krebs, Acc. Chem. Res., 2002, 35, 183 CrossRef CAS.
- T. Klabunde, C. Eicken, J. C. Sacchettini and B. Krebs, Nat. Struct. Biol., 1998, 5, 1084 CrossRef CAS.
- (a) Representative examples: N. Oishi, Y. Nishida, K. Ida and S. Kida, Bull. Chem. Soc. Jpn., 1980, 53, 2847 Search PubMed; (b) U. Casellato, S. Tamburini, P. A. Vigato, A. De Stefani, M. Vidali and D. E. Fenton, Inorg. Chim. Acta, 1983, 69, 45 CrossRef CAS; (c) M. R. Malachowski and M. Davidson, Inorg. Chim. Acta, 1989, 162, 199 CrossRef CAS; (d) M. R. Malachowski, H. B. Huynh, L. J. Tomlinson, R. S. Kelly and J. W. Furbee, J. Chem. Soc., Dalton Trans., 1995, 31 RSC; (e) B. Srinivas, N. Arulsams and P. S. Zacharias, Polyhedron,, 1991, 10, 731 CrossRef CAS; (f) D. A. Rockcliffe and A. E. Martell, J. Mol. Catal. A, 1996, 106, 211 CrossRef CAS; (g) J. Manzur, A. M. Garcia, V. Rivas, A. M. Atria, J. Valenzuela and E. Spodine, Polyhedron, 1997, 16, 2299 CrossRef CAS; (h) F. Zippel, F. Ahlers, R. Werner, W. Haase, H.-F. Nolting and B. Krebs, Inorg. Chem., 1996, 35, 3409 CrossRef CAS; (i) J. Reim and B. Krebs, J. Chem. Soc., Dalton Trans., 1997, 3793 RSC; (j) M. R. Malachowski, B. T. Borsey, M. J. Parker, M. E. Adams and R. S. Kelly, Polyhedron,, 1998, 17, 1289 CrossRef CAS; (k) E. Monzani, L. Quinti, A. Perotti, L. Casella, M. Gullotti, L. Randaccio, S. Geremia, G. Nardin, P. Faleschini and G. Tabbi, Inorg. Chem., 1998, 37, 553 CrossRef CAS; (l) E. Monzani, G. Battaini, A. Perotti, L. Casella, M. Gullotti, L. Santagostini, G. Nardin, L. Randaccio, S. Geremia, P. Zanello and G. Opromolla, Inorg. Chem., 1999, 38, 5359 CrossRef CAS; (m) R. Wegner, M. Gottschaldt, H. Görl, E.-G. Jäger and D. Klemm, Chem. Eur. J., 2001, 7, 2143 CrossRef CAS; (n) S. C. Cheng and H. H. Wei, Inorg. Chim. Acta, 2002, 340, 105 CrossRef CAS; (o) A. Neves, L. M. Rossi, A. J. Bortoluzzi, B. Szpoganicz, C. Wiezbicki, E. Schwingel, W. Haase and S. Ostrovsky, Inorg. Chem., 2002, 41, 1788 CrossRef CAS; (p) J. Ackermann, F. Meyer, E. Kaifer and H. Pritzkow, Chem. Eur. J., 2002, 8, 247 CrossRef CAS; (q) M. Kodera, T. Kawata, K. Karr, Y. Tachi, S. Itoh and S. Kojo, Bull. Chem. Soc. Jpn., 2003, 76, 1957 CrossRef CAS; (r) K. Selmeczi, M. Réglier, M. Giorgi and G. Speier, Coord. Chem. Rev., 2003, 245, 191 CrossRef CAS; (s) M. C. Mimmi, M. Gullotti, L. Santagostini, A. Saladino, L. Casella, E. Monzani and R. Pagliarin, J. Mol. Catal. A, 2003, 204-205, 381 CrossRef; (t) C. T. Yang, M. Vetrichelvan, X. Yang, B. Moubaraki, K. S. Murray and J. J. Vittal, Dalton Trans., 2004, 113 RSC.
- (a) Y. Gultneh, A. Farooq, K. D. Karlin, S. Liu and J. Zubieta, Inorg. Chim. Acta, 1993, 211, 171 CrossRef CAS; (b) D. Kovala-Demertzi, S. K. Hadjikakou, M. A. Demertzis and Y. Deligiannakis, J. Inorg. Biochem., 1998, 69, 223 CrossRef CAS; (c) M. U. Triller, D. Pursche, W. Y. Hsieh, V. L. Pecoraro, A. Rompel and B. Krebs, Inorg. Chem., 2003, 42, 6274 CrossRef CAS.
- (a) C. N. Verani, S. Gallert, E. Bill, T. Weyhermüller, K. Wieghardt and P. Chaudhuri, Chem. Commun., 1999, 1747 RSC; (b) S. Mukherjee, T. Weyhermüller, K. Wieghardt and P. Chaudhuri, Dalton Trans., 2003, 3483 RSC , and references therein..
- S. Mukherjee, E. Rentschler, T. Weyhermüller, K. Wieghardt and P. Chaudhuri, Chem. Commun., 2003, 1828 RSC.
- (a) FRG Pat., 110 4522, p. 1959 Search PubMed; (b) Chem. Abstr., 1962, 56, p. 5887 Search PubMed; (c) L. A. Maslovskaya, D. K. Petrikevic, V. A. Timoschchuk and O. I. Shadyro, Russ. J. Gen. Chem., 1996, 66, 1842; (d) P. Chaudhuri, C. N. Verani, E. Bill, E. Bothe, T. Weyhermüller and K. Wieghardt, J. Am. Chem. Soc., 2001, 123, 2213 CrossRef CAS.
- H. Chun, P. Chaudhuri, T. Weyhermüller and K. Wieghardt, Inorg. Chem., 2002, 41, 790 CrossRef CAS.
- (a) Comprehensive Coordination Chemistry, ed. G. Wilkinson, Pergamon, Oxford, 1987, vol. 4 Search PubMed; (b) T. K. Paine, T. Weyhermüller, E. Bothe and P. Chaudhuri, Eur. J. Inorg. Chem., 2003, 4299 CrossRef CAS.
- A. Dei, D. Gatteschi, C. Sangregorio, L. Sorace and M. G. F. Vaz, Chem. Phys. Lett., 2003, 368, 162 CrossRef CAS.
- G. M. Eisenberg, Anal. Chem., 1943, 15, 327 CAS.
- T. K. Paine, T. Weyhermüller, K. Wieghardt and P. Chaudhuri, Dalton Trans, 2004, 2092 RSC.
- P. R. Wells, S. Ehrenson and R. W. Taft, inProg. Phys. Org. Chem., ed. A. Streitweiser, Jr., and R. W. Taft, John-Wiley & Sons, New York, 1968, vol. 6, p. 222 Search PubMed.
- G. M. Sheldrick, SHELXL-97 and SHELXS-97, University of Göttingen, Germany, 1997 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Figs. 10 and 11. Analytical IR, NMR and mass spectrometric data for H2LCH3, H2LCl, H2LOCH3 and corresponding data for complexes 4–7. See http://www.rsc.org/suppdata/dt/b4/b410842f/ |
| This journal is © The Royal Society of Chemistry 2004 |