Syntheses, structural properties and catecholase activity of copper(II) complexes with reduced Schiff base N-(2-hydroxybenzyl)-amino acids†
Chang-Tong
Yang
a,
Muthalagu
Vetrichelvan
a,
Xiandong
Yang
a,
Boujemaa
Moubaraki
b,
Keith S.
Murray
b and
Jagadese J.
Vittal
*a
aDepartment of Chemistry, National University of Singapore, 3 Science Drive 3, Singapore 117543. E-mail: chmjjv@nus.edu.sg
bSchool of Chemistry, Monash University, P.O. Box 23, Victoria, 3800, Australia
First published on 1st December 2003
Abstract
A number of dicopper(II) complexes of reduced Schiff base ligands, N-(2-hydroxybenzyl)-amino acids [Cu2L2(H2O)x]·yH2O (L = Sgly (1), D-Sala (2), L-Sala (3), DL-Sala (4), Sab2 (5), Sbal (6), Sab4 (7), Sval (8), Shis (9), Styr (10) and Stryp (11), x = 0–2 & y = 0–2) have been synthesized, and the solid-state structures of 4, 5,10 and 11 have been determined. The compounds 4 and 5 are binuclear in which the Cu(II) centres have square-pyramidal geometry with apical sites occupied by aqua ligands. In 10 and 11 one axial site is occupied by water and the other by an oxygen atom of the carboxylate group from the adjacent dimer through oxygen atoms to form 1D helical polymer. Variable temperature magnetic measurements of the dimer 4 and helical polymer 10 showed that they are typical for moderately strong antiferromagnetic coupling. All the complexes show significant catalytic activity on the oxidation of 3,5-di-tert-butylcatechol. The activity measured in terms of Kcat in the range 199–3800 h−1 has been found to follow the order: 7 > 6 > 8 > 3 > 5 ∼ 2 ∼1 > 4 > 10 >9 > 11. The catalytic activity is found to increase with increasing the length of the methylene side chain of the amino acid in the reduced Schiff base ligands.
Introduction
The copper containing proteins are classified into three types: I, II and III. Type III copper proteins such as hemocyanin, tyrosinase have a strongly coupled binuclear copper active site, which is antiferromagnetically coupled and therefore EPR silent in the oxidized state.1–5 Catechol oxidase is another copper type III enzyme which only catalyses the oxidation of catechol to quinone without acting on tyrosinase.6 This reaction is of great importance in medical diagnosis for the determination of the hormonally active catacholamines adrenaline, noradrenaline and dopa.7,8 The copper in the isolated catechol oxidases was found to be EPR-silent and was assigned to an antiferromagnetically spin-coupled Cu(II)–Cu(II) pair.9The catecholase activity of synthetically prepared copper compounds with different structural parameters has been investigated to some extent.10–22 Modeling the enzyme features requires the control of the interaction of the two metal centres. As a consequence, the design of the binucleating ligand has to satisfy a number of conditions: metal–metal distance, steric, electronic and bridging ligand features, etc. The crystal structure of catechol oxidase recently reported by Krebs and co-workers reveals that these proteins have binuclear copper center and have similar spectroscopic behaviour, and show close functional relationships.23 The crystal structure of catechol oxidase have enhanced the understanding of the mechanism of catecholase activity of tyrosinase and/or catechol oxidase which was proposed by Solomon and others.23–26
In this paper, we report the synthesis of a series of binuclear copper(II) complexes as potential structural and functional models for the active sites of catechol oxidase. The ligands considered in this paper are as follows: H2Sgly, D-H2Sala, L-H2Sala, D,L-H2Sala, H2Sbal, H2Sab2, H2Sab4, H2Sval, H2Shis, H2Styr and H2Stryp (Scheme 1). These ligands contain correct terminal and endogenous bridging ligand types that strongly favour the formation of bimetallic species. The structures of binuclear complexes of H2Sgly, D-H2Sala, L-H2Sala, D,L-H2Sala, H2Sbal, H2Sab2, H2Sab4, H2Styr and H2Stryp have shown to have Cu⋯Cu distances in the range of 3 Å. The solid-state structures of 4, 5, 10 and 11 have been determined by X-ray crystallography. The catecholase activity of these binuclear compounds have been measured and compared. The results of our investigations are described in this paper in detail.
![Reduced Schiff base ligands. The numbers of the corresponding Cu(ii) complexes [Cu2L2(H2O)x]·yH2O of these ligands used in this paper are given in brackets.](/image/article/2004/DT/b310262a/b310262a-s1.gif) | ||
| Scheme 1 Reduced Schiff base ligands. The numbers of the corresponding Cu(II) complexes [Cu2L2(H2O)x]·yH2O of these ligands used in this paper are given in brackets. | ||
Results and discussion
Compounds 1–3 have been reported before.27,28 and the structure of 8 will be published elsewhere.29 A few years ago the optically active L-H2Sala and D-H2Sala ligand have been used to construct multi-dimensional hydrogen bonded network structures with chiral channels and these hydrated network structures undergo solid state supramolecular transformation to coordination polymeric network compounds.27 The basic building block consists of noncentrosymmetric dimers. In order to understand the effect of chirality of the ligands used in the building blocks on the packing, we have employed the D,L-Sala ligand, in which both enantiomers are present in equal amounts and the Sab2 ligand, which is optically inactive but closely related to the D,L-Sala ligand. Complexation of D,L-Sala with Cu(II) ions is expected to furnish centrosymmetric dimer [Cu2(D,L-Sala)2(H2O)2].The IR absorption band of the newly synthesized compounds 4–7 and 9–11 found in the range 3389–3471 cm−1 confirms the presence of lattice or coordinated water for the compounds.30 This is further supported by the weight loss observed in TG (see experimental section). The sharp band observed in the region 2895–2976 cm−1 may be assigned to ν(NH).31 The band in the region 1572–1615 cm−1 is assigned to the asymmetric vibration of coordinated carboxylate group [νas(COO−)] and the band in the region 1325–1397 cm−1 is attributed to the symmetric stretching vibration of carboxylate group [νs(COO−)]. The large difference between [νas(COO−)] and [νs(COO−)] in frequencies (Δν > 200 cm−1) in all complexes is indicative of a monodentate coordination of carboxylate groups.32–34 This observation was confirmed by X-ray crystal structures of 4 and 5. There are two kinds of [νs(COO−)] exhibited in IR spectrum, respectively. The small difference (Δν = 154 < 200 cm−1) between [νas(COO−)] and [νs(COO−)] in frequencies and large difference (Δν = 220 > 200 cm−1) between [νas(COO−)] and [νs(COO−)] indicate bridging coordination mode of the carboxylate group and terminal coordination mode for the carboxylate group, respectively, as found in 10 and 11. The band around the 1259–1285 cm−1 can be assigned to ν(C–O) of phenolic group in complexes.31
The electronic spectral data for the complexes as Nujol mull transmittance as well as the molar conductivity data are given in Table 1. The UV absorption band observed exhibits a charge transfer transition (CT) that falls in the range 404–360 nm for Cu(II) complexes, which may be assigned to ligand to metal transition. The CT band in those Schiff base complexes of salicylaldehyde with Gly and acetyllysine35 and Ala, Val, Phe, and His36 shows a shift to higher energy attributed to the delocalization of charge in the conjugated Schiff base ligands. The d–d transitions generally fall below 700 nm and are more consistent with square-pyramidal geometries about certain related Cu(II) complexes.37 Compound 6 is assumed to have square-planar geometry with a d–d transition at 620 nm similar to those observed for the structurally well characterized square-planar Cu(II) complexes.37 The molar conductance values in DMSO and CH3OH indicate that all complexes are non-electrolytes.38
Description of crystal structures
The solid-state structures of 4, 5, 10 and 11 have been described in this section in detail. No suitable single crystal for data collection has been obtained for 6, 7 and 9.[Cu2(D,L-Sala)2(H2O)2]·2H2O, 4
An ORTEP diagram with numbering scheme is shown in Fig. 1. The complex is a centrosymmetric binuclear compound with both D and L-Sala ligands coordinated to the Cu(II) centres in a mer fashion through phenolate, carbonyl oxygen and the imine nitrogen atoms. In the dimer, the phenolic oxygen atoms O1 and O1A bridge the two Cu(II) centres giving a Cu⋯Cu distance of 2.9742(7) Å. The apical coordination sites are occupied by water molecules in trans fashion. The coordination geometry of Cu(II) ion can be described as an ideal square pyramid as indicated by the angular structural parameter, τ = 0.013.39–41 The trans geometry of the two aqua ligands has been found in a number of binuclear Cu(II) compounds including [{Cu(HSalacet)H2O}2]·(NO3)242 (Salacet = salicylaldehyde acetylhydrazone) and [Cu2(bhsNO2)2(H2O)2]43 (bhsNO2 = 2-hydroxy-5-nitro-benzaldehyde benzoylhydrazone) resulting in Cu⋯Cu of 3.006(3) and 3.041(4) Å, respectively. Equatorial bond lengths [Cu(1)–O(1) 1.953(2) Å, Cu(1)–O(2) 1.952(2) Å, Cu(1)–N(1) 1.975(3) Å, Cu(1)–O(1)A 1.940(2) Å] and weakly bound axial bond length [Cu(1)–O(4) 2.326(2) Å] agree well with those reported for [{Cu(HSalacet)H2O}2]·(NO3)242 and [Cu2(bhsNO2)2(H2O)2].43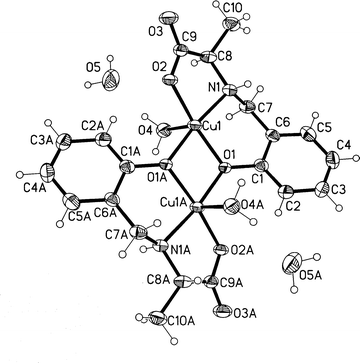 | ||
| Fig. 1 An ORTEP diagram of 4. | ||
The neutral dimers are packed in the solid state such that the carbonyl oxygen atoms are very weakly interacting with Cu with Cu⋯O, 3.076(4) Å which is further assisted by complementary N–H⋯O bonds as well as O–H⋯O hydrogen bonds. This leads to one dimensional hydrogen bonded polymers. The two lattice water molecules are also involved strong O–H⋯O bonding to form a 2D sheet as shown in Fig. 2.
 | ||
| Fig. 2 Packing of 4 showing the intermolecular hydrogen bonds. | ||
[Cu2(Sab2)2(H2O)2], 5
The complex is again a centrosymmetric dimer with the dianionic Sab22− ligand bound to Cu(II). The phenolate oxygen bridges the two Cu(II) centres giving a Cu⋯Cu distance of 3.0049(4) Å. These four donors are approximately planar with Cu ion displaced slightly towards a more weakly bound water molecule [Cu(1)–O(4), 2.603(4) Å], which completes the coordination sphere at the axial site. The Cu(II) geometry can be described as an ideal square pyramid with τ = 0.003.39–41 The structural conformation of 5 is similar to 4 as indicated by the superimposed structure of 5 and 4 (see ESI†).The packing of 5 in the solid state is similar to that of 4 as expected. The dimer is slip-stacked together through weak Cu⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds complemented by O–H⋯O hydrogen bonds to form 1D hydrogen bonded polymer. The imine H atom is not participating in the hydrogen bond to O atom of carboxylate group. In the absence of two lattice water molecules, unlike 4, these hydrogen-bonded polymers interact with adjacent strands through carbonyl oxygen and hydrogen atoms of the aqua ligand to form 2D hydrogen bonded sheets.
C bonds complemented by O–H⋯O hydrogen bonds to form 1D hydrogen bonded polymer. The imine H atom is not participating in the hydrogen bond to O atom of carboxylate group. In the absence of two lattice water molecules, unlike 4, these hydrogen-bonded polymers interact with adjacent strands through carbonyl oxygen and hydrogen atoms of the aqua ligand to form 2D hydrogen bonded sheets.
[Cu2(Styr)2(H2O)]·2H2O, 10
An ORTEP diagram of 10 with numbering scheme is shown in Fig. 3. The structure consists of the basic dimeric building block Cu2(Styr)2 in which both Cu(II) have approximate square pyramidal geometry (τ = 0.075 for Cu(1) and 0.016 for Cu(2))39–41 similar to 4 and 5. However 10 contains two unique Cu(II) centres. In Cu(2), the axial site is occupied by a water molecule (site A) while the Cu(1) (site B) is bridged by an adjacent dimer through one of the carboxylate oxygen atoms to form helical polymer along the a-axis as shown in Fig. 4. The coordination geometry of Cu(II) centres and structure arrangement are similar to that found in [Cu2(L-Sala)2(H2O)]n.27 These coordination polymers are further supported by O–H⋯O and N–H⋯O hydrogen bonds in the crystal structure. The water molecule and the carboxylate group are on the same side of the molecule, and form H–O–H⋯O–C hydrogen bonds. | ||
| Fig. 3 An ORTEP diagram with the atom labeling scheme of 10. | ||
 | ||
| Fig. 4 Packing of 10 showing the 1D helical coordination polymers. | ||
The N–H hydrogen atom of the ligand from site A forms a hydrogen bond to the O(11) of the lattice water. The proton in the side chain of the ligand O(8) is weakly hydrogen bonded to the other lattice water O(10). One of the hydrogens in the lattice water O(11) is involved in strong intramolecular hydrogen-bonding to the O(9) of the coordinated water. Both hydrogens in the other lattice water O(10) are involved in strong hydrogen-bonding to the carbonyl oxygen atom of the adjacent carboxylate group O(2) and lattice water O(11), respectively. One of the hydrogens in the coordinated water forms a intramolecular hydrogen-bond to the CO oxygen atom of the carboxylate group (O6) and the other hydrogen forms an intermolecular hydrogen-bond to the O(3) of adjacent carboxylate group as depicted in Fig. 5.
 | ||
| Fig. 5 A perspective view of hydrogen bonding network in 10. | ||
[Cu2(Stryp)2(H2O)], 11
The crystal structure reveals that the complex is a one-dimensional coordination polymer with the binuclear copper fragment Cu2(Stryp)2 as a building block. Both copper(II) atoms adapt a distorted square pyramidal geometry as indicated by the angular structural parameters τ = 0.096 for Cu(1) and 0.052 for Cu(2).39–41 The only difference in the coordination environment between Cu(1) and Cu(2) is the fifth donor site; a water molecule is coordinated to Cu(1) while the neighboring carbonyl oxygen atom is coordinated to Cu(2) to form a helical polymer along a-axis as shown in Fig. 6. Both axial substituents are on the same side of the dimer. Like [Cu2(Styr)2(H2O)]·2H2O complex, the coordination geometry of Cu(II) centres and structural arrangement are similar to that found for in [Cu2(L-Sala)2(H2O)]n.27 | ||
| Fig. 6 A view of 11 showing the 1D helical coordination polymers. | ||
The coordination polymers are further supported by the O–H⋯O and N–H⋯O hydrogen bonds in the crystal structure. Both hydrogens in the coordinated water O(7) are involved in strong hydrogen-bonding to oxygen atom of the adjacent carboxylate group O(2) and O(6), respectively. The N–H hydrogen atom of the side arm of ligand N(13A) forms a hydrogen bond to the oxygen atom of the carboxylate group O(5).
Thermal dehydration reactions
The TG of 4 exhibited two weight loss regions; the first one in the range 50–120 °C is due to the dehydration and the second above 245 °C is attributed to the decomposition of the dimer. The observed weight loss of 11.8% for the dehydration process matches well with the weight loss 12.2% calculated for the loss of all the four water molecules. It is noted here that two of the water molecules lost by 120 °C are indeed bound to two Cu(II) atoms. The loss of metal bound aqua ligands below 120 °C along with two lattice waters is very similar to those observed during solid state supramolecular transformations.27,44 The formation of new bonds between Cu and carbonyl oxygen atoms after dehydration is presumed to be the driving force for this behaviour, which is expected to form a 1D coordination polymer based on the packing in the solid state. However, the anhydrous 4 can easily be rehydrated on standing in air, as revealed by TG.44 Although the structure of 5 is quite similar to 4 with respect to the aqua ligand, all of the water could only be removed after heating the sample above 150 °C (ESI†). This behaviour is different from 4. It is worth noting that the 1D coordination polymers are formed by noncentrosymmetric binuclear building blocks obtained from either L-Sala or D-Sala.27 The formation of such coordination polymers appears to be enantiomerically unfavourable for the centrosymmetric dimers. The TG curve of 10 shows a weight loss of 7.2% occurred from room temperature to 150 °C (ESI†). This corresponds to the loss of one aqua ligand and two lattice water molecules (wt. loss calcd, 7.2%). The anhydrous sample is stable up to 245 °C. The TG curve of 11 shows that the weight loss of 2.5% occurred from room temperature to 180 °C corresponds to the loss of aqua ligand (calcd. wt. loss, 2.4%). The anhydrous sample is stable up to 253 °C.Magnetic properties
The room-temperature magnetic moments, per Cu, for the Cu(II) complexes are in the range 1.12∼1.50 µB which indicates moderately strong antiferromagnetic coupling is occurring in these complexes.45 The magnetic data are compatible with the structures reported herein. Magnetic susceptibilities were measured for 4 and 10 over the temperature 4.2–300 K. The data for 4 gave a maximum in susceptibility close to room temperature as shown in Fig. 7 and a good fit to the Bleaney–Bowers (−2JS1S2) model46 for a S = ½ binuclear compound. The best fit parameters are g = 2.01, 2J = −326 cm−1, TIP = 0.00006 cm3 mol−1, % monomer = 0.2. Compound 10 is even more strongly coupled and the best fit values are g = 2.10, 2J = −474 cm−1, TIP = 0.00006 cm3 mol−1, % monomer = 2.2. The larger amount of monomer ‘impurity’ in 10 is evident at low temperatures where a large increase in susceptibility is observed. The J values are in the range commonly observed for phenoxo bridged copper(II) compounds.47 The larger value of J for 10 is in agreement with the larger Cu–O–Cu angle and larger Cu⋯Cu separation. The very strong antiferromagnetic exchange coupling has also been observed in structurally closely related dicopper(II) centres bridged by phenoxo ligands.48,49 | ||
| Fig. 7 Plots of χ (□) and μeff (○), per Cu, vs. temperature for 4 in a field of 1 T. The solid lines are the calculated values using the parameters given in the text. | ||
Catecholase-mimetic activities
Model studies of synthetic analogues have achieved notable advances in the understanding of the structural and chemical properties of type III protein.10–22,50–56 In these studies, mono- or multinuclear complexes have been employed and the properties of the chelating ligands have been varied with respect to architecture, number and nature of the donor atoms. Binuclear copper(II) complex [Cu2bbpen2](ClO4)2·3MeOH (Hbbpen = 1,5-bis(2-benzimidazolyl)-3-pentanol) was studied by Krebs et al. as structural and functional model for catechol oxidase.12 Its reactions with 3,5-DTBC were investigated using UV-vis and XAS spectroscopic studies in MeOH solution (as illustrated in Scheme 2). The oxidation of 3,5-DTBC to the corresponding o-quinone was followed by the development of strong absorption band of the product at about 390 nm.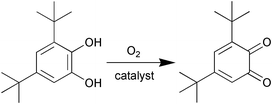 | ||
| Scheme 2 Oxidation of 3,5-DTBC to 3,5-DTBQ. | ||
Before going to the detailed kinetic study, it is necessary to get an estimation of the ability of the complexes to oxidize catechol. For this purpose, 10−4 mol dm−3 solutions of 1–11 were treated with 50 equivalents of 3,5-DTBC under the aerobic condition. The course of the reaction is followed by UV-vis spectroscopy. The UV-vis spectra of the original solution directly after the addition and after 5, 15, 20 min and up to 2 h were recorded and corrected for volume changes. The increase in the absorption at 390 nm, all the complexes 1–11 showed considerable catecholase activity. The course of oxidation of 3,5-DTBC by 7 with the increase in quinone absorption band at 390 nm is shown in Fig. 8. Since 3,5-DTBQ showed a characteristic absorption band at 390 nm and the content of copper(II) complex is very small, we monitored the increase in absorbance vs. time at this wavelength. The reactivity studies were performed in MeOH solution due to the good solubility of the substrate, 3,5-DTBC and of its product, DTBQ.
 | ||
| Fig. 8 Absorbance at 390 nm of the solution containing 7 and 3,5-DTBC (in methanol, 25 °C). The spectra have been recorded at various intervals of time from 10 to 120 min. | ||
The kinetics of the oxidation of 3,5-DTBC was determined by the method of initial rates by monitoring the growth of the 390 nm band of the product 3,5-DTBQ. In order to determine the kinetic parameters, the Michaelis–Menten approach which originally developed for enzyme kinetics, was applied.8,13,23 A linear relationship for the initial rates and the complexes concentration is obtained (ESI†) for 1–11, which show a first order dependence on the catalyst concentration for these systems. An example of Lineweaver–Burk plot is given in Fig. 9 for 7. Table 2 contains the results evaluated from Lineweaver–Burk plots. Although the real mechanism of the reaction is complicated, the data obtained from Lineweaver–Burk plot is sufficient for a comparison of the catalytic activity.
![A plot of 1/νvs. 1/[S] for 7.](/image/article/2004/DT/b310262a/b310262a-f9.gif) | ||
| Fig. 9 A plot of 1/νvs. 1/[S] for 7. | ||
| Complex | k cat/h−1 | K m/mM | V max 106/M s−1 |
|---|---|---|---|
| 1, [Cu2(Sgly)2(H2O)]·2H2O | 563 | 23.8 | 31.1 |
| 2, [Cu2(D-Sala)2(H2O)]·H2O | 564 | 20.9 | 29.4 |
| 3, [Cu2(L-Sala)2(H2O)]·H2O | 666 | 25.4 | 34.8 |
| 4, [Cu2(D,L-Sala)2(H2O)2]·2H2O | 365 | 14.1 | 17.3 |
| 5, [Cu2(Sab2)2(H2O)2] | 564 | 19.3 | 27.7 |
| 6, [Cu2(Sbal)2(H2O)2] | 1287 | 10.4 | 6.5 |
| 7, [Cu2(Sab4)2(H2O)2]·0.5H2O | 3800 | 8.7 | 193.3 |
| 8, [Cu2(Sval)2(H2O)3] | 678 | 24.0 | 26.6 |
| 9, [Cu2(Shis)2(H2O)]·H2O | 244 | 20.0 | 9.7 |
| 10, [Cu2(Styr)2(H2O)]·2H2O | 265 | 20.2 | 9.8 |
| 11, [Cu2(Stryp)2(H2O)] | 199 | 17.3 | 8.9 |
The catecholase activity follows the order: 7 > 6 > 8 > 3 > 5 ∼ 2 ∼ 1 > 4 > 10 >9 > 11. It has been assumed that geometry around the copper ions and intermetallic distance are the two key factors that determine the catalytic activity of the complexes.50–54 However, it should be borne in mind that the solid-state structure need not be retained in solution, and a direct correlation between the solid-state structure and catalytic activity need not be expected. For all Cu(II) complexes, the Cu⋯Cu distance is likely to be around 3.0 Å which allows a bridging catechol coordination compatible with the distance between the two o-diphenol oxygen atoms.50–52,54,55
From the data obtained in Table 2, among all the complexes, 7 shows the highest activity and 11 shows the lowest activity, from which one can infer that the catecholase activity is decreasing with increasing bulky substituents and functional groups with coordinating ability in the side chain of the reduced Schiff base ligand. It is argued that the square-planar geometry of copper complex is not ideal for possible catechol-oxidase catalyst.7–13 The high activity along with a lower KM for 7 can be explained by assuming that the Cu geometry in 7 should not be square-planar. The compounds bis[1,2-O-isopropylidene-6-N-(3-acetylbut-3-en-2-one)amino-6-deoxy-glucofuranoso] dicopper(II)] and bis[1,2-O-isopropylidene-6-N-(2,2-bisethylcarboxy ethylene)amino-6-deoxyglucofuranoso] dicopper(II)] with similar geometry also show very high catalytic activity.55
Recently Mukherjee and Mukherjee reported that the copper(II) complexes are reduced to copper(I) complexes during catalysis.54 It is more apparent by seeing the colour of the solution changes from dark green to yellow and then to brownish yellow. Their investigation confirmed that the electron transfer from catechol to the copper(II) complex begins after the formation of a copper–catechol intermediate which could be prevented by the competitive formation of copper–quinone complex.57 All the neutral binuclear compounds 1–11 contain one four-membered ring, Cu2O2 and two six-membered rings. Further, two more rings formed by the side arms of the reduced Schiff base ligands depend on the length of the alkyl chain in the amino acids. Except 6 and 7, five-membered rings have been formed by all the complexes. It is obvious that the catalytic activity increases as the length of the side arm of the reduced Schiff base ligands increases. Two seven-membered rings and two six-membered rings have been proposed for 6 and 7, respectively. Karlin and Casella have proposed a mechanism for the oxidation of catechols to O-quinones by binuclear copper(II) complexes.55,58–60 According to the proposed mechanism, the second step is the reduction of a dicopper(II) to a dicopper(I) system. Due to seven membered rings in the case of 6 and 7, the formation of a dicopper(I) system might be faster than the other complexes and thus account for the higher catecholase activities of 6 and 7. Further, the compounds with functional groups with coordinating ability (9–11) show lower activity as compared to those with non-reactive bulky groups since these functional groups can occupy the axial positions (inter or intramolecularly in solution) and thus prevent 3,5-DTBC to react with Cu(II) centres. The trend in Kcat among various Sala complexes is very unexpected and follows the order 3 > 2 > 4, i.e., L > D > D,L. The difference in activity between optically active and inactive Cu(II) complexes may be attributed to the difference in the symmetry associated with the structures. But, it is rather surprising to find that the handedness of the ligand has some influence on the rate of oxidation of 3,5-DTBC.
Summary
A series of Cu(II) complexes with reduced Schiff base ligands N-(2-hydroxybenzyl)-amino acids have been synthesized and characterized. Structural, spectroscopic properties of these compounds have been investigated. These neutral binuclear Cu(II) complexes have Cu⋯Cu distance of about 3 Å and antiferromagnetically coupled at room temperature. All the neutral dicopper(II) complexes show medium to significantly high catalytic influence on the oxidation of 3,5-DTBC to the corresponding O-quinones. The compounds are both structural and functional models for type III copper proteins. Our present systematic investigation shows that the increase in the length of the side arm of the reduced Schiff base ligand has a profound effect on the catecholase activity.Experimental
All reagents were commercially available and were used as received. Reagents used for the physical measurements were of spectroscopic grade. The yields are reported with respect to the metal salts. All the syntheses were carried out at room temperature in air.The 1H NMR spectra were recorded on a Bruker ACF 300FT-NMR spectrometer using TMS as an internal reference at 25 °C in DMSO and the infrared spectra (KBr pellet) were recorded using FTS165 Bio-Rad FTIR spectrophotometer in the range 4000–450 cm−1. The electronic transmittance spectra were recorded on a Shimadzu UV-2501/PC UV-vis spectrophotometer in nujol mull. Conductance measurements were made using a Kyoto Electronics CM-115 conductivity meter using 1 mM solutions. The elemental analyses were performed in the Microanalytical Laboratory, Department of Chemistry, National University of Singapore. Water present in the compounds was determined using a SDT 2980 TGA Thermal Analyzer with a heating rate of 10 °C min−1 in a N2 atmosphere using a sample size of 5–10 mg per run. Room-temperature magnetic susceptibility measurements were carried out on a Johnson-Matthey Magnetic Susceptibility balance with Hg[Co(SCN)4] as standard. Corrections for diamagnetism were made using Pascal's constants. The reported magnetic moments are per Cu(II) ion. Variable temperature magnetic measurements were made using a Quantum Design MPMS 5 SQUID magnetometer operating in an applied field of 1 T. The samples were contained in a gel capsule and held in a soda straw which was fixed to the end of the sample rod.
H2Sbal, H2Sab4 were synthesized according to the reported literatures.61–62 The syntheses of ligands, N-(2-hydroxybenzyl)-amino acids preparation, D,L-H2Sala, H2Sab2, H2Shis·0.5H2O and H2Styr·1.5H2O are described below.
To a solution of the amino acid in H2O (10 mL) containing NaOH (0.040 g, 1.00 mmol) was added salicylaldehyde (0.122 g, 1.00 mmol) in EtOH (10 mL). The yellow solution was stirred for 30 min at room temperature prior to cooling in an ice bath. The intermediate Schiff base solution was carefully adjusted to pH = 6.0–7.0 with HOAc, then excess solid NaBH4 (0.460 g, 1.20 mmol) was added in portions with gentle and stirring while the yellow colour slowly discharged. After 10 min the solution was evaporated and extracted with dry MeOH, then acidified with concentrated HCl to pH of 5.0–6.0. The resulting colourless solid was filtered off, washed with dry MeOH and Et2O, dried, and recrystallized from H2O/EtOH (1 ∶ 1).
D,L-H2Sala
Yield: 78 mg (40%). m.p. 241–242 °C. Anal. Calcd for C10H13NO3: C, 61.6; H, 6.6; N, 7.2. Found: C, 61.5; H, 6.7; N, 7.2%. 1H NMR (dmso-d6): δ 1.3 (d, 3H, J = 7.1 Hz), 3.2 (q, 1H, J = 7.1 Hz), 3.9 and 4.0 (AB system, 2H, JAB = 13.5 Hz), 6.7–6.8 (m, 2H), 7.1–7.2 (m, 2H). IR (KBr, cm−1): ν(OH) 3445, ν(NH) 3110, νas(COO−) 1607, νs(COO−) 1380, ν(C–O) (phenolic) 1264.H2Sab2
Yield: 0.136 g (65%). m.p. 231–232 °C. Anal. Calcd for C11H15NO3: C, 63.2; H, 7.2; N 6.7. Found: C, 63.0; H 7.4; N, 6.8%. 1H NMR (dmso-d6): δ 1.3 (s, 6H), 3.9 (s, 2H), 6.7–6.8 (m, 2H), 7.2–7.3 (m, 2H). IR (KBr, cm−1): ν(OH) 3445; ν(NH) 2919, νas(COO−) 1614, νs(COO−) 1458, ν(phenolic, CO) 1278.H2Shis·0.5H2O
Yield: 0.199 g (77%). m.p. 219–220 °C. Anal. Calcd for C12H16N3O3.5: C, 57.9; H, 5.7; N 15.5; H2O, 3.5. Found: C, 57.9; H 5.6; N, 15.6; H2O, 3.5%. 1H NMR (D2O): δ 2.92 (d, 2H, J = 7.0 Hz), 3.00 (t, 1H, J = 7.0 Hz), 3.79 and 3.99 (AB system, 2H, JAB =13.6 Hz), 6.8 (s, 1H), 6.8–6.9 (m, 2H), 7.1–7.2 (m, 2H), 7.56 (s, 1H). IR (KBr, cm−1): ν(OH) 3420; ν(NH) 3116, νas(COO−) 1603, νs(COO−) 1380, ν(phenolic, CO) 1275.H2Styr·1.5H2O
Yield: 0.172 g (55%). m.p. 230–232 °C. Anal. Calcd for C16H20NO5.5: C, 61.2; H, 6.4; N 4.4; H2O, 8.6. Found: C, 61.3; H 6.2; N, 4.2; H2O, 8.7%. 1H NMR (D2O): δ 2.9 (d, 2H, J = 7.0 Hz), 3.4 (t, 1H, J = 7.0 Hz), 3.6 and 3.8 (AB system, 2H, JAB =13.6 Hz), 6.7–6.8 (m, 4H), 7.0–7.1 (m, 4H). IR (KBr, cm−1): ν(OH) 3420; ν(NH) 3167, νas(COO−) 1617, νs(COO−) 1374, ν(phenolic, CO) 1255.H2Stryp
Yield: 0.211 g (68%). m.p. 235–237 °C. Anal. Calcd for C18H18N2O3: C, 69.7; H, 5.8; N 9.0. Found: C, 69.9; H 5.9; N, 7.9%. 1H NMR (dmso-d6): δ 2.93 (d, 2H, J = 7.0 Hz ), 3.37 (t, 1H, J = 7.0 Hz), 3.7 and 3.9 (AB system, 2H, JAB =13.6 Hz), 6.6–6.7 (m, 4H), 6.9–7.4 (m, 5H). IR (KBr, cm−1): ν(OH) 3426; ν(NH) 3111, νas(COO−) 1604, νs(COO−) 1370, ν(phenolic, CO) 1275.[Cu2(D,L-Sala)2(H2O)2]·2H2O, 4
To the solution of D,L-H2Sala (0.195 g, 1.00 mmol) and LiOH (0.048 g, 2.00 mmol) in water (10 mL) was added a solution Cu(OAc)2·H2O (0.200 g, 1.00 mmol) in water (10 mL). The dark green reaction mixture was filtered and allowed to stand in air at room temperature for several days to obtain the needle-like green crystals of 4. Yield: 0.214 g (73%). Anal. Calcd for C20H30N2O10Cu2: C, 41.0; H, 5.1; N 4.8; H2O, 12.3. Found: C, 41.3; H, 5.0; N, 4.6; H2O, 12.1%. IR (KBr, cm−1): ν(OH) 3447, ν(NH) 2935, νas(COO−) 1612, νs(COO−) 1386, ν(phenolic, CO) 1259. µB = 1.32 BM.[Cu2(Sab2)2(H2O)2], 5
H2Sab2 (0.209 g, 1.00 mmol) and LiOH (0.048 g, 2.00 mmol) were stirred in water for 15 min, and then the reaction mixture was filtered. To the filtrate was added a solution of Cu(OAc)2·H2O (0.200 g, 1.00 mmol) in water (10 mL). The reaction mixture turned deep blue immediately. After filtration, the solution was allowed to stand in air at room temperature for several days to furnish needle-like green crystals of 5. Yield: 0.196 g (68%). Anal. Calcd for C22H30N2O8Cu2: C, 45.7; H, 5.2; N 4.8; H2O, 6.2. Found: C, 45.9; H, 5.3; N, 4.8; H2O, 6.3%. IR (KBr, cm−1): ν(OH) 3421, ν(NH) 2933, νas(COO−) 1615, νs(COO−) 1386, ν(phenolic, CO) 1260. µB = 1.35 BM.[Cu2(Sbal)2(H2O)2], 6
The addition of a solution of H2Sbal (0.200 g, 1.00 mmol) and LiOH (48 mg, 2.00 mmol) in water (15 mL) to a solution of Cu(OAc)2·H2O (0.199 mg, 1.00 mmol) in 15 mL of MeOH gave the light green product 6, which was filtered off and washed with H2O, MeOH and Et2O before drying under vacuum. Yield: 0.18 g (66%). Anal. Calcd for C20H26N2O8Cu2: C, 43.9; H, 4.8; N 4.8; H2O, 6.6. Found: C, 43.7; H, 4.7; N, 5.1; H2O, 6.7%. IR (KBr, cm−1): ν(OH) 3448, ν(NH) 2931, νas(COO−) 1572, νs(COO−) 1355, ν(phenolic, CO) 1260. µB = 1.37 BM.[Cu2(Sab4)2]·0.5H2O, 7
To the solution of H2Sab4 (0.209 g, 1.00 mmol) and LiOH (0.048 g, 2.00 mmol) in water (10 mL) was added a solution Cu(OAc)2·H2O (0.200 g, 1.00 mmol) in water (10 mL). The dark green reaction mixture was filtered and allowed to stand in air at room temperature for several days to obtain dark green fine crystals of 7. Yield: 0.162 g (59%). Anal. Calcd for C22H27N2O6.5Cu2: C, 47.8; H, 4.8; N 4.9; H2O, 1.6. Found: C, 48.0; H, 4.9; N, 5.1; H2O, 1.5%. IR (KBr, cm−1): ν(OH) 3421, ν(NH) 2936, νas(COO−) 1599, νs(COO−) 1397, ν(phenolic, CO) 1267. µB = 1.36 BM.[Cu2(Shis)2(H2O)2]·H2O, 9
To the solution of H2Shis (0.255 g, 1.00 mmol) and LiOH (0.048 g, 2.00 mmol) in water (10 mL) was added a solution Cu(OAc)2·H2O (0.200 g, 1.00 mmol) in water (10 mL). The light green reaction mixture was filtered and allowed to stand in air at room temperature for several days to get a light green powder of 9. Yield: 0.199 g (57%). Anal. Calcd for C26H32N6O9Cu2: C, 44.4; H, 4.6; N 12.0; H2O, 7.7. Found: C, 44.6; H, 4.6; N, 12.0; H2O, 7.9%. IR (KBr, cm−1): ν(OH) 3401, ν(NH) 2895, νas(COO−) 1595, νs(COO−) 1382, ν(phenolic, CO) 1250. µB = 1.50 BM.[Cu2(Styr)2(H2O)]·2H2O, 10
This complex was prepared as same procedure as described for 7 using H2Styr·1.5H2O instead of H2Sbal·H2O. Dark green needle crystals of 10 were obtained. Yield: 0.286 g (76%). Anal. Calcd for C32H36N2O11Cu2: C, 51.1; H, 4.8; N 3.7; H2O, 7.2. Found: C, 51.3; H, 4.9; N, 3.6; H2O, 7.2%. IR (KBr, cm−1): ν(OH) 3418, ν(NH) 3160, νas(COO−) 1605, νs(COO−) 1451 and 1385, ν(phenolic, CO) 1275. µB = 1.12 BM.[Cu2(Stryp)2(H2O)], 11
This complex was prepared as same procedure as described for 5 using H2Styr·1.5H2O instead of H2Stryp. Dark green needle crystals of 11 were obtained. Yield: 0.274 g (72%). Anal. Calcd for C36H34N4O7Cu2: C, 56.7; H, 4.5; N 7.4; H2O, 2.4. Found: C, 56.8; H, 4.4; N, 7.4; H2O, 2.5%. IR (KBr, cm−1): ν(OH) 3451, ν(NH) 3219, νas(COO−) 1619, νs(COO−) 1484 and 1381, ν(phenolic, CO) 1271. µB = 1.27 BM.X-ray crystallography
The diffraction experiments carried out on a Bruker AXS SMART CCD diffractometer. The program SMART63 was used for collecting frames of data, indexing reflection and determination of lattice parameter, SAINT64 for integration of the intensity of reflections and scaling, SADABS64 for absorption correction and SHELXTL65 for space group and structure determination, least-squares refinements on F2. There are two lattice water molecules found in the lattice of 1 and 2, respectively. All the hydrogen atom positions of water molecules were located and their positional parameters were refined in the least-squares cycles. Selected crystallographic data and refinement details are displayed in Table 3.CCDC reference numbers 209712–209715 (4, 5, 10 and 11).
See http://www.rsc.org/suppdata/dt/b3/b310262a/ for crystallographic data in CIF or other electronic format.
| Complex | 4 | 5 | 10 | 11 |
|---|---|---|---|---|
| a R 1 = Σ‖Fo| − |Fc‖/ΣFo|. b wR 2 = [Σw(Fo2 − Fc2)2/Σw(Fo2)2]1/2. The absolute structure parameters for 10 and 11 are 0.03(1) and −0.009(15), respectively. | ||||
| Formula | C20H30N2O10Cu2 | C22H30N2O8Cu2 | C32H36N2O11Cu2 | C36H34N4O7Cu2 |
| Formula weight | 585.54 | 577.56 | 751.72 | 761.75 |
| T/K | 223(2) | 223(2) | 223(2) | 293(2) |
| Crystal system | Triclinic | Triclinic | Monoclinic | Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P21 | P21 |
| a/Å | 7.2289(3) | 7.6081(6) | 10.4143(5) | 10.3549(3) |
| b/Å | 8.9892(3) | 8.3925(7) | 11.0346(5) | 10.5453(2) |
| c/Å | 9.8982(4) | 9.7927(8) | 14.7206(7) | 15.9521(4) |
| α/° | 73.497(2) | 112.186(1) | 90 | 90 |
| β/° | 70.294(1) | 93.542(2) | 105.881(1) | 104.367(1) |
| γ/° | 82.701(2) | 104.951(2) | 90 | 90 |
| V/Å3 | 580.24 | 550.45(8) | 1627.09(1) | 1687.42(7) |
| Z | 1 | 1 | 2 | 2 |
| µ/mm−1 | 1.891 | 1.986 | 1.370 | 1.316 |
| Reflns collected | 3349 | 5868 | 9364 | 9642 |
| Independent reflns | 2047 | 1924 | 5049 | 4908 |
| R int | 0.0528 | 0.0362 | 0.0259 | 0.0307 |
| Final R [I > 2σ], R1,awR2b | 0.0423, 0.1161 | 0.0318, 0.0770 | 0.029, 0.0668 | 0.0387, 0.0846 |
Acknowledgements
This research was supported by grant R-143-000-153-112 to JJV from the National University of Singapore and by an Australian Research Council Discovery grant to K.S.M.References
- K. D. Karlin and J. Zubieta, Copper Coordination Chemistry: Biochemical & Inorganic Perspectives, Adenine Press Inc., Guilderland, New York, 1983, p. 371 Search PubMed.
- Bioinorganic Chemistry of Copper, ed. K. D. Karlin and Z. Tyeklár, Chapman & Hall, New York, 1993, p. 251 Search PubMed.
- W. Kaim and J. Rall, Angew. Chem., Int. Ed. Engl., 1996, 35, 43 CrossRef CAS.
- S. J. Lippard, J. M. Berg, Principles of Bioinorganic Chemistry, University Science Books, Mill Valley, CA, 1994, p. 283 Search PubMed.
- D. P. Kessissoglou, Bioinorganic chemistry: an inorganic perspective of life, Kluwer Academic Publishers, Dordrecht–Boston, 1995, p. 371 Search PubMed.
- M, Trèmolières and J. B. Bieth, Phytochem., 1984, 23, 501 Search PubMed.
- D. Meiwes, B. Ross, M. Kiesshauer, K. Cammann, H. Witzel, M. Knoll, M. Borchardt and C. Sandermaier, Lab. Med., 1992, 15, 24.
- R. Than, A. A. Feldman and B. Krebs, Coord. Chem. Rev., 1999, 182, 211 CrossRef.
- A. Rompel, H. Fisher, K. Büldt-Karentzopoulos, D. Meiwes, F. Zippel, H.-F. Nolting, C. Hermes, B. Krebs and H. Witzel, J. Inorg. Biochem., 1995, 59, 715 CrossRef.
- N. Oishi, Y. Nishida, K. Ida and S. Kida, Bull. Chem. Soc. Jpn., 1980, 53, 2847 CAS.
- P. Gentschev, N. Möller and B. Krebs, Inorg. Chim. Acta, 2000, 300, 442 CrossRef.
- F. Zippel, F. Ahlers, R. Werner, W. Haase, H.-F. Nolting and B. Krebs, Inorg. Chem., 1996, 35, 3409 CrossRef CAS.
- J. Reim and B. Krebs, J. Chem. Soc., Dalton Trans., 1997, 3793 RSC.
- L. M. Berreau, S. Mahapatra, J. A. Halfen, R. P. Houser, V. G. Young and W. B. Tolman, Angew. Chem., Int. Ed. Engl., 1999, 38, 207 CrossRef CAS.
- E. Monzani, G. Battaini, A. Perotti, L. Casella, M. Gullitti, L. Santigostini, G. Nardin, L. Randaccio, S. Geremia, P. Zanello and G. Opromolla, Inorg. Chem., 1999, 38, 5359 CrossRef CAS.
- E. Monzani, L. Quinti, A. Perotti, L. Casella, M. Gullitti, L. Randaccio, S. Geremia, G. Nardin, P. Faleschini and G. Tabbi, Inorg. Chem., 1998, 37, 553 CrossRef CAS.
- S. Parimala, K. N. Gita and M. Kandaswamy, Polyhedron, 1998, 17, 3445 CrossRef CAS.
- M. R. Malachowski, B. T. Dorsey, M. J. Parker, M. E. Adams and R. S. Kelly, Polyhedron, 1998, 17, 1289 CrossRef CAS.
- M. R. Malachowski, H. B. Huynh, L. J. Tomlinson, R. S. Kelly and J. W. Jr. Furbee, J. Chem. Soc., Dalton Trans., 1995, 31 RSC.
- M. R. Malachowski, L. J. Tomlinson, M. G. Davidson and M. J. Hall, J. Coord. Chem., 1992, 25, 171 CAS.
- H. Börzel, P. Comba and H. Pritzkow, Chem. Commun., 2001, 97 RSC.
- M. Gupta, P. Mathur and R. J. Butcher, Inorg. Chem., 2001, 40, 878 CrossRef CAS.
- C. Gerdemann, C. Eicken and B. Krebs, Acc. Chem. Res., 2002, 35, 183 CrossRef CAS.
- E. I. Solomon, U. M. Sundaram and T. E. Machonkin, Chem. Rev., 1996, 96, 2563 CrossRef CAS.
- A. Sánchez-Ferrer, J. N. Rodriguez-Lopez, F. García-Cánovas and F. García-carmona, Biochim. Biophys. Acta, 1995, 1247, 1 CAS.
- C. Eicken, B. Krebs and J. C. Sacchettini, Curr. Opin. Struct. Biol., 1999, 9, 677 CrossRef CAS.
- M.-Q. Chen, J.-X. Xu and H.-L. Zhang, J. Struct. Chem. (Chin.), 1991, 1, 1 Search PubMed.
- (a) J. D. Ranford, J. J. Vittal, D. Wu and X. Yang, Angew. Chem., Int. Ed., 1999, 38, 3498 CrossRef CAS; (b) S. Pakhomova, J. Ondrácek and F. Jursík, Collect. Czech. Chem. Commun., 1998, 63, 356 CrossRef CAS.
- J. J. Vittal and X. Yang, to be submitted for publication.
- J. R. Ferraro, Low-frequency Vibrations of Inorganic and Coordination Compounds, Plenum Press, New York, 1971 Search PubMed.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, 4th edn., John Wiley & Sons, New York, 1986, p. 191 Search PubMed.
- C. Djordjevic, M. Lee and E. Sinn, Inorg. Chem., 1989, 28, 719 CrossRef CAS.
- G. B. Deacon and R. Philips, Coord. Chem. Rev., 1980, 33, 227 CrossRef CAS.
- M. Tsaramyrsi, M. Kaliva, A. Salifoglou, C. P. Raptopoulou, A. Terzis, V. Tangorlis and J. Giapintzakis, Inorg. Chem., 2001, 40, 5772 CrossRef CAS.
- M. R. Wager and F. A. Walker, Inorg. Chem., 1983, 22, 3021 CrossRef CAS.
- L. Casella and M. Guillotti, Inorg. Chem, 1986, 25, 1293 CrossRef CAS.
- A. B. P. Lever, Inorganic Electronic Spectroscopy, Elsevier, Amsterdam, 1984, p. 553 Search PubMed.
- W. J. Geary, Coord. Chem. Rev., 1971, 7, 81 CrossRef CAS.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349 RSC.
- G. Murphy, C. O. Sullivan, B. Murphy and B. Hathaway, Inorg. Chem., 1998, 37, 240 CrossRef CAS.
- D. S. Marlin, M. M. Olmstead and P. K. Mascharak, Inorg. Chem., 2001, 40, 7003 CrossRef CAS.
- S. C. Chan, L. L. Koh, P.-H. Leung, J. D. Ranford and K. Y. Sim, Inorg. Chim. Acta, 1995, 236, 101 CrossRef CAS.
- N. R. Sangeetha, K. Baradi, R. Gupta, C. K. Pal, V. Manivannan and S. Pal, Polyhedron, 1999, 18, 1425 CrossRef CAS.
- J. J. Vittal and X. Yang, Cryst. Growth Des., 2002, 2, 259 CrossRef CAS.
- L. K. Koh, J. D. Ranford, W. T. Robinson, J. O. Svensson, A. L. C. Tan and D. Wu, Inorg. Chem., 1996, 35, 6466 CrossRef CAS.
- B. Bleaney and K. D. Bowers, Proc. R. Soc. London, Ser. A, 1952, 214, 451.
- R. L. Martin, in New Pathways in Inorganic Chemistry, ed. E. A. V. Ebsworth, A. G. Maddock and A. G. Sharpe, Cambridge University Press, Cambridge, 1968, p. 175 Search PubMed.
- R. Gupta, S. Mukherjee and R. Mukherjee, J. Chem. Soc., Dalton Trans., 1999, 4025 RSC.
- H. Saimiya, Y. Sunatsuki, M. Kojima, S. Kashino, T. Kambe, M. Hirotsu, H. Akashi, K. Nakajima and T. Tokii, J. Chem. Soc., Dalton Trans., 2002, 3737 RSC.
- S. Torrelli, C. Belle, I. Gautier-Luneau, J. L. Pierre, E. Saint-Aman, J. M. Latour, L. Le Pape and D. Luneau, Inorg. Chem., 2000, 39, 3526 CrossRef CAS.
- S. Albedyhl, M. T. Averbuch-Pouchot, C. Belle, B. Krebs, J. L. Pierre, E. Sanit-Aman and S. Torelli, Eur. J. Inorg. Chem., 2001, 1457 CrossRef CAS.
- C. Belle, C. Beguin, I. Gautier-Luneau, S. Hamman, C. Philouze, J. L. Pierre, F. Thomas, E. Saint-Aman and M. Bonin, Inorg. Chem., 2002, 41, 479 CrossRef CAS.
- A. Neves, L. M. Rossi, A. J. Bortoluzzi, B. Szpoganicz, C. Wiezbicki, E. Schwingel, W. Haase and S. Ostrovsky, Inorg. Chem., 2002, 41, 1788 CrossRef CAS.
- J. Mukherjee and R. Mukherjee, Inorg. Chim. Acta, 2002, 337, 429 CrossRef CAS.
- R. Wegner, M. Gottschaldt, H. Görls, E-G. Jäger and D. Klemm, Chem. Eur. J., 2001, 7, 2143 CrossRef CAS.
- C.-H. Kao, H.-H. Wei, Y.-H. Liu, G.-H. Lee, Y. Wang and C.-J. Lee, J. Inorg. Biochem., 2001, 84, 171 CrossRef CAS.
- S. Casellato, S. Tamburimi, P. A. Vigato, A. de Stefani, M. Vidali and D. E. Fenton, Inorg. Chim. Acta, 1983, 69, 45 CrossRef CAS.
- K. D. Karlin, M. S. Nasin, B. I. Cohen, R. W. Cruse, S. Kaderli and A. D. Zuberbühler, J. Am. Chem. Soc., 1994, 116, 1324 CrossRef CAS.
- K. D. Karlin, N. Wei, B. Jung, S. Kaderli, P. Niklaus and A. D. Zuberbühler, J. Am. Chem. Soc., 1993, 115, 9506 CrossRef CAS.
- G. Battaini, E. Monzani, L. Casella, L. Santagostini and R. Pagliarin, J. Biol. Inorg. Chem., 2000, 5, 262 CrossRef CAS.
- C. T. Yang, B. Moubaraki, K. S. Murray, J. D. Ranford and J. J. Vittal, Inorg. Chem., 2001, 40, 5934 CrossRef CAS.
- C. T. Yang and J. J. Vittal, Inorg. Chim. Acta, 2003, 344, 65 CrossRef CAS.
- SMART & SAINT Software Reference Manuals, Version 6.22, Bruker AXS Analytic X-Ray Systems, Inc., Madison, WI, 2000 Search PubMed.
- G. M. Sheldrick, SADABS, Software for Empirical Absorption Correction, University of Göttingen, Germany, 2000 Search PubMed.
- SHELXTL Reference Manual, Version 5.1, Bruker AXS, Analytic X-Ray Systems, Inc., Madison, WI, 1997 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Thermogravimetry curves, UV/vis spectra, a plot of 1/νvs. 1/[S], structure diagrams, plots of χ and μeff, and H-bond distances. See http://www.rsc.org/suppdata/dt/b3/b310262a/ |
| This journal is © The Royal Society of Chemistry 2004 |