Heterogeneous enantioselective catalysts: strategies for the immobilisation of homogeneous catalysts
Paul
McMorn
and
Graham J.
Hutchings
*
Department of Chemistry, Cardiff University, P.O. Box 912, Cardiff, UK CF10 3TB. E-mail: hutch@cf.ac.uk
First published on 29th January 2004
Abstract
Enantioselective formation of C–H, C–C, C–O and C–N bonds has been extensively studied using homogeneous asymmetric catalysts for many years. However, these catalysts have yet to make a significant impact in the industrial synthesis of fine chemicals. A central reason is that homogeneous asymmetric catalyst design requires relatively bulky ligands and catalyst re-use through recovery and recycle often causes problems. One mechanism to overcome this problem is to immobilise the asymmetric catalyst onto a support and the resulting heterogeneous asymmetric catalyst can, in principle, be readily re-used. This tutorial review covers the different methodologies for immobilisation, including: adsorption, encapsulation, tethering using a covalent bond and electrostatic interaction and is aimed at both researchers new to the field and those with a wider interest in the immobilisation of homogeneous catalysts. Most importantly, recent studies will be highlighted that demonstrate that immobilised catalysts can give higher enantioselection when compared with their non-immobilised counterparts and the question of how high enantioselection can be achieved is addressed.
 Paul McMorn Paul McMorn | Dr Paul McMorn graduated from the University of Liverpool in 1993 with a BSc (Hons) in chemistry, and in 1996 with a PhD. Between 1996 and 1998 he was appointed as a post-doctoral research associate at Liverpool University studying catalytic asymmetric aziridination of styrene using microporous materials. Between 1998 and 2002 he held a number of post-doctoral positions studying the design, preparation and application of immobilised catalysts. In 2002 he was appointed as a Cardiff Research Fellow. |
 Graham J. Hutchings Graham J. Hutchings | Professor Graham Hutchings is currently Head of Department, School of Chemistry, Cardiff University, a position he has held since 1997. He has worked in the field of immobilisation of enantioselective catalysis for the last 10 years working mainly on zeolite immobilised catalysts. He has worked in the field of heterogeneous catalysis since 1975, working initially in industry at ICI Petrochemicals Division on Teeside and AECI Ltd. in Modderfontein. In 1984, he was appointed to the academic staff at the University of Witwatersrand, where he is currently a visiting Professor. In 1987, he returned to the UK to help establish the Leverhulme Centre for Innovative Catalysis at Liverpool University. |
Introduction
The enantioselective formation of C–H, C–C, C–O and C–N bonds represents one of the biggest challenges in synthetic chemistry. Significant progress has been achieved in the last thirty years with homogeneous catalysts and the main emphasis has been on ligand design to control the reactant approach to the active centre, so that the reaction occurs at only one face of the approaching prochiral substrate. This has led to the design of libraries of chiral ligands with varying degrees of steric bulk that can control the enantioselection, e.g. C2 symmetric ligands : salen 1, bis(oxazoline) 2, BINAP 3.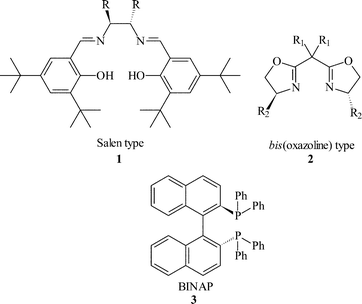
In 2001, the significant achievements in the design and application of asymmetric homogeneous catalysts were recognised by the award of the Chemistry Nobel Prize to W. S. Knowles and R. Noyori for enantioselective hydrogenation catalysis and K. B. Sharpless for enantioselective oxidation catalysis.1 However, these elegant homogeneous processes are not making as much impact in the arena of fine chemicals synthesis as may have been expected.2 One reason for this state of affairs is that the ligands used to create the chiral active centre (e.g. 1, 2, 3) are relatively expensive and must be recovered and re-used if the catalytic process is to be viable. Often this proves to be difficult. A method for overcoming this difficulty is to immobilise the asymmetric catalyst on a non-soluble support, thereby creating a chiral heterogeneous catalyst that can be readily recovered from reaction mixtures. Immobilisation of a chiral catalyst is just one method of creating a chiral heterogeneous catalyst and Davies3 has considered the many different ways in which chirality can be introduced into a catalyst system to ensure a chiral transition state is achieved, i.e. chirality can be introduced by using a chiral surface, a chiral solvent, a chiral reactant or a chiral modifier. The topic of immobilisation has been the subject of several hundred publications and several previous reviews4,5 and the most recent by Song and Lee6 has focused on the extensive range of reactions exemplified by immobilised asymmetric catalysts. In view of this, we do not aim to present a comprehensive overview of this field rather, in this review, we focus on the different methodologies used to create immobilised heterogeneous asymmetric catalysts and aim to address the question of how high enantioselectivity can be achieved with immobilised catalysts.
Immobilisation strategies
Four distinct methodologies have been developed for the heterogenisation of homogeneous catalysts or the creation of heterogeneous chiral catalysts. The following sections contain representative examples of given methodologies. In addition, where comparisons with the equivalent homogeneous catalyst have been made, these are given.Adsorption
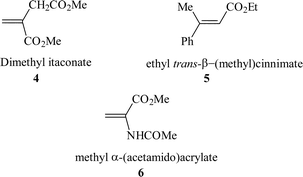
Immobilisation of chiral ligands onto surfaces has been one of the major pathways to achieving a heterogeneous asymmetric catalyst. For example, the adsorption of cinchona alkaloids onto metal surfaces has been extensively studied as enantioselective hydrogenation catalysts for the conversion of α-ketoesters to lactates. This body of work has been reviewed recently7–9 and this topic will not be included in this review.Catalysts immobilised by this method can sometimes rely only on van der Waals interaction between the catalyst and the support. As this is only a weak interaction, the catalyst will readily leach into solution as the catalyst will reach equilibrium between the surface absorbed species and solution species. The stability of the supported catalyst can be improved by modifying the catalyst and support to allow hydrogen bonding to occur. Chiral rhodium phosphine catalysts have been immobilised on silica in this way where the phosphine ligand was modified to incorporate sulfonic acid groups which hydrogen bonded with the silanols present on the silica surface (Fig. 1).10 Catalysts which contain a sulfonic acid counterion can also be immobilised in this way. In this case, the sulfonate counterion interacts with both the support and the catalyst giving rise to an immobilisation strategy more in keeping with electrostatic immobilisation, an approach that is described later in the review. In this specific method, grafting was carried out in dichloromethane and repeated washing of the catalyst with anhydrous dichloromethane was reported not to cause any catalyst loss. However if protic, polar solvents were used for washing (methanol and ethanol), total loss of the catalyst into solution was observed. This has important implications concerning the long term storage and re-use of these catalysts as polar, protic compounds need to be avoided, including any water which may be absorbed from the air. To study the applicability of this system, the hydrogenation of dimethyl itaconate 4, ethyl trans-β-(methyl) cinnimate 5 and α-(acetamido) acrylate 6 was carried out with the aim of comparing the homogeneous reaction (in methanol) with the heterogenised catalysts (in n-heptane). Low to moderate e.e.s. were obtained overall and the e.e.s obtained in the heterogeneous system were generally comparable to those obtained for the homogeneous reactions, although they were lower in some cases. Heterogenisation did give a significant advantage for the hydrogenation of ethyl trans-β-(methyl) cinnimate as the heterogenised reaction was faster than the homogeneous counterpart, which is unusual as immobilisation tends to introduce pore diffusion limitations, which results in lower rates of reaction. Multiple re-use experiments were reported to show no loss of catalyst activity, but no data was given on this aspect.
![Immobilisation of Rh+
[R–R–BDPBzPSO3]− complex by adsorption.10 (Reproduced with permission from Wiley-VCH.)](/image/article/2004/CS/b200387m/b200387m-f1.gif) | ||
| Fig. 1 Immobilisation of Rh+ [R–R–BDPBzPSO3]− complex by adsorption.10 (Reproduced with permission from Wiley-VCH.) | ||
The principles of carrying out homogeneous reactions under biphasic conditions (where the catalyst is in a different liquid phase to the reactants and products) have been applied to the immobilisation of homogeneous catalysts on inorganic supports. Two different types of biphasic catalytic systems have been investigated, (a) two immiscible solvent phases, and (b) one solvent phase which contains a surfactant. In the latter case, the catalyst is normally contained within the micelles formed by the surfactant. Examples of immobilisation by these two methods are outlined below.
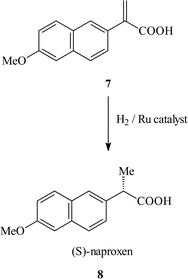
For the two immiscible solvent biphasic method, the catalyst is dissolved in a suitable solvent and this mixture is used to form a thin film on the surface of the support. The reaction is then carried out in a solvent which is immiscible with the solvent which contains the catalyst. Effectively this is catalysis in a biphasic system where one of the liquid phases is immobilised on the support material. Davis et al.11 reported the immobilisation of RuBINAP complexes for the asymmetric hydrogenation of 2-(6′-methoxy-2′-napthyl)acrylic acid 7 to naproxen 8 (Scheme 1) on controlled pore glass beads. The catalyst was immobilised within the ethylene glycol layer on the surface of the beads which was immersed in an organic solvent which contains the reactants and products. The catalyst remains in the polar ethylene glycol phase and, for this reason, little leaching is observed (17–40 ppm of Ru following reaction). It was also reported that, by careful catalyst preparation and by controlling the solvent polarity by using solvent mixtures, the amount of Ru in solution could be reduced to 32 ppb without adversely affecting the e.e. As is typically observed, immobilisation results in a decrease in the rate of reaction (TOF ca. 50 h−1) compared to the homogeneous reaction (TOF 100 h−1). A biphasic homogeneous reaction was also carried out using the same solvent combinations used for the immobilised catalyst and this was found to have a much lower rate of reaction (TOF 15 h−1). This difference is more likely to be due to greater surface contact between the two phases in the immobilised system rather than an increase in the activity of the immobilised catalyst. The e.e.s of the immobilised system were found to be dependent upon the amount of ethylene glycol, but once optimised an e.e. of 96% was reported which is identical to the maximum e.e. of 96% reported for the homogeneous catalyst.
 | ||
| Scheme 1 | ||
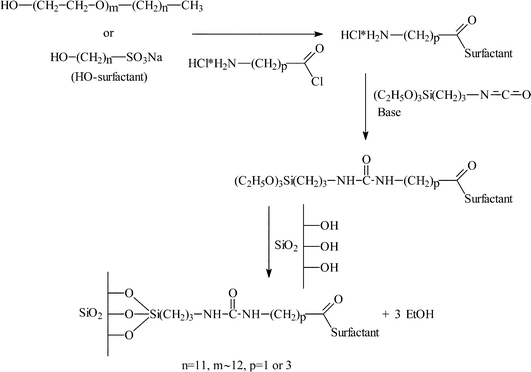
The application of the solvent/surfactant biphasic reaction system to catalyst immobilisation has been reported by Flach et al.12 using a range of surfactants. The surfactant was typically reacted with an aliphatic amino acyl chloride (Fig. 2), and this compound was reacted with 3-(triethoxysilyl) propyl isocyanate. The surfactant was physisorbed onto the support surface and the presence of the surfactant molecules on the solid support results in the support exhibiting micelle type qualities and, hence, it can be used to immobilise catalysts which would normally be used in the solvent/surfactant biphasic system. The catalyst to be immobilised, (Rh[BPPM]BF4), was then stirred with the surfactant modified support and investigated for the asymmetric hydrogenation of (Z)-α-acetamidocinnamate 9. Excellent e.e.s were obtained for the non-immobilised systems (83–95%) and only a small reduction in e.e. was observed for the equivalent immobilised system (78–93%). Immobilisation had a far greater effect on reaction rate where all but one of the immobilised systems gave a lower rate compared to that of the homogeneous reaction. The re-use of these immobilised systems was also reported and, although the half life of the reaction increased on re-use, which was considered to be the result of ligand loss, there was no change in the degree of enantioselection.
 | ||
| Fig. 2 Covalent attachment of surfactants over silica.12 (Reproduced with permission from Springer-Verlag.) | ||
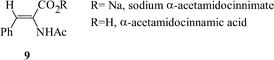
Jamis et al.13 adsorbed Ru-BINAP into hexagonal mesoporous silica (HMS) which had been pre-treated with a variety of silanisation agents and studied the hydrogenation of sodium α-acetamidocinnimate 9. Moderate e.e.s (37–57%) could be obtained, the catalysts showed some stability under the reaction conditions as re-use of the catalyst invariably gave a small reduction in e.e. and conversion. However, the authors report the loss in activity was not considered to be due to leaching of metal from the catalyst.
Encapsulation
Encapsulation is the only catalyst immobilisation process which does not require any interaction between the catalyst and the support, and because of this it is the only method which attempts to mimic the homogeneously catalysed reaction process. Other methods, by necessity, lead to changes in the catalyst. For example, covalent tethering relies on modification of the ligand (which may influence electronic character of the ligand and/or the conformation of the ligand), and physisorption and ion-exchange methods result in the catalyst being in close proximity to the support which may also effect electronic properties and ligand conformation.To satisfy the condition of encapsulation, the catalyst must be larger than the pores of the support material to prevent loss of the catalyst into solution during the course of the reaction. As the catalyst complex is larger than the pores of the support, techniques such as impregnation can not be used to synthesise these catalysts. The supported catalyst can be prepared by either i) assembling the catalyst within the pores of the support or ii) assembling the support around the catalyst, and both of these methods have been used to encapsulate catalysts. The route used for the catalyst encapsulation is dictated by the chemistry of the support and the catalyst complex. If the catalyst is to be assembled within the pores of the support, then the support needs to be stable under the reaction conditions used. Fortunately, the materials used as supports tend to be inert so this is generally not a problem. The chemical processes used to prepare the catalyst are of crucial importance and side reactions need to be avoided and each step must have high yields. If many different species are present alongside the catalyst (due to side reactions in the ligand synthesis), it is likely that reduced yields and enantioselection will be obtained, and because these moieties are encapsulated they are difficult to remove selectively from the support. If the support is assembled around the catalyst then the catalyst needs to be stable to the synthesis conditions of that support. Hence, if the catalyst can be made easily in a small number of steps then assembly within the pores is preferred. If the catalyst is difficult to make but is stable then formation of the support around the catalyst is preferred.
Encapsulation by catalyst assembly within pores
This method is typically used to immobilise metal Salen complexes within the supercages of faujasite type zeolites.14,15 The manganese complex of trans-(R,R)-1,2-bis(salicyideneamino)-cyclohexane was immobilised within the pores of zeolite Y. Initially, the zeolite was partially ion exchanged with Mn2+ followed by addition of the building units of the chiral ligand (salicylaldehyde and trans-(R,R)-1,2-diaminocyclohexane) as shown in Fig. 3. Hölderich and co-workers. later extended this study to other salen type ligands with a variety of transition metals as the active centre.16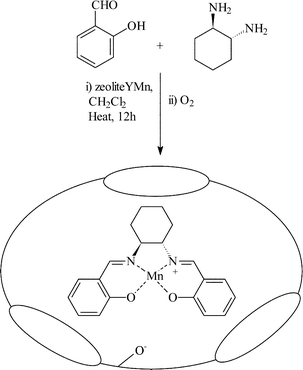
The catalyst reported by Corma et al.15 was studied with five different alkenes (Table 1) using NaOCl as the terminal oxidant for both the immobilised and homogeneous catalysts. The rates of the reaction for the heterogeneous catalysts were lower than the rates of the homogeneous catalysts, which is unsurprising considering the increase in the barrier to diffusion which immobilisation will always bring. Despite this, comparable conversions were obtained for indene and styrene epoxidation. In all cases a reduction in e.e. was observed upon immobilisation, although moderate e.e.s could be obtained for cis-β-methyl styrene (e.e. of 58% for the trans epoxide) and indene (e.e. of 50%). Uncatalysed epoxidation reactions (which were not studied) and uncomplexed Mn ions were cited as reasons for the decrease in enantioselectivity; however, the confinement effect of the zeolite may also play a role. As the zeolite is a rigid structure and the catalyst is a close fit within the supercage, this may subtly effect the conformation of the ligand and reaction intermediates by preventing changes to ligand geometry during the reaction. If this were to occur, significant differences in e.e. may be obtained between the immobilised and homogeneous systems. To overcome the problem of unfavourable interactions between the complex and the support, Hölderich and co-workers16 partially de-aluminated the zeolite to increase the size of the supercage prior to assembling the salen complex, together with extraction, to remove any immobilised species from the surface of the crystallites. Ideally for these systems, the complex will be immobilised within the pores of the zeolite, however, it is possible to immobilise the complex on the exterior surface of the catalyst. By using a polymeric iodosylbenzene, which cannot diffuse into the zeolite pores, the absence of active catalysts on the exterior surface and a lack of any active solution species was demonstrated. This also agreed with the analysis of the liquid phase after reaction which demonstrated that Mn was not leached into solution. An alternative strategy would be to use a substrate which was too large to enter the pores.
| Alkenea | Catalyst | Conversion %b | Epoxide selectivity | Eec |
|---|---|---|---|---|
| a Reactions were run at 5 °C in CH2Cl2. b Reaction time 2–3 h for (salen)MnIIICl and 12–15 h for (salen)MnIIIY. c Ee of trans epoxide. d Homogeneous catalyst. e Heterogeneous catalyst. | ||||
|
(salen)MnIIICld | 37 | 65 | 8 |
| (salen)MnIIIYe | 11 | 65 | 5 | |
|
(salen)MnIIICl | 47 | 80 | 27 |
| (salen)MnIIIY | 40 | 61 | 20 | |

|
(salen)MnIIICl | 28 | 100 | 74 |
| (salen)MnIIIY | 5 | 76 | 58 | |
|
(salen)MnIIICl | 23 | 90 | 41 |
| (salen)MnIIIY | 11 | 100 | 24 | |
|
(salen)MnIIICl | 30 | 100 | 60 |
| (salen)MnIIIY | 20 | 96 | 50 | |
Hölderich and co-workers16 used a similar approach to study the immobilisation of transition metal salen complexes for the diastereoselective epoxidation of α-pinene and limonene using oxygen. In certain cases the immobilised and homogeneous catalysts gave comparable yields, selectivities and d.e.s for the reaction of α-pinene, but for limonene the d.e.s were lower than those obtained from the homogeneous reaction. This approach has also been used to immobilise a palladium-salen complex for the enantioselective hydrogenation of 3-methyl-2-cyclohexanone, however, low e.e.s were obtained (ca. 7%).17 A possible reason for the low e.e.s may be due to the size of the ligand in relation to the host pore diameter, as the Jacobsen–Katsuki salen ligand was used, which contains four tBu groups. Corma and co-workers15 calculated that complexes which contain this ligand could not be accommodated within the supercage of the support, therefore, it would be expected that few of the Pd centres are properly modified by the ligand. This would give rise to a mainly racemic reaction, which is observed, despite the high ligand content (one ligand every 4 supercages) used in the study.
Encapsulation by forming the support around the catalyst
Encapsulation of complexes by this method has mainly been achieved through the use of silica based supports. These can be divided into pure silica supports and polydimethylsiloxane (PDMS) films. Using sol–gel processes, the support is polymerised around the pre-formed complex thus leading to encapsulation. One drawback of this method is the pore diameters and pore openings are poorly defined compared to supports such as zeolites. This can lead to a number of different types of encapsulation sites, where the space around the catalyst can vary significantly. If the catalyst is immobilised in a dense part of the support then the space between the walls of the support and the catalyst may be small, which in turn may influence the conformations of the catalyst – substrate complex that can be accommodated. This can have either a positive or negative effect on the e.e. but will always have a strong negative effect on the rate of reaction because of the introduction of severe diffusion limitations. If the catalyst is immobilised in a less dense region of the support, then behaviour similar to the homogeneous reaction is to be expected with perhaps only a slight decrease in the reaction rate. Since the pore diameters can vary, care must be taken to ensure that any catalyst not properly immobilised must be removed prior to reaction to ensure no homogeneous reaction takes place.PDMS films have been used to immobilise Rh-MeDuPHOS for the enantioselective hydrogenation of methyl-2-acetamidoacrylate (in water) and methylacetoacetate (in methanol).18 For both of these reactions high e.e.s were obtained with the heterogenised catalyst (90–96%), but this is still slightly reduced compared to the homogeneous reactions which typically give e.e.s of 99%. The measured TOF for the immobilised catalysts show an order of magnitude reduction of rate compared to the homogeneous reaction, which is entirely due to the requirement of small pores for successful encapsulation. The leaching of Rh-MeDuPHOS from the PDMS membrane has been studied by contacting the immobilised catalyst with a range of solvents. Those which are effective solvents for the dissolution of the complex (methanol and dichloromethane) resulted in 31 and 52% leaching respectively. Others such as xylene, heptane and water resulted in ≤3% leaching.
Jamis et al.13 studied the immobilisation of rhodium BPPM and ruthenium BINAP complexes by entrapment within a silica matrix by using sol–gel chemistry, and used these to study the hydrogenation of sodium α-acetamidocinnimate 4. Moderate e.e.s (41%) could be obtained; however, the catalysts were not stable and re-use of the catalyst invariably gave a large decrease in e.e. and usually a decrease in conversion. The loss in activity was not considered to be due to leaching of the catalyst as the metal content of the reaction filtrates were reported to be negligible.
Covalent tethering
Immobilisation of complexes using covalent tethering techniques is, at present, the most favoured approach to designing stable heterogeneous asymmetric catalysts. Different strategies have been developed depending upon the reaction being catalysed.Carbon–carbon bond formation
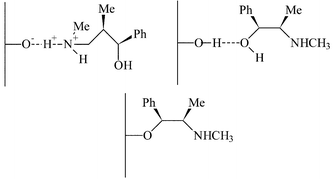 | ||
| Fig. 4 Possible interactions of (−)-ephedrine with the surface of mesoporous micelle templated silicas.20 | ||
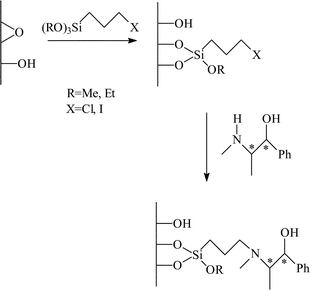 | ||
| Fig. 5 Immobilisation of (−)- or (+)- ephedrine over mesoporous micelle templated silicas by covalent tethering.20 | ||
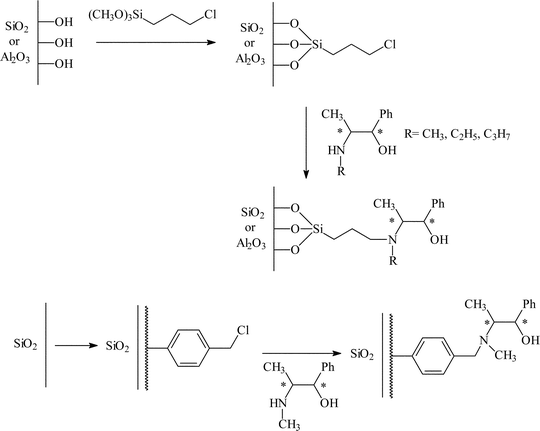
One of the most widely studied systems using this approach is the immobilisation of chiral auxiliaries for the addition of diethyl zinc to aldehydes to synthesise chiral secondary alcohols. This was first reported by Soai et al.19 for the immobilisation of a series of chiral N-alkylnorephedrines on silica gel, alumina and silica gel coated with chloromethylated polystyrene (Fig. 6). The first step was the grafting of (3-chloropropyl)triethoxy silane onto the surface hydroxy groups of alumina and silica. This gave a chlororopropyl modified material which was then reacted with the chiral auxiliary where the auxiliary displaces the Cl group and becomes covalently bonded to the support via the Si-propyl tether. A slightly different approach was used for silica gel coated with chloromethylated polystyrene. Rather than using (3-chloropropyl)triethoxy silane as the linker between auxiliary and support, (chloromethyl)styrene and divinyl benzene were copolymerised onto the surface of silica. This resulted in a surface rich in C6H4CH2Cl groups which were then reacted with the chiral auxiliary. Reasonable yields and moderate e.e.s were obtained using these catalysts. A range of aldehydes were studied, although the study focused mainly on benzaldehyde, which gave the highest overall e.e. (59%). The alumina supported catalysts tended to give higher e.e.s (40–59%) compared to the silica gel immobilised catalyst (25–39%), although little information concerning catalyst re-use was provided. A more in depth study was carried out into the silica gel/chloromethylated polystyrene immobilised catalyst. With this system, e.e.s up to 56% were reported with yields of 64%. Catalysts immobilised in this way could be recycled without the loss of enantioselectivity.
The surface grafting of ephedrine for the alkylation of benzaldehyde was studied in detail by Lasperas and co-workers,20 with particular emphasis on optimising the support for the reaction. The mesoporous MCM-41 was used as the support rather than silica gel as this has well defined pore dimensions, typically in the region of 40Å, which helped increase the rate of diffusion of the reactants to the active site. Initially, the same approach was used to graft the ephedrine chiral auxiliary to the support by modifying the support with (3-halopropyl)triethoxy silanes (Cl and I were studied) and then attaching the auxiliary to the support by displacement of the halide. Enhanced yields of the alkylation product were observed (>80%, calculated from conversion and selectivity) when compared to the studies of Soai et al.19 The e.e.s were comparable for both studies (26–37%) but were inferior to the e.e.s obtained in the equivalent homogeneous reaction (67%). The unmodified support was also found to be active for the racemic reaction, although the rate was much lower than when the ephedrine was present. The residual activity of the support was concluded to be responsible for the lower e.e.s in the immobilised systems.
This raises a key point for the comparison of homogeneous systems with immobilised systems. The chiral auxiliary utilised in the homogeneous reaction (ephedrine) is not the same auxiliary used in the immobilised system since, in the immobilised system, the N moiety is more highly substituted. This may play a role in determining the geometry of the transition state (and hence the e.e.) and also the rate of the reaction. To gain a more accurate idea of the maximum expected e.e., the structure of the auxiliary tested in the homogeneous reaction should be as close to the immobilised auxiliary as possible, even though the ultimate aim is comparison with the existing homogeneous system.
It was subsequently discovered that the ephedrine could directly interact with the surface either through the interaction of silanol groups with the N or O moiety, or by the formation of Si–O–C moieties formed by condensation between silanols and the OH group present in ephedrine.20 This resulted in similar e.e.s to those obtained by the covalently bound ephedrine. Re-use experiments revealed the same decrease in enantioselectivity observed for the covalently bonded systems, suggesting that, even in the covalently bonded systems, the surface bonded ephedrine constitutes an important part of the reaction system.
An alternative approach to the immobilisation of ephedrine was reported by Chung and Rhee.21 Silica was treated with 3-aminopropyltriethoxysilane, which was used as an anchoring site for a dendrimer. After dendrimer formation the ephedrine was immobilised onto the end of the dendrimer (Fig. 7). This immobilised catalyst gave comparable e.e.s to other ephedrine immobilised systems and could be re-used three times without any loss of selectivity or e.e., which is a significant advantage over other methods mentioned above.
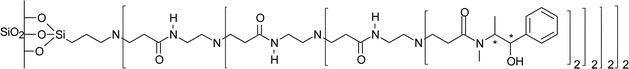 | ||
| Fig. 7 Immobilisation of dendritic chiral amino alcohol over silica.21 | ||
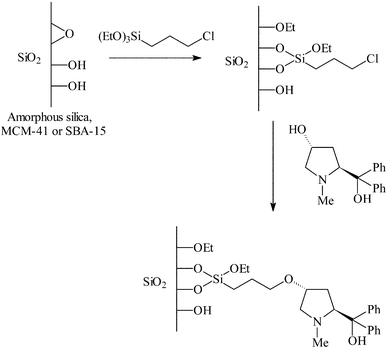
Proline (an alternative amino alcohol to ephedrine) has been immobilised on mesoporous silicas for the above reaction.22 3-Chloropropyltriethoxysilane was first grafted onto the silica surface and the proline immobilised via an ether linkage as shown in Fig. 8. To study the effect of pore diameter on the reaction, amorphous silica (84 Å), MCM-41 (23–30 Å) and SBA-15 (85 Å) were used as supports. Two methods were used to carry out the reactions: method (a) where the catalyst was vacuum dried prior to reaction, and method (b) where the catalyst was vacuum dried and pre-treated with n-butyl lithium prior to reaction. The pre-treatment with butyl lithium ensured all of the silanols on the catalyst surface were capped and hence species related to those in Fig. 4 could not form, and all traces of water were removed from the system. Generally, enhanced e.e.s were obtained using method (b). The e.e.s obtained for the MCM-41 and silica gel immobilised systems are broadly consistent with those obtained in the ephedrine system (20–50%). However, immobilisation on SBA-15 resulted in slightly higher e.e.s (64 and 75%) but these are still lower than the e.e.s obtained for the homogeneous system (ca. 90%).
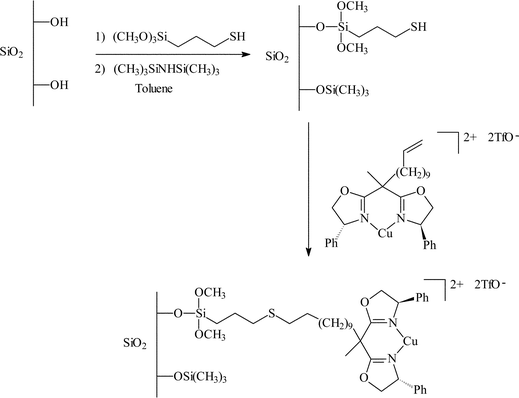 | ||
| Fig. 9 Chiral copper(II) bisoxazoline covalently anchored to silica and MCM-41 for the enantioselective Friedel–Crafts hydroxyalkylation.23 | ||
There are several key points described in this study which may contribute to the promising results obtained with the immobilised catalysts. The moiety used as a linker to attach the bis(oxazoline) to the mercaptopropyl modified support contained 9 CH2 units. This was to ensure that support–complex interactions were minimised. Secondly, the number of immobilised species was kept low to prevent any complex–complex interactions which may reduce the e.e., and finally the residual silanol groups were capped by reaction with hexamethyldisilazane, as these were reported to reduce the e.e. obtained with the immobilised catalysts.
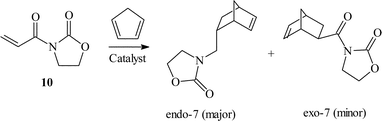 | ||
| Scheme 2 | ||
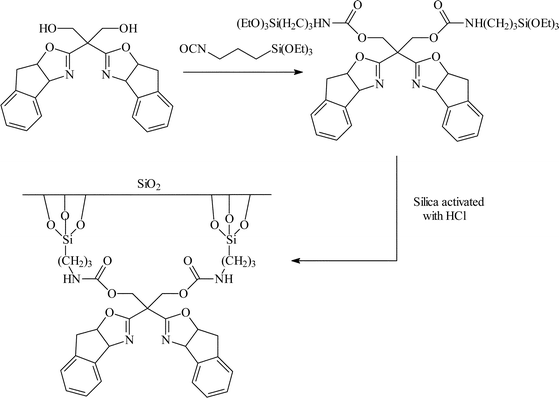 | ||
| Fig. 10 Chiral copper(II) bisoxazoline covalently anchored to silica the enantioselective Diels–Alder reaction.24 | ||
To understand the role of the silica support further, the homogeneous reaction (with the unmodified ligand) was studied in the presence and the absence of the silica support. In the absence of the silica an e.e. of 80% was obtained. This decreased to 66% when the silica was present. The authors suggested that the reduction in e.e. was due to the complexation of the copper directly to the silanol groups on the surface of the silica. This process gives rise to racemic sites which will be expected to give a reduction in e.e. Other Lewis acidic metals were also studied for the immobilised system; Sc and Yb gave racemic products, Co gave 16% e.e. and Zn gave 11% e.e.
3-Membered ring synthesis
Li and co-workers25 reported the immobilisation of a variant of the Sharpless epoxidation catalyst on MCM-41 and silica. This was achieved by reacting tartaric acid with 3-aminopropyltriethoxysilane using a number of steps, and the triethoxysilane moiety used to attach the modified tartramide to the silica surface. The final step was addition of the titanium centre in the form of titanium tetraisopropoxide to give the catalyst as shown in Fig. 11. The catalyst once formed was not isolated but used immediately for the epoxidation of allyl alcohol. The performance of the immobilised catalyst compared well with data obtained for titanium isopropoxide/diethyltartrate (which was studied as a homogeneous analogue). The turnover numbers were 10–14 (15 for the homogeneous reaction) and the e.e.s 78–86 (83% for the homogeneous reaction). The catalysts immobilised on silica gave slightly higher turnover numbers and e.e.s than those obtained for those immobilised on MCM-41. Unfortunately no catalyst re-use data was reported, and there was no reported attempt to study the catalyst stability under the reaction conditions.
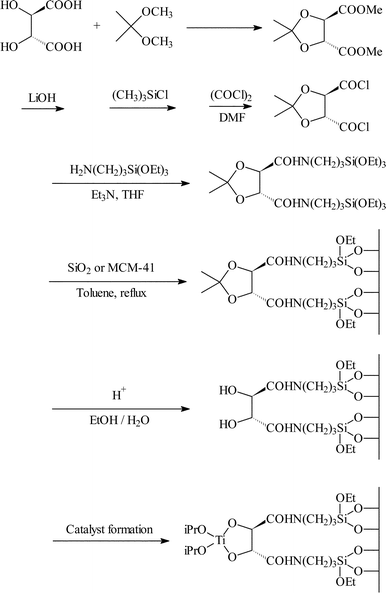 | ||
| Fig. 11 Covalent immobilisation of a chiral titanium–tartaric acid derivative over silica and MCM-41 for the asymmetric epoxidation of allyl alcohol.25 | ||
The immobilisation of the Jacobsen–Katsuki salen complexes by covalent tethering has also been studied. Pini et al.26 studied the immobilisation of Mn(III) salen on silica by forming a sulfide link between ligand and organically modified silica using the two methods outlined in Fig. 12. Both of the methods outlined led to identical materials. 1,2-Dihydronapthalene, indene and 1-phenylcyclohexene were epoxidised with e.e.s of 31, 38 and 58% respectively.
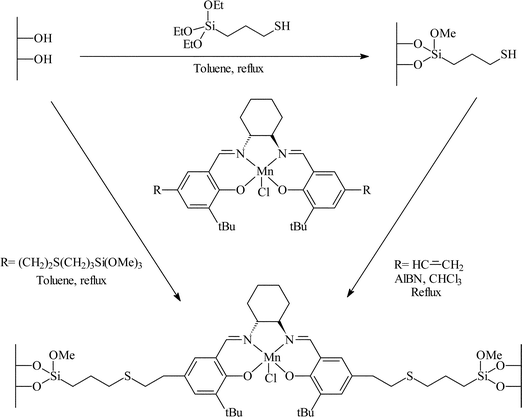 | ||
| Fig. 12 Covalent immobilisation of Mn(II) salen complex over silica for the asymmetric epoxidation of alkenes.26 (Reproduced with permission from Elsevier.) | ||
Choudary et al.27 reported the immobilisation of a chiral salen complex on silica gel. Several strategies were studied as outlined in Fig. 13, where iodosylbenzene and m-CBPA were used as terminal oxidants. Styrene, α-methyl styrene and both cis and trans stilbene were studied with a number of these catalysts. The e.e.s of the immobilised catalysts were poor, where most of the catalysts gave only a racemic product. For styrene epoxidation, only one catalyst gave an e.e. comparable to that obtained for the homogeneous reaction (15 and 16% respectively). Re-use of this catalyst showed a significant decrease in epoxide yield but only a small decrease in e.e. over three reaction cycles.
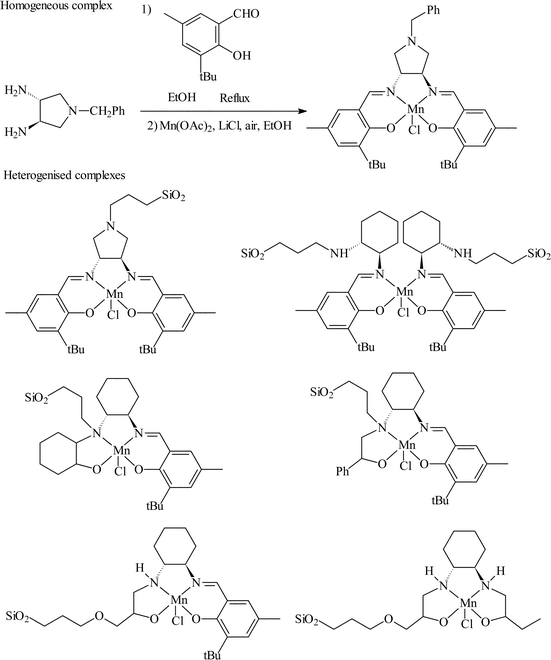 | ||
| Fig. 13 Covalent immobilisation of Mn(II) salen complexes over silica for the asymmetric epoxidation of alkenes.27 | ||
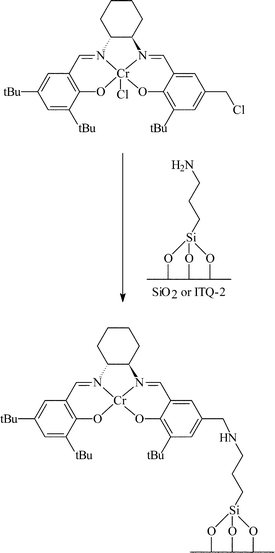
A combination of covalent attachment and electrostatic interaction has been used to immobilise Cr salen complexes for the enantioselective hydrolysis of epoxides with trimethylsilyl azide.28 The surface of silica, MCM-41 and ITQ-2 were initially functionalised with 3-aminopropyltriethoxysilane. For the electrostatic immobilisation, this was reacted with a chromium salen complex and immobilisation was achieved by an electrostatic interaction between the Cr salen complex and the NH2 group of the aminopropyl silane as shown in Fig. 14. Although this method gave e.e.s of 50–70%, considerable leaching was observed and the e.e. was likely to be due to the solution species acting as a homogeneous catalyst. The Cr salen complex was also immobilised by attaching the ligand to the NH2 group as shown in Fig. 15. The stability of the complex with respect to leaching was much improved, however, the e.e.s obtained were low (8–18%). The decreased e.e. was reportedly due to the fact that in the homogeneous reaction, two Cr salen molecules are required to give high e.e.s. As the Cr salen complex is anchored in place in the immobilised catalyst, it is difficult to form the transition state involving two Cr salen molecules, especially since it would be difficult to get two salen complexes in close proximity due to the combination of the steric bulk of the salen ligand and the short covalent tether used.
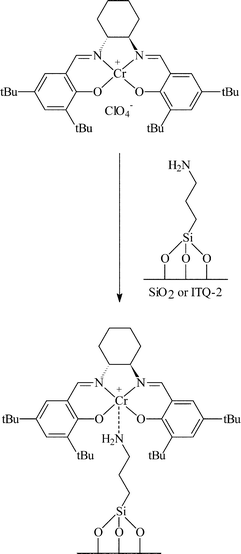 | ||
| Fig. 14 Covalent immobilisation of Mn(II) salen complexes over silica for the asymmetric ring opening of epoxides. Anchoring via the metal centre.28 | ||
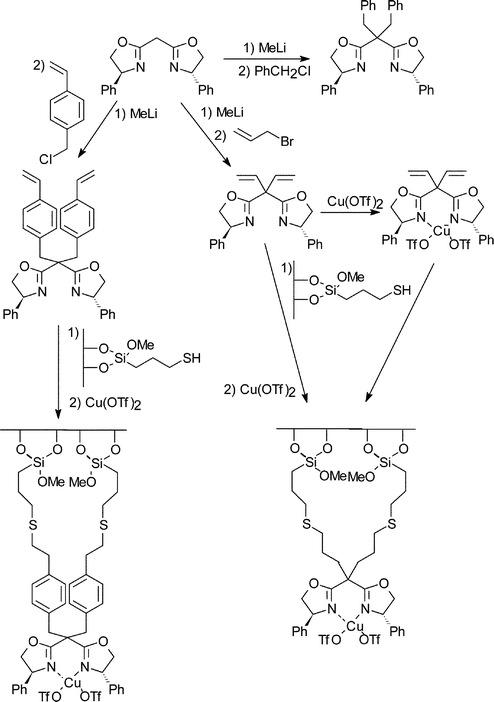 | ||
| Fig. 16 Covalent immobilisation of chiral copper(II) bisoxazoline over silica for the enantioselective cyclopropanation of alkenes.29 | ||
Enhanced e.e.s were observed in the vinyl benzene modified ligand (and the benzyl modified ligand used as the homogeneous equivalent). The immobilised catalysts gave e.e.s in the range of 6–65%, however this was still reduced with respect to the homogeneous reaction which gave e.e.s in the range of 40–86%. The yield and e.e.s of the cyclopropanes formed were also influenced by the preparation method. Direct immobilisation of the copper bis(oxazoline) complex gave decreased e.e.s when compared to the material synthesised sequentially by first immobilising the ligand and then forming the copper complex.
Clarke and Shannon30 also studied the immobilisation of copper bis(oxazoline) complexes for the cyclopropanation of styrene. Again the standard methodology was employed where the grafting moiety was attached to the methylene bridge as shown in Fig. 17. Both MCM-41 and MCM-48 were used as support materials.
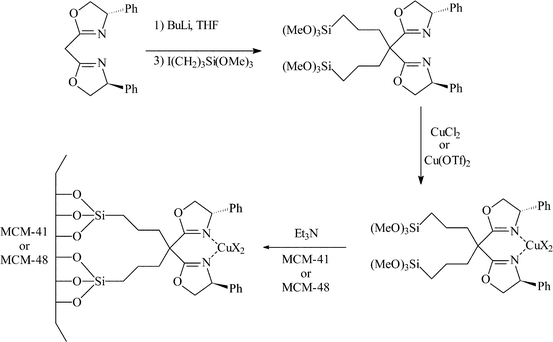
Hydrogenation of alkenes
The covalent immobilisation strategy has been used for the hydrogenation of ethyl nicotinate as reported by Raynor et al.31 Ferrocenyl acetate was modified to incorporate a trimethoxysilane tether as shown in Fig. 18. This moiety was then surface grafted into the pores of MCM-41 (to ensure the catalyst was only immobilised within the pores, the exterior surface of the MCM-41 was treated with Ph2SiCl2). Hydrogenation resulted in a product with a e.e. of 17% (conversions >50%). The e.e. observed was shown to be due to the incorporation of the catalyst within the pore by tethering the complex to an incompletely condensed silsesquioxane cube which, when tested as a catalyst, gave a racemic product. Thus, this work is of exceptional interest since the true nature of the non-immobilised complex is tested relative to the immobilised complex. Filtrate analysis after reaction revealed that only 3 ppb of palladium was leached from the immobilised catalyst.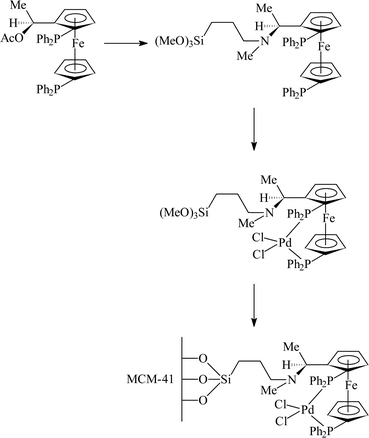 | ||
| Fig. 18 Covalent immobilisation of chiral palladium ferrocenyl complex for the enantioselective hydrogenation of ethyl nicotinate.31 | ||
Adima et al.32 reported the immobilisation of alkoxysilylated derivatives of (−)-(1R,2R)-1,2-diaminocyclohexane for the rhodium catalysed hydrogenation of prochiral ketones. The silylated ligands were reacted with varying amounts of tetraethylorthosilicate (TEOS) under sol–gel conditions to give the immobilised ligand, which was subsequently reacted with [Rh(cod)Cl]2 to give the final rhodium complex (Fig. 19). Both the mono- and di-silylated ligands were investigated. Data for the equivalent surface grafted material were also studied. Unfortunately no catalyst stability studies were carried out and, hence, we are unable to comment on this aspect. These catalysts were tested for the hydrogenation of acetophenone. The homogeneous equivalent reactions gave much higher conversions than those obtained for the immobilised catalyst (95 and 60–70% respectively). For the monosilylated ligand the e.e.s were generally low for both the homogeneous (14%) and for the heterogeneous reactions (22–25%). The homogeneous reaction carried out with the bis-silylated ligand gave the highest e.e. (58%), which was far superior to the immobilised examples (10–15%). The authors also observed that carrying out the sol–gel processing of the ligand in the absence of the TEOS resulted in a material which gave e.e.s close to that of the homogeneous reaction (54%, Table 2). These catalysts were also used for the hydrogenation of a variety of ketones. Moderate to excellent e.e.s (up to 98%) were obtained, where the stereochemistry of the products were inverted with respect to the starting ligand in all but one case. However, the data for the corresponding reactions with the homogeneous system were, unfortunately, not reported.
 | ||
| Fig. 19 Covalent immobilisation of chiral bis(amine) ligands for the rhodium catalysed reduction of acetophenone.32 | ||
| Entry | Si auxiliary ratio | Auxiliarya | Reaction time (d) | Conversion | ee |
|---|---|---|---|---|---|
| a M is ligand tethered at one nitrogen, B is ligand tethered at both nitrogens. b Auxiliary formed into gel without the addition of TEOS. | |||||
| 1 | 0∶1 | M | 5 | 95 | 14 |
| 2 | 2∶1 | M | 7 | 70 | 22 |
| 3 | 5∶1 | M | 5 | 80 | 25 |
| 4 | 0∶1 | B | 5 | 95 | 26 |
| 5 | 0∶1b | B | 5 | 75 | 58 |
| 6 | 1∶1 | B | 7 | 60 | 10 |
| 7 | 3∶1 | B | 8 | 20 | 15 |
Electrostatic interaction
Many porous solids, including zeolites, zeotypes, ordered mesoporous silicates and layered materials, including clays, hydrotalcites, can act as ion exchangers. This presents a relatively facile mechanism for the immobilisation of metal cations and complexes through electrostatic interaction. At first sight, many would consider that this interaction does not have sufficient strength to ensure the catalyst remains truly heterogeneous, yet the approach has been used to produce stable heterogeneous enantioselective catalysts that can exhibit significantly improved catalytic performance when compared with the homogeneous counterparts.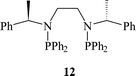
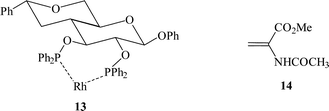
Mazzei et al.33 were the first to use this approach for the immobilisation of a Rh complex with PNNP 12 into the interlayers of a range of clays. The interlayer distances of the clays were increased by treatment with water or alcohols to permit the ion exchange of the bulky Rh complex. The immobilised catalyst gave up to 72% e.e. for the hydrogenation of (Z)-α-acetamidocinnamic acid 4. The catalyst could be re-used but the activity and selectivity decreased. Selke et al.34 subsequently immobilised cationic Rh chelates of 2,3-bis(O-diphenylphosphino)-β-D-glucopyranoside 13 on silica using electrostatic interaction. The enantioselectivity for the hydrogenation of α-acetamidoacrylic acid ester 14 with the immobilised catalyst was slightly higher (95% e.e.) than that for the homogeneous catalyst (91% e.e.). Catalysts were recycled up to 20 times and, while the enantioselectivity was retained, appreciable Rh leaching was noted. However, pre-treatment of the silica support minimised leaching and, in some cases, leaching was eliminated.
We have used this immobilisation strategy to design asymmetric heterogeneous catalysts for alcohol dehydration,35 aziridination,36 and epoxidation37 of alkene, and carbonyl and imino ene reactions.38 The design approach involves the ion exchange of suitable cations into the intracrystalline pores of zeolites, e.g. zeolite Y, or mesoporous solids, e.g. MCM-41 and the subsequent modification of the cation with a chiral ligand prior to reaction. Our initial studies used the acidic form of zeolite Y with chiral dithiane 1-oxides as chiral ligands as a catalyst for the enantioselective dehydration of butan-2-ol. Although only one active centre per zeolite supercage was modified, the modified sites were several orders of magnitude more active than the non-modified sites. As a result, the catalysts were effective for the enantioselective and catalysed dehydration reaction. At low conversion, the chiral catalyst modified with the R-dithiane 1-oxide preferentially reacted S-butan-2-ol in the presence of both enantiomers. Unfortunately, the enantioselective reaction was short-lived,39 but the enhancement in the rate of reaction with the modified catalyst was long-lived. Detailed spectroscopic studies of the active catalyst showed that the extra framework alumina, present in the pores of the zeolite, were an integral part of the active site. The rate enhancement was proposed to be due to activation of the Brønsted OH group of the zeolite through interaction with the oxygen of the dithiane 1-oxide and the extra framework alumina moiety (Fig. 20). These studies were considered as a proof-of-concept since, although the enantioselectivity was short-lived, it was shown that cations within zeolites could be modified to form chiral catalysts.
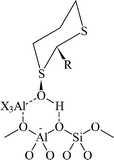 | ||
| Fig. 20 Proposed active site for the enantioselective dehydration of 2-butanol catalysed by zeolite USY modified with dithiane oxide.39 | ||
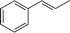
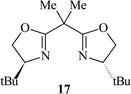
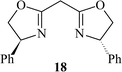
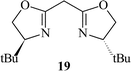
Subsequently, we immobilised copper(II) bis(oxazoline) complexes with zeolite Y and Al-MCM-41.36 Initially, the zeolite or mesoporous solid was partially ion-exchanged to give the Cu2+ form with 3–5 wt% Cu. The aziridination of alkenes was investigated using [N-(p-tolylsulfonyl)imino]phenyliodinane (PhI![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NTs) and [N-(p-nitrophenylsulfonyl)imino]phenyliodinane (PhINNs) as nitrene donors (Fig. 21). By careful control of the nitrene donor/substrate rates, e.e. in excess of 95% could be achieved with these catalysts (Table 3) with good yields. One of the key features with the immobilised catalysts is that they did not require excess substrate to drive the reaction to completion, a feature observed with the homogeneous catalyst. The catalysts were found to be very stable and, although some Cu2+ was lost from the catalyst during use, the leached Cu2+ was inactive for the aziridination reaction.40 This is due to the reaction by-products, iodobenzene and sulfonamide, inhibiting the aziridination reaction. Variation in the bis(oxazoline) showed some interesting effects (Table 4). Typically, the heterogeneously catalysed reaction gave much higher e.e.s compared with the homogeneous catalyst. However, less hindered bis(oxazolines) which were particularly ineffective for the homogeneously catalysed process gave high e.e. with the heterogeneous catalyst. This, in effect, was considered to be due to a containment effect within the pores of the zeolite. Detailed epr spectroscopy41 showed that the Cu2+ is chelated by the bis(oxazoline) in the zeolite pores as a square planar complex. Consequently, the zeolite cage structure places additional constraints on the approach of the substrate, an effect not possible in the homogeneously catalysed process. As a result, the immobilised complex is capable of higher enantioselection than the non-immobilised complex. An interesting observation with the copper-bis(oxazoline) catalysed aziridination reaction is that the enantioselection increases with the alkene conversion. This is an uncommon observation since, for most immobilised catalysts, the e.e. remains constant or declines with increased extent of reaction. The effect is considered to be due to the interaction of the chiral aziridine product with the active copper-nitrene intermediate as shown in Fig. 22.
NTs) and [N-(p-nitrophenylsulfonyl)imino]phenyliodinane (PhINNs) as nitrene donors (Fig. 21). By careful control of the nitrene donor/substrate rates, e.e. in excess of 95% could be achieved with these catalysts (Table 3) with good yields. One of the key features with the immobilised catalysts is that they did not require excess substrate to drive the reaction to completion, a feature observed with the homogeneous catalyst. The catalysts were found to be very stable and, although some Cu2+ was lost from the catalyst during use, the leached Cu2+ was inactive for the aziridination reaction.40 This is due to the reaction by-products, iodobenzene and sulfonamide, inhibiting the aziridination reaction. Variation in the bis(oxazoline) showed some interesting effects (Table 4). Typically, the heterogeneously catalysed reaction gave much higher e.e.s compared with the homogeneous catalyst. However, less hindered bis(oxazolines) which were particularly ineffective for the homogeneously catalysed process gave high e.e. with the heterogeneous catalyst. This, in effect, was considered to be due to a containment effect within the pores of the zeolite. Detailed epr spectroscopy41 showed that the Cu2+ is chelated by the bis(oxazoline) in the zeolite pores as a square planar complex. Consequently, the zeolite cage structure places additional constraints on the approach of the substrate, an effect not possible in the homogeneously catalysed process. As a result, the immobilised complex is capable of higher enantioselection than the non-immobilised complex. An interesting observation with the copper-bis(oxazoline) catalysed aziridination reaction is that the enantioselection increases with the alkene conversion. This is an uncommon observation since, for most immobilised catalysts, the e.e. remains constant or declines with increased extent of reaction. The effect is considered to be due to the interaction of the chiral aziridine product with the active copper-nitrene intermediate as shown in Fig. 22.
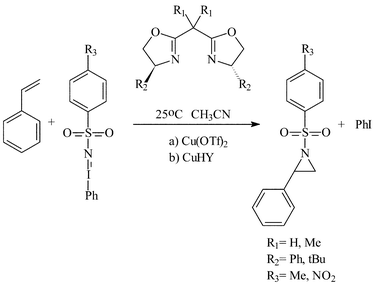 | ||
| Fig. 21 Reaction scheme for the enantioselective aziridination of styrene catalysed by the copper/bis(oxazoline) catalysts.40 | ||
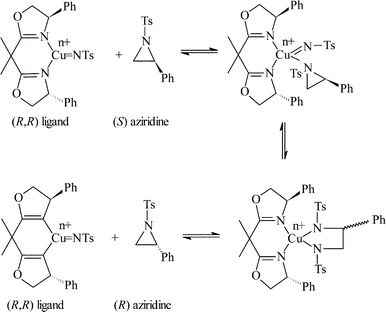 | ||
| Fig. 22 Proposed mechanism for the interconversion of R and SN-tosyl aziridine. | ||
| Styrene∶PhINNs mol ratio | Yield %b | Aziridine ee (%) | |
|---|---|---|---|
| Aziridine | Benzaldehyde | ||
| a CH3CN, 25 °C, 2,2-bis[(4R)-4-phenyl-1,3-oxazolin-2yl]propane as chrial modifier, reaction time 16h. Data in parentheses are for the homogeneous catalyst Cu(OTf)2 (0.015mmol) under identical conditions. b Yields based on styrene | |||
| 1∶1 | 68.3 (27.3) | 7.2 | 49.4 (65.2) |
| 1∶1.1 | 84.7 (82.8) | 11.3 | 81.2 (76.6) |
| 1∶1.2 | 87.8 (72.2) | 11.2 | 89.4 (87.6) |
| 1∶1.3 | 82.0 (99.5) | 17.9 | 91.0 (79.8) |
| 1∶1.4 | 76.6 (99.4) | 20.1 | 95.2 (78.8) |
| 1∶1.5 | 67.2 (83.1) | 21.2 | 87.8 (86.8) |
| Bis(oxazoline) | PhINTs |
PhINNs |
||
|---|---|---|---|---|
| Yield (%) | Ee (%) | Yield (%) | Ee (%) | |
|
a Styrene∶nitrene donor ratio = 1∶1.5, 25 °C, CuHY (0.3g); figures in parenthesis are for the homogeneous catalyst Cu(OTf)2
(0.015mmol) under identical conditions. All reactions followed by dissolution of the nitrene donor, reaction times for all oxazolines similar for Cu(OTf)2: 1 and 1.5 h for PhINTs and PhINNs respectively: for CuHY: 3 and 5 h for PhINTs and PhINNs respectively.
|
||||
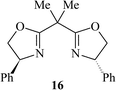
|
78 (91) | 76 (73) | 78 (96) | 85 (81) |
|
58 (78) | 24 (35) | 68 (79) | 82 (43) |
|
70 (74) | 77 (28) | 80 (85) | 82 (54) |
|
80 (85) | 28 (8) | 72 (42) | 77 (31) |
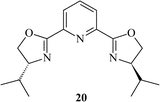
|
55 (70) | 5 (11) | 29 (42) | 6 (15) |
Recently, we have extended the use of immobilised copper bis(oxazoline) catalysts to carbonyl-ene and imino-ene reactions.38 For these reactions, the catalyst re-use was demonstrated without loss of yield or enantioselection. In addition, the immobilised catalysts were found to be effective for substrates that were unreactive with the homogeneously catalysed reaction. This approach was also used for the immobilisation of manganese-salen complexes for the aziridination of alkenes37 within the pores of Al-MCM-41. As observed with other attempts at immobilising the salen ligand, this approach did not enhance the stability of the catalyst and, in general, lower e.e. and yields were observed with the immobilised catalysts.
Comparison of methodologies and advantages of immobilisation
Of the four immobilisation strategies developed to date for the preparation of asymmetric heterogeneous catalysts, two, namely immobilisation by covalent tethering and electrostatic interaction, have been shown to form reasonably stable catalysts that are capable of re-use. Adsorption methodologies offer a relatively facile method of immobilisation but tend to produce non-stable catalysts. Encapsulation of complexes during the preparation methods provides a very elegant methodology but is relatively complex compared with the more recently developed covalent tethering methods. Covalent tethering will undoubtedly produce stable catalysts and can be considered the preferred methodology for catalysts which do not lend themselves to immobilisation through electrostatic interaction. Immobilisation via ionic interaction is conceptually simple and is a facile method of immobilising ionic catalysts. The approach has been shown to produce stable catalysts that are capable of re-use, but it has been noted recently by Rechavi and Lemaire that methodology of catalyst synthesis is crucial if stable catalysts are to be obtained.42Many early studies concerned with immobilisation of asymmetric homogeneous catalysts showed that, for the most part, lower yields and enantioselection were achieved with the immobilised catalysts. Hence, the immediate advantage of using a heterogeneous catalyst, namely the facile recovery and re-use of the expensive catalyst/ligand system, could not be realised. However, recent studies have shown this need not be the case and that immobilised catalysts can give higher e.e. and yields than non-immobilised catalysts. Indeed, some immobilised systems have been shown to be effective with immobilised systems for substrates that are non-selective with their homogeneous counterparts. Two reasons for this change of fortunes for the immobilised catalysts are apparent. First, site isolation is achievable through the appropriate design of immobilised catalysts. This prevents the polymerisation of some chiral ligands which limits their effectiveness when used as homogeneous catalysts. Second, and most importantly, is the observation by several research groups of the containment effect, i.e. that the immobilised asymmetric catalyst is constrained through interaction with the supporting matrix.43 The additional constraints placed upon the chiral catalyst can induce increased enantioselection compared with the homogeneous catalyst, and several examples have been given in this review. However, this effect presents both a clear problem and a clear opportunity with respect to the design of immobilised catalysts. The usual starting point for the design of immobilised catalysts is typically the homogeneous catalyst complex that has been demonstrated to give high enantioselection. The additional constraint of the immobilised catalyst means that in some cases simpler chiral catalysts will be more effective and, hence, the use of ligands specifically designed for use in homogeneously catalysed processes will not, necessarily, be the best for use with the immobilised catalysts. This, therefore, presents us with a major opportunity to design more effective asymmetric heterogeneous catalysts that take full account of the highly effective containment effect. However, it can be expected that the containment effect observed with both the tethering and electrostatic interaction can be used effectively to design a new generation of immobilised asymmetric catalysts capable of both high yields and enantioselection. The effect provides a clear methodology by which high enantioselection can be obtained.
References
- For details see Nobel lectures, Angew. Chem. Int. Ed., 2002, 41, p. 998 Search PubMed.
- H.-U Blaser, Chem. Commun., 2003, 293 RSC.
- M. Davies, Microporous Mesoporous Mater., 1998, 21, 173 CrossRef CAS.
- H.-U. Blaser and B. Pugin, in Chiral Reactions in Heterogeneous Catalysis, Plenum, New York, 1995, p 33 Search PubMed.
- D. C. Sherrington, Catal. Today, 200, 57, 87 Search PubMed.
- C. E. Song and S.-G. Lee, Chem. Rev., 2002, 19, 3495 CrossRef CAS.
- A. Baiker, J. Mol. Catal. A, 1997, 115, 473 CrossRef CAS.
- H.-U. Blaser, H. P. Jalett, M. Müller and M. Studer, Catal. Today, 1997, 37, 441 CrossRef CAS.
- P. B. Wells and A. G. Wilkinson, Topics Catal., 1998, 5, 39 CrossRef CAS.
- C. Bianchini, P. Barbaro, V. Dal Santo, R. Gobetto, A. Meli, W. Oberhauser, R. Psaro and F. Vizza, Adv. Synth. Catal., 2001, 343, 41 CrossRef CAS and references cited therein.
- K. T. Wan and M. E. Davis, J. Catal., 1995, 152, 25 CrossRef CAS and references cited therein.
- H. N. Flach, I. Grassert, G. Oehme and M. Capka, Colloid Polym. Sci., 1996, 274, 261 CAS.
- J. Jamis, J. R. Anderson, R. S. Dickson, E. M. Campi and W. R. Jackson, J. Organomet. Chem., 2001, 627, 37 CrossRef CAS.
- S. B. Ogunwami and T. Bein, Chem. Commun., 1997, 901 RSC.
- M. J. Sabatier, A. Corma, A. Domenech, V. Fornes and H. Garcia, Chem. Commun., 1997, 1285 RSC.
- C. Schuster, E. Mollmann, A. Tompos and W. F. Hölderich, Catal. Lett., 2001, 74, 69 CrossRef CAS and references cited therein.
- S. Ernst, E. Fuchs and X. Yang, Microporous Mesoporous Mater., 2000, 35–36, 137 CrossRef CAS.
- A. Wolfson, S. Janssens, I. Vankelcom, S. Geresh, M. Gottlieb and M. Herkowitz, Chem. Commun., 2002, 388 RSC and references cited therein.
- K. Soai, M. Watanabe and A. Yamamoto, J. Org. Chem., 1990, 55, 4832 CrossRef CAS.
- S. Abramson, N. Bellocq and M. Lasperas, Top. Catal., 2000, 13, 339 CrossRef CAS and references cited therein.
- Y.-M. Chung and H.-K. Rhee, Chem. Commun., 2002, 238 RSC.
- J. S. Bae, S.-W. Kim, T. Hyeon and B. M. Kim, Chem. Commun., 2000, 32 Search PubMed.
- A. Corma, H. Garcia, A. Moussaif, M. J. Sabatier, R. Zniber and A. Redouane, Chem. Commun., 2002, 1058 RSC.
- D. Rechavi and M. Lemaire, J. Mol. Catal. A, 2002, 182–183, 239 CrossRef CAS.
- S. Xiang, Y. Zhang, Q. Xin and C. Li, Angew. Chem. Int. Ed., 2002, 41, 821 CrossRef CAS.
- D. Pini, A. Mandoli, S. Orlandi and P. Salvadori, Tetrahedron: Asymmetry, 1999, 10, 3883 CrossRef CAS.
- B. M. Choudary, N. S. Chowdari, M. L. Kantam and P. L. Santhi, Catal. Lett., 2001, 76, 213 CrossRef CAS.
- C. Baleizao, B. Gigante, M. J. Sabatier, H. Garcia and A. Corma, Appl. Catal. A, 2002, 288, 279 CrossRef.
- M. I. Burguete, J. M. Fraile, J. I. Garcia, E. Garcia-Verdugo, C. I. Herrerias, S. V. Luis and J. A. Mayoral, J. Org. Chem., 2001, 66, 8893 CrossRef CAS.
- R. J. Clarke and I. J. Shannon, Chem. Commun., 2001, 1936 RSC.
- S. A. Raynor, J. M. Thomas, R. Raja, B. F. G. Johnson, R. G. Bell and M. D. Mantle, Chem. Commun., 2000, 1925 RSC.
- A. Adima, J. J. E. Moreau and M. W. C. Man, Chirality, 2000, 12, 411 CrossRef CAS.
- M. Mazzei, W. Marconi and M. Riocci, J. Mol. Catal., 1980, 9, 381 CrossRef CAS.
- R. Selke and M. Capka, J. Mol. Catal., 1990, 63, 319 CrossRef CAS.
- S. Feast, D. Bethell, P. C. B. Pate, F. King, C. H. Rochester, M. R. H. Siddiqui, D. J. Willock and G. J. Hutchings, J. Chem. Soc., Chem. Commun., 1995, 2499 RSC.
- S. Taylor, J. Gullick, P. McMorn, D. Bethell, P. C. Bullman Page, F. E. Hancock, F. King and G. J. Hutchings, J. Chem. Soc., Perkin 2, 2001, 1714 RSC.
- P. Piaggio, P. McMorn, D. Murphy, D. Bethell, P. C. Bullman Page, F. E. Hancock, C. Sly, O. J. Kerton and G. J. Hutchings, J. Chem. Soc., Perkin 2, 2000, 2008 RSC.
- F. E. Hancock, G. J. Hutchings and N. A. Caplan, Patent WO 03/018191 (2003).
- S. Feast, M. R. H. Siddiqui, R. P. K. Wells, D. J. Willock, F. King, C. H. Rochester, D. Bethell, P. C. B. Page and G. J. Hutchings, J. Catal., 1997, 167, 533 CrossRef CAS.
- S. Taylor, J. Gullick, P. McMorn, D. Bethell, P. C. Bullman Page, F. E. Hancock, F. King and G. J. Hutchings, J. Chem. Soc., Perkin 2, 2001, 1714 RSC.
- Y. Traa, D. M. Murphy, R. D. Farley and G. J. Hutchings, Phys. Chem. Chem. Phys., 2001, 3, 1073 RSC.
- D. Rechavi and M. Lemaire, Chem. Rev., 2002, 102, 3467 CrossRef CAS.
- J. M. Thomas, T. Maschmeyer, B. F. G. Johnson and D. S. Shephard, J. Mol. Catal. A, 1999, 141, 139 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2004 |