Noble metal decoration of single crystal platinum surfaces to create well-defined bimetallic electrocatalysts
J. S.
Spendelow
and
A.
Wieckowski
*
Department of Chemistry and Department of Chemical and Biomolecular Engineering, University of Illinois at Urbana-Champaign, Urbana, IL 61801, USA. E-mail: andrzej@scs.uiuc.edu; Fax: +1-217-244-8068; Tel: +1-217-333-7943
First published on 20th October 2004
Abstract
Electrocatalytic studies on bimetallic single crystal electrodes present a unique opportunity to explore the reactivity of complex surfaces with known structure and composition. Such electrochemical studies, together with measurements in ultra-high vacuum, provide the theoretical and experimental basis for rational design of more active electrocatalysts. Pure platinum, though the most active single-component electrocatalyst for many reactions, is still not active enough for some applications, particularly in fuel cell technology, and cannot be considered as a true catalyst for most electrocatalytic processes. Therefore, a concerted effort has been made in the last 40 years to enhance the electrocatalytic activity of Pt via modification by a second metal. Most early work used polycrystalline alloys, but in recent years, many workers have begun modifying (decorating) Pt single crystals by deposition of a second noble metal, which is usually ruthenium, palladium, rhodium, osmium, or silver. A critical review of electrocatalysis on such well-defined bimetallic surfaces is offered, and a brief analysis of the inverted approach, in which single crystal noble metal surfaces are modified by Pt deposition, is also presented.
1. Introduction
Electrocatalysis has been an area of active research for more than four decades now, but it was not until the year 1980, which saw the development of specific methods for producing and maintaining well-defined single crystal surfaces of noble metals,1–4 that the study of electrocatalytic reactions on surfaces of known composition and geometry became possible. Although there were earlier attempts to study such single crystal surfaces, beginning with the work by Will in the 1960s,5 these studies found only limited success due to the complications imposed by contamination and/or surface disordering. Many fundamental studies of electrocatalytic reactivity on single crystal surfaces were performed in the 1980s, including some studies in which metal surfaces were modified by underpotential deposition (UPD) of a second (non-noble) metal. Such studies are on-going, but in the last decade attention has been shifting to the use of bimetallic single crystal noble metal electrodes, which are of interest from a fundamental as well as applied science perspective.In the broadest terms, the noble metal deposition methods included in this review can be divided into two classes: “wet” deposition and vacuum deposition. Wet deposition can be further divided into electrodeposition and electroless deposition, while vacuum deposition may be subdivided into physical deposition and chemical deposition.
Electrodeposition relies on application of well-known electrochemical thermodynamics to the problem of metal deposition: by manipulating the potential of the Pt electrode in a solution containing ions of a noble metal, deposition of the second noble metal can be made thermodynamically favorable. Several factors complicate the otherwise simple interpretation: (i) even in a thermodynamically favorable regime, kinetic limitations may in some cases prevent deposition from occurring; (ii) the presence of complexing agents (e.g. halides) in solution changes both the thermodynamics and kinetics of metal electrodeposition; (iii) lattice mismatch and chemical interaction between the admetal and the substrate makes deposition of ultrathin pseudomorphic layers of one metal onto another metal (heteroepitaxy) energetically dissimilar to homoepitaxy. The amount of deposited metal may be determined from the charge passed during deposition, although such a calculation will be inexact when the Faradaic efficiency is not 100%. Admetal nucleation during electrodeposition typically occurs at steps and other defects,6 which may be a disadvantage when a uniform coverage is desired.
In addition to traditional bulk electrodeposition, the possibility of highly localized deposition of metal clusters has been demonstrated by using scanning tunneling microscopy (STM), although the mechanism of deposition is not clear in all cases. In one formulation, the STM tip, which is loaded with the metal to be deposited, is pulsed to a sufficiently high potential to dissolve some of the metal, which then may locally electrodeposit on the substrate.7,8 This technique is not limited to electrodeposition, however; a similar approach applied in UHV produces field emission from the tip and subsequent deposition on the substrate.9 Alternatively, application of a large tip-sample bias in solution, when combined with the local double layer structure modifications caused by the very small tip-sample separation, can promote electrodeposition of metal ions already in solution, i.e. not generated at the tip.10 Direct mechanical contact between the tip and the substrate, either in electrolyte, air or, vacuum, may also be applied to transfer atoms from the tip to the substrate.11,12
Electroless deposition eliminates the need for potential control, and can be subdivided into forced and spontaneous deposition. In the case of forced deposition, a chemical reducing agent is used to drive deposition of the metal. This agent may be either in solution or adsorbed on the surface. The first applications of this technique used UPD hydrogen as the reducing agent,13 while UPD layers of Cu or Pb have been used more recently.14 Noble metal ions in solution oxidize the UPD layer, and are simultaneously reduced and adsorbed. The amount of noble metal deposited may be determined from the coverage of the sacrificial UPD layer. Solution phase hydrogen has also been used as the reducing agent.15 In this case, a drop of solution containing a known amount of noble metal ions may be attached to the working surface of the Pt single crystal, followed by exposure to hydrogen gas to reduce and adsorb the metal ions. Mechanisms of forced deposition have not been thoroughly investigated, but in some cases (such as forced deposition by H2) the role of the reductant is simply to adjust the potential of the electrode on which metal is being deposited, in this way mimicking electrodeposition.
Spontaneous deposition is performed by immersing a noble metal substrate in a solution containing noble metal ions. The mechanism of deposition is not well understood, and it is likely that many mechanisms exist for different combinations of substrate and adsorbed metal. If the standard potential for deposition of the admetal is more positive than that of the substrate metal, spontaneous deposition may proceed through the cementation process, in which reduction of the solution-phase metal is driven by oxidation, and in some cases dissolution, of the substrate metal. This phenomenon may occur, for instance, during spontaneous deposition of Pt on Ru substrates. In the case of deposition of a less-noble metal on a more-noble metal, which is typical of spontaneous deposition of metals on Pt surfaces, deposition proceeds through adsorption of hydrous oxides or complex ions containing the depositing metal (e.g. Ru), possibly also with an anion adsorption component. A predominantly metallic deposit is obtained only after reduction of this adsorbate.
Vacuum deposition techniques used to augment Pt(hkl) with other noble metals can be subdivided into several classes: physical deposition by evaporation (physical vapor deposition) or sputtering, and chemical vapor deposition. Evaporation is the most widely used vacuum method for the preparation of thin films of noble metals on well-defined noble metal substrates. Unlike in the wet deposition methods, evaporation can be carried out over a wide range of substrate temperature, allowing precise control of surface diffusion in the surface structure formation process. Sputtering has not been used extensively for the modification of Pt(hkl) surfaces, but it has some advantages that may be exploited in the future, such as the ability to selectively and non-destructively deposit metal clusters of a given mass.16 Chemical vapor deposition (CVD), in which adsorbed metal-containing precursors undergo chemical decomposition to produce the metallic adsorbate, has also not received much attention compared to evaporation. One complication presented by this procedure is possible contamination of the surface by products of the decomposition. Some annealing and oxygen treatment may be required to clean the surface following CVD, resulting in modifications of the surface structure and composition. An advantage of all the vacuum deposition techniques is that the resultant film can be characterized during or directly after the deposition using UHV analytical techniques such as Auger electron spectroscopy (AES) and low-energy electron diffraction (LEED).
All of the surface decoration techniques described above may be further modified by lithography. In addition to the traditional photolithography and electron-beam lithography, the development of scanning probe techniques has allowed nanoscale lithography by the selective removal of “resist,” which for this purpose is often a self-assembled monolayer.17 Surface modification in this way allows unprecedented control of the resulting deposit structure, and may be a profitable method to investigate structure and island size effects.
Metal-on-metal heteroepitaxial growth has been traditionally divided into three thermodynamic growth modes.18 The operative mode is determined by the surface energy of the substrate metal, the surface energy of the film metal, and the interface energy, which includes effects from strain and from chemical interactions. Although these models, as originally formulated, describe equilibrium states, they are often used in the literature to describe kinetically limited cases. Frank–van der Merwe growth involves layer by layer deposition in which nucleation of a layer occurs only following completion of the previous layer.19 Volmer–Weber growth, in contrast, involves nucleation of the second (and subsequent) layers before completion of the first layer, resulting in the creation of three-dimensional clusters.20 In Stranski–Krastanov growth, an intermediate mode, the first monolayer is completed before nucleation of the second layer, but further growth produces three-dimensional clusters.21 Each of the three growth modes is observed at least in approximate form during deposition of the various noble metals discussed here on Pt(hkl). A common characteristic of all admetals included in this review is that they tend to form pseudomorphic islands on Pt(hkl) at submonolayer coverage. This phenomenon results from the attractive lateral interactions between adatoms, which may be ascribed to the relatively large cohesive energies of the admetals and small adatom dipole moments.22 The islands tend to be pseudomorphic, as would be expected from the small lattice mismatch (less than 5% in all cases). As the coverage increases, islands grow two-dimensionally and begin to coalesce for the admetals with low surface energy (e.g. Pd), conforming to a Frank–van der Merwe or Stranski–Krastanov growth mode, while admetals with high surface energy (e.g. Ru) begin growing in three dimensions (Volmer–Weber growth).
An alternative paradigm for the study of well-defined bimetallic single-crystal surfaces is the use of single-crystal bulk alloys. These alloy surfaces may contribute to the understanding of electrocatalytic processes since: (i) they typically produce surfaces with a random or nearly-random dispersion of two elements, rather than the nanoisland formation that is universally observed with noble metal adatom-modified electrodes; (ii) the resulting surface contains both metals in the same terrace, in contrast to the situation resulting from adatom deposition. Although these attributes can be advantageous for the understanding of electrocatalytic mechanisms, the ideal bimetallic surfaces taken as cross-sections of bulk crystal structure are rarely obtained due to segregation processes. Unfortunately, surface segregation is difficult to track by using the most popular analytical techniques in UHV such as AES and X-ray photoelectron spectroscopy (XPS), although a combination of these techniques and low energy ion scattering spectroscopy (LEISS) has been successfully employed in this pursuit.23–26 Of course, similar effects may be obtained through island deposition followed by annealing to create a surface alloy, a technique that may be applied in some cases even for metals with low bulk solubility.
One of the greatest advantages of using well-defined single crystal electrodes is that reproducibility is much greater than for other electrodes (e.g. polycrystalline, alloy, codeposited), although differences in the exact preparation procedure, method and conditions of noble metal deposition, and testing procedure complicate efforts to compare results from laboratory to laboratory. Nevertheless, in this review, results from different laboratories are compared when appropriate. For this purpose, all potentials quoted in this review are given with respect to the reversible hydrogen electrode (RHE).
This review is organized according to Pt(hkl)/Me combination. The first two sections cover Pt(hkl)/Ru and Pt(hkl)/Pd, the two most important Pt(hkl)/Me combinations for electrocatalysis. Subsequently, briefer sections on the other Pt/Me combinations are included. Lastly, an inverted formula, Ru(ijkl)/Pt, is discussed. Within each section, methods of noble metal deposition and characterization of the resulting surface structure are discussed, followed by a review of the electrocatalytic properties of such surfaces.
2. Pt(hkl)/Ru surfaces
The first work on polycrystalline Pt/Ru alloy electrocatalysts was reported in the early 1960s,27–30 and since that time numerous electrocatalytic studies on Pt/Ru electrodes have been performed. This interest stems from the known advantages of Pt/Ru catalysts for the electrooxidation of CO, methanol, and other organic molecules, and the desire to exploit these advantages in the design of CO-tolerant hydrogen reformate polymer electrolyte membrane fuel cells (PEMFC) and direct-oxidizing fuel cells such as the direct methanol fuel cell (DMFC). In the case of methanol oxidation, it is well known that Pt is required to catalyze the methanol dehydrogenation reaction, as methanol does not adsorb to a significant extent on Ru sites at temperatures below 80 °C.31 The role of Ru in promoting methanol oxidation is more complicated. It has been proposed that the function of Ru is primarily to provide adsorbed oxygen-containing species which facilitate the oxidation of methanol oxidation intermediates (primarily CO) to CO2 (the bifunctional mechanism).32 In addition, some authors have found evidence that Ru enhances methanol oxidation through an electronic effect on neighboring Pt atoms (the ligand effect).33–35 In this mechanism, Ru may accelerate the adsorption and dehydrogenation of methanol on Pt sites at low potentials, or it may weaken the Pt–CO bond, allowing oxidation of CO at lower potentials. Since ample evidence exists for both modes of enhancement, both the bifunctional mechanism and the ligand effect can be said to be operative; indeed, some progress has been made in efforts to measure the relative contribution of the two enhancement modes.36,37 Ru has also been shown to modify the selectivity of Pt-based catalysts for methanol oxidation, with Pt/Ru electrocatalysts yielding more CO2 and less formate and formic acid than pure Pt.38,39Although research in this field has been ongoing for more than four decades, it is only in the last decade that some workers have turned their attention to studies of well-defined model bimetallic catalysts of controlled surface composition and geometry.40–44 Studies on Pt single crystals modified by adsorbed Ru have shed new light on the mechanism of Ru enhancement, and have demonstrated the importance of Ru deposit geometry and substrate crystallography on the methanol and CO electrooxidation reactions.
2.1 Electrodeposition
Electrodeposition of Ru is typically performed using baths of RuCl3 or Ru(NO)(NO3)3, with Ru coverage frequently measured using the deposition charge. Tremiliosi-Filho et al. demonstrated, however, that the relationship between Ru coverage and deposition charge is not a simple one.45 Namely, because of spontaneous deposition (see below), some deposition of Ru occurs before the onset of electrolysis (Fig. 1). In contrast, no spontaneous deposition occurs from Ru(NO)(NO3)3 solutions. Furthermore, although the nucleation process and initial island growth is facile using either precursor, Ru deposition becomes unfavorable at a coverage of ca. 0.2 and 0.3 monolayers (ML) for deposition from RuCl3 and Ru(NO)(NO3)3 solutions, respectively, resulting in a low Faradaic efficiency at higher coverage, as indicated by the deviation from linearity in Fig. 1.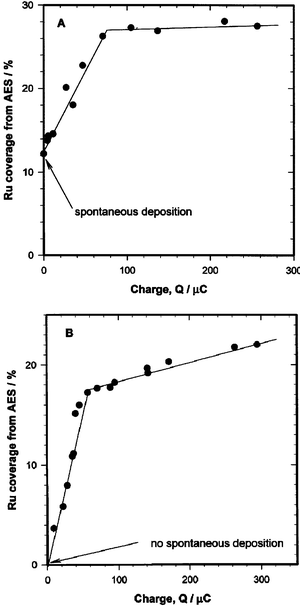 | ||
| Fig. 1 Deposition of Ru at 0.30 V from 0.5 mM RuCl3 solution in 0.1 M HClO4 (A) or 0.2 mM Ru(NO)(NO3)3 in 0.5 M H2SO4 (B): the dependence of Ru coverage (in ML) on the deposition charge. Reprinted from ref. 45, © Elsevier 1999, with permission. | ||
Stimming, Friedrich and collaborators examined Pt(hkl)/Ru surfaces obtained via constant potential electrodeposition using FTIR spectroscopy, scanning tunneling microscopy (STM), XPS, and surface X-ray scattering (SXS).44,46–50 The emphasis in these studies was on CO adsorption and electrooxidation. Distinct stretching frequencies were observed corresponding to separate CO adsorption on Pt and Ru surface phases (Fig. 2), in contrast to previous IR studies on Pt/Ru alloys, where only one vibrational band of CO was observed,51 demonstrating that Ru was deposited as distinct surface islands. Island formation was confirmed by scanning tunneling microscopy (STM), which revealed primarily monatomic Ru islands with a diameter of 2–5 nm and a Ru coverage as high as 0.7 ML (Fig. 3).47 SXS measurements showed that the Ru islands formed on a Pt(100)/Ru surface are monatomic and commensurate with the platinum surface.46
 | ||
| Fig. 2 IR spectra of adsorbed CO on different Ru-modified Pt(111) electrodes with varying Ru coverage in 0.1 M HClO4 at 0.40 V. (A) Pt(111) without Ru. (B) Pt(111) with 0.25 ML of Ru. (C) Pt(111) with 0.45 ML of Ru. (D) Pt(111) with 0.6 ML of Ru. Reprinted from ref. 50, © Elsevier 2002, with permission. | ||
 | ||
| Fig. 3 STM images of Ru-modified Pt(111) electrodes recorded in 0.1 M HClO4 at 0.5 V. (A) Substrate without Ru. (B) After deposition from 0.1 M H2SO4 + 5 mM RuCl3 at 0.60 V for 5 min. (C) Higher resolution image of surface in (B). (D) Deposition was performed for 30 min. Reprinted from ref. 50, © Elsevier 2002, with permission. | ||
Lin et al.52 investigated electrodeposition of Ru on Pt(111) electrodes using UHV surface science techniques, as well as cyclic voltammetry (CV), in a closed (air isolated) UHV/electrochemical transfer system. By using electron diffraction and AES (see also refs. 41 and 53), it was determined that the electrodeposition of Ru created monatomic islands at lower coverage, and then was subject to Volmer–Weber growth at higher coverage values.
The electrodeposition of Ru on vicinal Pt(111) surfaces was investigated in two papers from the Baltruschat laboratory.54,55 Although STM studies of Ru decorated Pt single crystals prepared by spontaneous deposition found no preference toward step edges for Ru deposition,56–62 Baltruschat, Samjeské and coworkers interpreted the suppression of voltammetric features corresponding to hydrogen adsorption at steps as evidence of step-decoration by Ru. Samjeské et al. point out that a monatomic row of Ru deposited along a step would not be resolvable by STM. Alternatively, the differing observations on step-decoration may be attributed to a fundamental difference between the Ru spontaneous and electrodeposits. STM images of Pt(111)/Ru surfaces prepared by Ru electrodeposition48–50 clearly show a distortion of the straight-line steps visible on the clean Pt(111) surface (Fig. 3), an observation that may be explained as a preferential deposition of Ru below steps; such a distortion is not observed on surfaces modified by spontaneous Ru deposition.
2.2 Electroless deposition
Oxides of Ru, in the form of aqua-anions, were proposed to be the predominant surface species following spontaneous deposition.40 This prediction was supported by an XPS study of the Pt(111)/Ru surface after rearranging (dehydrating) in UHV, which showed Ru to be predominantly in the form of oxides (Fig. 4).64 After spontaneous deposition and transfer to Ru-free solution, the first negative-going scan was found to produce a higher cathodic current than was observed in the subsequent stable CV.40 Therefore, it was concluded that the Ru oxides initially formed by spontaneous deposition are reduced, at least partially, by scanning or stepping the potential to less positive values. This interpretation was confirmed by the XPS results,64,65 which showed mainly metallic Ru after potentiostatic or potentiodynamic treatment at sufficiently cathodic potentials, followed by emersion and transfer to UHV (Fig. 4). Primarily metallic Ru was also observed at a potential of 0.1 V using in situ XANES on a supported Pt/Ru alloy catalyst,66 and on a similar catalyst acting as the anode in a hydrogen fuel cell,67 confirming the ex situ single crystal results.64
 | ||
| Fig. 4 (a) Ru 3d5/2 spectrum for Ru(0001), with binding energy 280.1 eV. (b) As in (a), but for 0.5 ML of Ru spontaneously deposited on Pt(111) followed by reduction at 0.08 V for 30 min. Reprinted with permission from ref. 65, © Springer-Verlag 2004 . | ||
Core level binding energy shifts for Ru spontaneously deposited on Pt(111), as well as several adsorbates on Pt(111)/Ru, were examined in an XPS study by Vericat et al.65 Binding energy shifts of I (3d5/2) and CO (C 1s) adsorbed on Pt(111)/Ru, as compared with Ru(0001), suggest an electron transfer from Pt to Ru, in agreement with earlier NMR68 and X-ray absorption spectroscopy (XAS)69 studies on Pt/Ru alloys. It is therefore somewhat surprising that metallic Ru on Pt(111) showed no significant shift in the Ru 3d3/2 binding energy compared with Ru(0001), but this discrepancy may be due to a strain effect within the Ru islands.
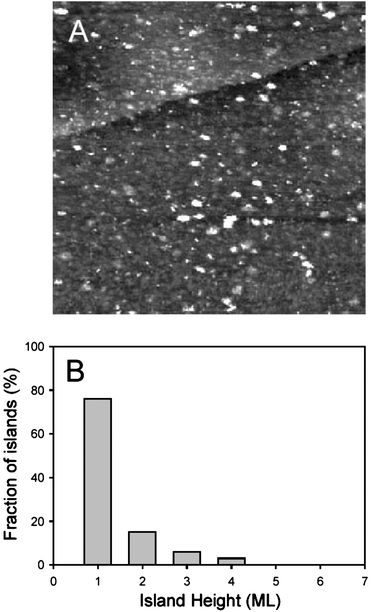
A number of STM studies, both in situ and ex situ, of the Pt(hkl)/Ru surface obtained by spontaneous deposition have been published.56–62 Homogeneous deposition of 2–5 nm ruthenium islands, with island formation insensitive to step or surface defect sites, was obtained on all three platinum single crystal faces. The islands that populated the surfaces were predominantly two-dimensional for short deposition times (Fig. 5), while longer deposition times and multiple depositions produced a majority of three-dimensional islands (up to 6 monolayers62), corresponding to a Volmer–Weber growth mode. Pt(100) had the highest and Pt(110) the lowest affinity for the spontaneous deposition, but Ru coverage higher than 0.25 ML could not be obtained on any of the surfaces through a single deposition. It is, however, possible to repeat the spontaneous deposition procedure sequentially, in order to yield Ru coverage higher than 0.25 ML.60 On Pt(111), such a repetitive deposition was studied using STM, and it was found that after 4 depositions, approximately 30–35% of the surface was covered with large and diverse (2–12 nm) ruthenium islands of varying heights. No changes in Ru island morphology on Pt(100)/Ru were observed during four voltammetric cycles up to 0.80 V in HClO4 solution.61 In contrast, oxidation of Ru islands on Pt(111) at 0.95 V in H2SO4 solution, followed by reduction at 0.35 V, results in disintegration of Ru islands into smaller islands, with diameter 1–3 nm.62
2.3 Vacuum deposition
 | ||
| Fig. 6 Ru island growth on Pt(111), preparation by Ru vapor deposition in UHV, STM images taken in the same chamber. (a) Ru coverage of ca. 0.25 ML. (b) Ru coverage of ca. 0.60 ML.74 | ||
Annealing of Pt(110)/Ru and Pt(111)/Ru surfaces obtained by vapor deposition results in a segregation of Ru to the subsurface layers at a temperature much lower than that at which dissolution of Ru into the bulk crystal occurs, as shown by a comparison of XPS and LEISS coverage measurements (Fig. 7).72,75 Flash annealing at 800 K results in a complete disappearance of Ru from the top layer of atoms, while the XPS measurement of the top four layers shows essentially unchanged Ru content. Similar conclusions were drawn for evaporated Ru on Pt(111) after annealing at 600 K. Although voltammetric features corresponding to surface Ru disappeared after annealing, AES measurements demonstrated no significant change in the Ru content in the near-surface region.73
 | ||
| Fig. 7 Effect of flash annealing on coverages determined from XPS (triangles) and LEISS (squares) on a Pt(110) surface following deposition of Ru by evaporation at 350 K. Reprinted from ref. 75, © Elsevier 2000, with permission. | ||
2.4 Electrocatalysis
Several studies of methanol oxidation have been performed using Pt single crystals modified by Ru.42,45,52,53,62,70,74,78,79 In the earliest investigation, Herrero et al.78 studied surface crystallographic effects on methanol oxidation on Pt(hkl)/Ru electrodes in which Ru was electrodeposited under voltammetric conditions. On the Pt(110) electrode, a surface redox couple involving Ru deposits was assigned to a “ruthenium enhancement” in methanol oxidation. This work was expanded upon in the systematic studies by Chrzanowski et al., which revealed the high methanol electrooxidation activity that may be obtained on Pt/Ru surfaces with optimized crystallography.42,45,79 The catalytic activity of the Pt(100)/Ru, Pt(111)/Ru, Pt(110)/Ru, and Pt(poly)/Ru surfaces prepared by Ru electrodeposition at 0.30 V was examined with the focus on optimum ruthenium coverage, rate of oxidation current decay, Tafel slopes and reaction turnovers, and the apparent activation energies. Fig. 8 shows a plot of the Pt(111)/Ru steady-state current density toward methanol electrooxidation as a function of ruthenium coverage. The optimum Ru coverage for methanol electrooxidation was observed to be 0.20 ML for the Pt(111) surface. Fig. 9 shows chronoamperometric curves at 0.31 V vs. RHE in a 0.6 M methanol solution at 65 °C for the three surfaces at their optimum Ru coverage values.45 These results provided the first demonstration of the remarkable structure-sensitivity of the Pt/Ru surface for methanol electrooxidation, with a unique reactivity on the Pt(111) surface covered by 0.20 ML of Ru. In contrast, the Pt(100)/Ru surface is highly inactive, and Pt(110)/Ru and Pt(poly)/Ru are moderately active. The Pt(111)/Ru electrode was found to be the most active laboratory scale fuel cell anode for methanol electrooxidation, and it was concluded that crystallographic variables should be exploited in syntheses of novel metal-alloy catalysts for fuel cell use. In addition to measurement of activation energies for methanol oxidation on each of the Ru-modified low index Pt surfaces, fast cyclic voltammetry (CV) was used to determine the CO coverage on the polarized electrodes in methanol solution.45 A CO coverage of ca. 0.5 ML (where 1 ML is defined as 1 molecule per surface Pt atom) was found on clean and Ru-modified Pt(111) at potentials between 0.25 V and 0.55 V for times greater than 1 s, with slightly lower CO coverage at 0.55 V attributed to incipient CO oxidation at this potential.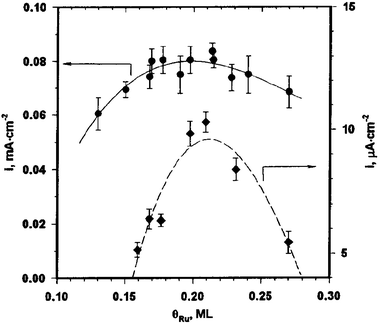 | ||
| Fig. 8 Methanol electrooxidation current densities as a function of surface ruthenium coverage at 0.49 V (circles) and at 0.31 V (diamonds) on the Pt(111)/Ru electrode. Reprinted with permission from ref. 42, © American Chemical Society 1998. | ||
 | ||
| Fig. 9 Current density vs. time plots for methanol oxidation at 65 °C and 0.31 V (left axis) and corresponding reaction turnover frequencies (right axis) for Pt(hkl)/Ru surfaces at the optimum Ru coverage (0.20, 0.15, and 0.30 ML for Pt(111), Pt(110), and Pt(100) respectively). Reprinted from ref. 45, © Elsevier 1999, with permission. | ||
Although Kim et al. demonstrated that Ru chemisorbed on Pt(111) is mainly metallic at low potential, when the surface was stepped to more positive potentials (0.40 V or higher, Fig. 10), progressively more Ru oxides reappeared.64 Since the ruthenium enhancement of methanol electrocatalysis is more prominent at low potentials, where metallic ruthenium is present, it was concluded that the presence of a Ru metallic phase is a prerequisite for effective methanol oxidation electrocatalysis. The Ru island size is also a factor in determining the activity for methanol oxidation. Oxidation and subsequent reductive splitting of Ru islands to form smaller (1–3 nm) islands results in a surface with a slightly lower activity for methanol oxidation than a similar surface prepared without Ru island oxidation/reduction, a difference that has been attributed to the lower concentration of Pt ensembles available for methanol dehydrogenation on the surface with higher Ru dispersion.62
 | ||
| Fig. 10 (a) X-ray photoelectron spectrum of the Ru 3d region after deposition from 2 mM Ru(NO)(NO3)3 in 0.5 M H2SO4 at 0.30 V for 1 min. (b) As in (a), but following the deposition at 0.30 V the electrode potential was stepped to 0.40 V for 1 min. (c) As in (a), but following the deposition the electrode potential was stepped to 0.50 V for 1 min. (d) As in (a), but following the deposition the electrode potential was stepped to 0.60 V for 1 min. No further change was found in the range from 0.6 to 0.9 V. Reprinted from ref. 64, © Elsevier 2001, with permission. | ||
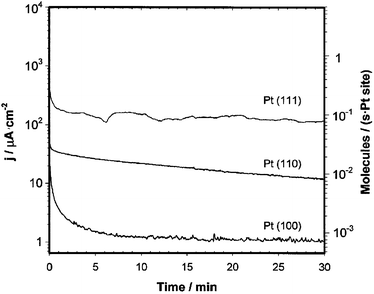
Electrodes modified by spontaneous Ru deposition were shown to have a higher activity for methanol oxidation than electrodes produced by forced deposition, and both had higher activity than Pt(111)/Ru produced by Ru evaporation (Fig. 11). This difference is apparently related to the larger Ru island size resulting from forced deposition and evaporation, as inferred from FTIR spectra of CO-covered electrodes and STM images, respectively.70 However, all Ru-modified electrodes had a lower activity than Pt/Ru alloy, a result in contrast to the work of Chrzanowski et al.,53 who found a much higher activity for the Pt(111) surface modified by electrodeposited Ru.
 | ||
| Fig. 11 Plot of the current density for methanol oxidation from current–time curves at 0.50 V as a function of Ru coverage. Data obtained at 300 s. ● = Pt(111)/Ru formed by spontaneous deposition, □ = Pt(111)/Ru prepared by Ru forced deposition with bubbling H2, × = Pt(111)/Ru prepared by Ru evaporation, and ○ = Pt/Ru alloys prepared in UHV. Reprinted with permission from ref. 70, © American Chemical Society 2000. | ||
Based on the results of Iwasita et al.70 and Strbac et al.,62 it appears that the optimum Ru island size for methanol oxidation is in the 2–5 nm range. Such an optimum is most likely due to the need to balance the number of Pt–Ru pairs, where CO oxidation occurs, with the number of Pt ensembles large enough for methanol adsorption.
Hoster et al. investigated the role of Pt–Ru pairs in methanol oxidation, and demonstrated that the activity for methanol activation could be increased by first sputtering the Pt(111) substrate to create defects, then depositing Ru by evaporation.74 On the defect-rich sputtered surface, Ru deposition occurs mostly through step decoration, rather than island formation, resulting in a higher number of Pt–Ru pairs. However, a part of the increased activity can be attributed to the higher fraction of low-coordination Ru. Surface alloys produced by Ru evaporation and subsequent annealing, in which roughly 20% of the surface atoms are Ru, show much lower activity for methanol oxidation than the surfaces produced by Pt sputtering followed by Ru deposition without annealing.74 The decrease in activity is largely due to the higher coordination number of Ru atoms in the annealed surface, although the smaller Pt domains on the annealed surface may also limit the adsorption of methanol.
The oxidation of ethanol on Pt(hkl)/Ru has also been studied.61 Colle et al. showed a Ru enhancement in the oxidation of ethanol on Pt(100)/Ru prepared by spontaneous deposition. The activity vs. deposition time curve was shown to reach a maximum at about 90 s, an observation attributed to a higher proportion of metallic Ru formed at this deposition time. This observation could also be explained in terms of the Ru coverage as a function of deposition time, similar to the case with methanol oxidation.42
In agreement with previous observations on Pt–Ru alloy surfaces, ruthenium-modified Pt single crystal surfaces exhibit a substantial electrocatalytic enhancement toward the oxidation of adsorbed CO, demonstrated in early studies by the shift of the CO oxidation peak to more negative values.25,44,52 FTIR spectra on the CO-covered electrodes indicate that the electrocatalytic activity of the Ru modified Pt(111) for CO oxidation is slightly better than that of a 50∶50 Pt/Ru alloy.52
Lu et al. studied the voltammetric and chronoamperometric oxidation of chemisorbed CO in sulfuric acid electrolytes on Pt(111)/Ru prepared by spontaneous deposition.80 Voltammetric CO stripping from Pt(111)/Ru at 50 mV s−1 yielded two well-resolved current peaks (Fig. 12), around 0.55 V and 0.67 V, confirming a similar tendency found in refs. 35, 54 and 72. The first peak originates from CO chemisorbed on and around ruthenium islands deposited on Pt(111), while the other peak comes from CO chemisorbed on the “pure” Pt(111) phase of the Pt(111)/Ru electrode, away from the Ru islands. This interpretation has been confirmed recently using sum frequency generation (SFG) spectroscopy,81 wherein the spectral feature at 1970 cm−1, corresponding to CO adsorbed on Ru sites, was removed by scanning the electrode potential through the region of the first voltammetric peak. During this process, the absorption band at 2070 cm−1 was not removed, indicating that the Pt–CO population remained (Fig. 13). Using double-potential step chronoamperometry, Lu et al. also investigated CO oxidation on these two surface phases. Apparently, the low potential oxidation of chemisorbed CO (on the Ru phase) occurs via the Langmuir–Hinshelwood mechanism. In contrast, CO chemisorbed on Pt sites not occupied by Ru diffuses to the Pt(111)/Ru edge, where it is oxidized at higher potential.82,83 The voltammetric peak splitting also occurs on platinum nanoparticle electrodes modified by ruthenium.35 NMR results carried out with such Pt/Ru samples confirm two distinct types of CO, and a lack of exchange between the two CO surface phases.35
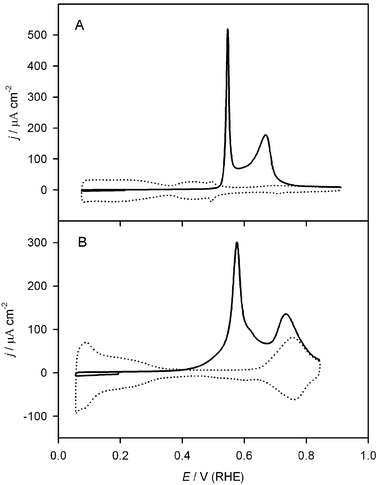 | ||
| Fig. 12 Voltammetric CO oxidation on Pt(111)/Ru in CO-free solution (solid line = CO stripping, dotted line = background). (A) 0.1 M H2SO4 solution (20 min Ar purge). (B) 0.1 M NaOH solution (5 min Ar purge). Scan rate = 50 mV s−1. Reprinted from ref. 84, © Elsevier 2004, with permission. | ||
 | ||
| Fig. 13 (A) In situ SFG spectra of chemisorbed CO on Pt(111)/Ru (ca. 0.18 ML of Ru) before (a) and after (b) stripping of Ru–CO, and (B) on Pt(111) before (a) and after (b) partial CO stripping in CO-free 0.1 M H2SO4.81 (Reprinted from ref. 81, © Elsevier 2004, with permission.) | ||
A similar peak splitting was observed during CO oxidation from Pt(111)/Ru in 0.1 M NaOH, as shown in Fig. 12.84 However, in the alkaline system, a much higher fraction of the CO is stripped in the lower potential peak (ca. 80%) than in the acid system (ca. 33%). This difference can be understood in terms of the contribution of the pure Pt(111) phase to the CO oxidation. Whereas in the acid media all CO oxidation occurs at the Pt/Ru edge, in the alkaline system the Pt(111) phase is also active for CO oxidation at low potential, so the larger CO stripping peak is a superposition of the Langmuir–Hinshelwood oxidation of CO on and around Ru islands and the oxidation of CO on Pt(111) far from Ru islands, which has been interpreted as either a Langmuir–Hinshelwood reaction involving Pt defects or an Eley–Rideal reaction.84
After selective Ru electrodeposition at steps on several vicinal Pt(111) surface, Baltruschat and coworkers54,55 observed a splitting of the voltammetric CO oxidation peak into two peaks for the Ru-modified Pt(111) and Pt(665) [12(111) × (111)] surfaces in sulfuric acid solution, but for the Ru-modified Pt(332) [6(111) × (111)] and Pt(755) [6(111) × (100)] surfaces CO oxidation occurred in a single peak. Galvanostatic experiments showed a similar result. These results were explained in terms of an electronic effect of Ru, as well as an OH spillover effect. On the surfaces with narrow terraces, the Pt–CO bond is weakened by an electronic effect, and all CO is oxidized at a low potential. On the larger terraces, CO bonded to Pt sites close to Ru atoms is weakened, but CO farther from Ru is unaffected. This CO is still oxidized at a lower potential than on clean Pt surfaces because of OH spillover from Ru sites. Essentially the same interpretation was proposed by Davies et al. to explain the presence of two peaks in the CO stripping voltammogram for Pt(111)/Ru produced by Ru evaporation.72 Alternatively, similar observations in other laboratories have been explained in terms of slow diffusion of CO to Ru islands83 or a higher overpotential for CO oxidation at the Pt/Ru edge after oxidation of CO from Ru sites and Pt sites adjacent to Ru atoms.80 The slow diffusion of CO to Ru islands is, however, in contrast to an EC-NMR study that shows uniquely fast diffusion on the Ru islands on Pt,35 and chronoamperometric studies on Pt(111) that suggest rapid CO diffusion.85 Despite these observations, there is some theoretical justification for a slow diffusion of CO on Pt to the Ru edge, since the weakening of the CO bond on Pt sites neighboring Ru (the ligand effect) would result in a slow “uphill diffusion” of CO to these sites.83 It should be noted that not all CO stripping studies on Pt(111)/Ru have found a split CO oxidation peak. The peak splitting is only manifested at low to moderate Ru coverage.65 Also, the CO coverage at the onset of CO stripping affects the degree of splitting.80
Based on CO temperature programmed desorption (TPD) and stripping measurements with Pt(110)/Ru crystals prepared by Ru evaporation and annealed to have various amounts of Ru in the top and subsurface layers, Davies et al. concluded that, although subsurface Ru weakens the Me–CO bond, it has little effect on the CO stripping potential.25,75 Pt(110)/Ru prepared without annealing shows improved CO stripping behavior relative to pure Pt(110), demonstrating a contribution from Ru adatoms, but the main contribution to improved CO oxidation catalysis comes from Ru incorporated in the surface layer of atoms.25,75 This result is in contrast to the surface Pt(111)/Ru alloy results of Hoster et al., which showed a dramatic reduction in methanol electrooxidation activity on surface alloys relative to the unannealed Pt(111)/Ru surfaces.74
CO TPD on Pt(110)-(1 × 2) modified by Ru CVD (Fig. 14)36,76 is similar to that reported for evaporated Ru on Pt(110) by the Hayden group.75 TPD spectra for hydrogen and water have also been presented.36 Based on the experimental results in vacuum, Lu and Masel made comparisons with electrochemical CO oxidation, including a conclusion that, though CO adsorption is weakened by Ru-modification (the ligand effect), the role of Ru in the activation of OH (the bifunctional mechanism) accounts for ca. 80% of the decrease in CO oxidation potential observed in the electrochemical environment.
 | ||
| Fig. 14 A series of TPD spectra taken by exposing either a clean 110 K Pt(110)-(1 × 2) sample (a) or a Ru-covered Pt(110)-(1 × 2) sample prepared by CVD (b) to various amounts of CO followed by heating at 15 K s−1. Reprinted with permission from ref. 36, © American Chemical Society 2001. | ||
3. Pt(hkl)/Pd surfaces
Compared to Pt, relatively few studies on the electrochemical properties of Pd single crystals have been published, although the studies by Soriaga and coworkers are notable.86,87 Pd, which has been shown to catalyze a number of reactions both in the gas phase and in solution, owes some of its properties to the unusual weakness of the Pd–Pd bond. This weak metallic bonding facilitates both adsorption of surface species and absorption of subsurface species. The relative scarcity of electrochemical studies on Pd(hkl) surfaces is primarily due to the difficulty in preparing such surfaces, since the flame annealing technique is difficult to apply to Pd, and bulk Pd electrodes absorb significant amounts of hydrogen at low potentials, disordering the surface and complicating efforts to study other electrode processes.88,89 For this reason, studies of thin Pd films deposited on Pt(hkl) and Au(hkl) have been performed by some workers as a substitute for bulk Pd single crystals. Of course, the combination of Pd with these other metals may have significant catalytic advantages, making these bimetallic surfaces interesting in their own right. The modification of Au(hkl) by Pd has been studied extensively, with important contributions by the Uosaki90,91 and Kolb92,93 groups on Pd electrodeposition, but for the purpose of this review discussion will be limited to Pd decoration of Pt single crystals. The advantages of alloying Pt with Pd for formic acid oxidation were recognized early on by Capon and Parsons,94 and since then Pt/Pd surfaces have attracted significant attention because of their unique activity for this reaction. On pure Pt, electrooxidation of formic acid proceeds through a parallel path mechanism,95,96 in which one path produces CO as a poisoning intermediate,97 while the other path proceeds directly to CO2. Addition of Pd to Pt shifts the selectivity toward the direct path, as discussed in greater detail below. In this respect, catalysis of formic acid oxidation on Pt/Pd shows a sharp contrast to methanol oxidation on Pt/Ru – whereas Ru promotes methanol oxidation primarily by expediting surface CO removal, Pd promotes formic acid oxidation by impeding the formation of CO.3.1 Electrodeposition
Following the first study of Pd deposition on Pt(111) by spontaneous deposition,98 Inukai and Ito reported the electrodeposition of Pd on Pt(111) and Pt(100) by cyclic voltammetry in sulfuric acid solutions containing Pd2+.99 A sharp, reversible peak was observed at 0.22 V on Pt(111)/Pd in 0.5 M H2SO4 with submonolayer coverage of Pd. This peak disappeared at higher coverage,100 being replaced by a peak at 0.26 V (Fig. 15), as previously reported for the forced deposition of Pd.15 The adsorption of (bi)sulfate on Pd islands, along with the simultaneous desorption of hydrogen, is the source of the sharp peak at 0.22 V,101 while the peak at 0.26 V may be assigned to a similar process occurring on Pd atoms adsorbed in the second (or higher) layer from the Pt surface,102–104 although some workers have assigned this peak to hydrogen adsorption/desorption coupled with anion desorption/adsorption at Pd steps.105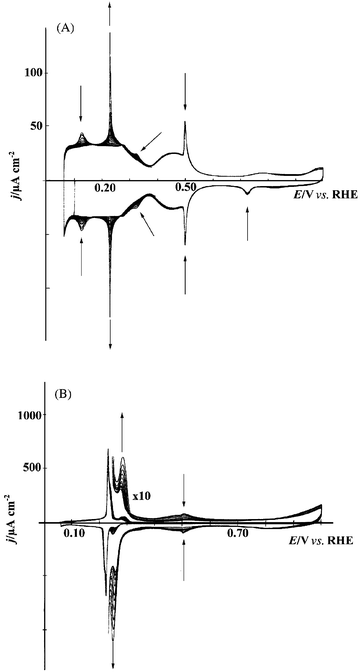 | ||
| Fig. 15 Consecutive voltammetric cycles recorded for a Pt(111) electrode in 0.1 M H2SO4 + 0.01 mM PdSO4 showing the progressive modification of the surface due to the Pd deposition at low Pd coverage (A) and Pd coverage near 1 ML (B). Sweep rate = 50 mV s−1.100 | ||
Inukai and Ito reported FTIR spectra of Pt(111)/Pd and Pt(100)/Pd electrodes with adsorbed CO at various stages of the Pd deposition, with features indicative of island or film formation by the adsorbed Pd, rather than adsorption as single adatoms.99 These observations have been reproduced in several other studies.103,106–109
The morphology and surface relaxations of Pd electrodeposits on Pt(111) and Pt(100) have been studied using in situ surface X-ray scattering (SXS) in H2SO4 solutions.105,110–112 Pd was found to form pseudomorphic films on both substrates. The Pt-Pd spacing on the Pt(111)/Pd surface is expanded by ∼2% relative to the bulk layer spacing in the presence of adsorbed hydrogen and adsorbed (bi)sulfate.110,111 Adsorption of CO causes no change in the surface relaxation; however, during bulk CO oxidation the surface layer was shown to be contracted by ∼1% relative to the bulk spacing.111
On Pt(111), SXS results show that Pd electrodeposition by CV in PdO/H2SO4 solutions creates a full Pd monolayer prior to growth of the second Pd adlayer, whereas additional Pd deposition forms islands, corresponding to a Stranski–Krastanov growth mode.105 Similar results were found for Pt(100)/Pd, although in this case the second layer growth starts at a coverage of 0.8 ML.112 Nevertheless, the first Pd monolayer on Pt(100) is essentially completed prior to the development of large islands in subsequent layers. These results are in good agreement with theoretical predictions of the thermodynamically favored growth mode, according to a mean-field model,113,114 which predicts Stranski–Krastanov growth for Pt(100)/Pd and either Stranski–Krastanov or Frank–van der Merwe growth for Pt(111)/Pd, but they contrast with the Frank–van der Merwe growth reported for evaporation of Pd onto Pt(111).102,115
STM imaging of Pd electrodeposition on Pt(111) in 0.1 M H2SO4 solutions further confirmed the Stranski–Krastanov growth mechanism, as shown in Fig. 16.104 Potentiostatic deposition from PdSO4 solution at 0.77 V was shown to result in very slow Pd deposition, in which island nucleation is apparently insensitive to surface structure. More than 30 min of deposition was required to complete the first monolayer. Deposition of additional Pd layers is induced at lower potential. At 0.71 V, Pd deposition in the form of clusters on top of the first Pd monolayer was observed, with nucleation occurring preferentially at steps. Palladium deposition from H2PdCl4 solution is more facile than deposition from chloride-free solutions. In this case, the first Pd layer may be formed at 0.85 V, while bulk deposition proceeds at 0.80 V. Since the reversible potential for Pd deposition in this solution is 0.82 V, deposition of the first Pd layer is a UPD process, in contrast to the deposition from chloride-free solution, where no Pd UPD is observed. This observation is in agreement with predictions, based on Monte Carlo simulations of an embedded atom model, of Pd UPD on Pt(hkl).116 However, the same simulations predicted no UPD of Pd on Au(hkl), in contrast with experimental observations,93,117,118 so the validity of these simulations, which did not consider anion adsorption or the presence of an electric field, is questionable. Pd deposition from the H2PdCl4 solution results in smoother deposits than are observed in chloride-free solutions, with essentially no 3D cluster formation prior to the completion of the second monolayer (Fig. 16), thus approaching the Frank–van der Merwe growth mode observed for Pd vapor deposition on Pt(111).102,115
 | ||
| Fig. 16 Sequence of STM images showing Pd deposition on Pt(111) from 0.1 M H2SO4 + 0.1 mM H2PdCl4. (a) Completion of the first layer; (b) growth of the second layer; (c) completed second layer; (d) Pt(111) covered by an equivalent of almost 4 Pd ML. Reprinted from ref. 104, © Elsevier 2003, with permission. | ||
It should be noted at this point that all FTIR, STM, and SXS studies showing Pd deposition in the form of large islands or films, rather than dispersed adatoms, have been performed in H2SO4 solutions, which contain the strongly adsorbing sulfate/bisulfate anion. Voltammetric data suggests a different morphology of Pd electrodeposits in HClO4 solutions. Whereas the “butterfly peak” at 0.49 V, associated with large, clean Pt(111) domains, is observed in 0.1 M H2SO4 solutions even at Pd coverage approaching one monolayer, in HClO4 solutions a similar feature at 0.79 V disappears at Pd coverage as low as 0.05 ML.100,119 Therefore, it appears that Pt(111)/Pd prepared in HClO4, or prepared in H2SO4 and transferred to HClO4, is characterized by small nanoislands of Pd or individual Pd adatoms, rather than the extensive films observed in H2SO4. This behavior shows full reversibility during transfer between different acid solutions, indicative of a high degree of Pd surface mobility. These conclusions, drawn only from voltammetric features, have not yet been investigated using surface structure-sensitive techniques such as in situ STM or SXS; such studies are recommended to confirm the interpretation. Although the presence of electrodeposited Pd on Pt(111) in HClO4 was found to disrupt the long-range surface order, even at low Pd coverage, voltammetric experiments indicate a high degree of surface ordering at a Pd coverage of exactly 1 ML.119 At this level of coverage, a “butterfly” feature appears at 0.69 V, very similar to the feature observed on clean Pt(111) at 0.79 V (Fig. 17). The feature disappears at slightly lower or slightly higher coverage. These observations further support the conclusion that Pd electrodeposited on Pt(111), at least under the conditions employed in these studies, forms an epitaxial film in which essentially no second-layer growth occurs until the first monolayer is completed.
 | ||
| Fig. 17 Stationary voltammetric profiles of different Pt(111)/Pd electrodes in 0.1 M HClO4. (a) 0.94 ML of Pd; (b) 1.02 ML of Pd; (c) 1.13 ML of Pd. The voltammetric profile of the Pt(111) electrode is also shown in (b) (dotted line). Sweep rate = 50 mV s−1. Reprinted from ref. 119, © Elsevier 2002, with permission. | ||
The potential of zero total charge (pztc), as determined by CO charge displacement experiments, is a useful quantity in determining coverage of adsorbates from voltammetric data. Alvarez et al. determined the pztc of Pt(111)/Pd as a function of Pd coverage for submonolayer coverage. In 0.1 M H2SO4 solution, the pztc was found to vary from 0.33 V (θPd = 0 ML) to 0.23 V (θPd = 1 ML).101 Interestingly, the pztc in 0.1 M HClO4 was found to be independent of Pd coverage and equal to the pztc of clean Pt(111), 0.33 V.100,120 Based on pztc measurements and CO stripping voltammograms, the maximum CO coverage under these conditions was shown to be 0.64 ML for the Pd-free Pt(111) surface in both electrolytes, decreasing to 0.56 ML and 0.59 ML for the surfaces with 1 ML of Pd in 0.1 M H2SO4 and 0.1 M HClO4, respectively. Also observed was an increase in the hydrogen coverage for the Pt(111)/Pd electrode, with a maximum coverage of Hads of close to 1 ML in both sulfuric and perchloric acid.100,101 This higher coverage of Hads relative to clean Pt(111) (θHads ≈ 0.66 ML121) is perhaps not surprising, since the sharp hydrogen adsorption voltammetric peak suggests that the repulsive interactions between adsorbed hydrogen on Pt(111) are less significant on the Pd monolayer. In contrast, a recent study found a much lower hydrogen/anion desorption/adsorption charge, suggesting a hydrogen coverage well below 1 ML.104 The reason for this discrepancy is unclear.
On clean Pt(111), SXS experiments demonstrated that adsorption of CO from CO saturated solutions results in a (2 × 2)-3CO adlayer at low potentials,122 with a transition to a (√19 × √19)R23.4°-13CO structure at higher potentials,123 confirming earlier STM and FTIR results.124,125 The (√19 × √19)R23.4°-13CO structure could also be deduced from the previously reported LEED patterns from a Pt(111) electrode covered with CO and then emersed to vacuum.126 In contrast, SXS measurements have not detected ordered CO adlayers on Pt(111) modified by a monolayer of Pd.111 However, FTIR studies have shed some light on the adsorption site preference of CO on Pt(hkl)/Pd surfaces. Pt(111) modified with electrodeposited Pd adsorbs CO in three-fold hollow sites at low CO coverage,106 shifting to bridge-bonded sites at higher coverage.99,103,106–109 Bridge-bonded CO has also been observed on Pd films deposited on Pt(100).127 Unlike Pt(111) and Pt(100), Pt(111)/Pd and Pt(100)/Pd show little tendency to adsorb CO on atop Pd sites, at least on terraces. A band observed near 2035 cm−1 on various vicinal Pt(111)/Pd surfaces has been assigned to CO adsorbed in the atop position on Pd atoms at step edges,103 although other workers have assigned this peak to atop CO on small Pt domains not covered by Pd.107,108 A splitting of the bridge-bonded CO on Pd, observed for Pd multilayers on Pt(111) as well as stepped surfaces with Pt(111) terraces covered by a Pd monolayer, may similarly be attributed to bridged CO adsorbed on steps and on terraces.103
3.2 Electroless deposition
The forced deposition technique was used to prepare multilayers of Pd on Pt(110) and Pt(100), which were examined using CO adsorption and FTIR, as well as Cu UPD, in two studies by Gomez et al.129,130 Whereas CO adsorbs on Pt(111)/Pd, Pt(100)/Pd, and Pt(110)/Pd primarily in the bridge-bonded configuration at high CO coverage,99,103,106–109,127,129,130 a small band at ca. 2050 cm−1 on Pt(110)/Pd has been attributed to atop CO.129 A similar feature on Pt(111)/Pd was later attributed to CO adsorbed in the atop position at steps.103 CO charge displacement measurements revealed that the currents observed at potentials slightly more positive than the hydrogen absorption region on Pt(100)/Pd and Pt(110)/Pd correspond to simultaneous adsorption/desorption of hydrogen and desorption/adsorption of (bi)sulfate anions,129,130 similar to results from the Pt(111)/Pd system.100,101
Cu UPD was also studied on Pt(111)/Pd prepared by forced deposition of Pd,131 and the dissolution of the Pd films in solution containing halide ions was observed. In sulfuric acid solutions containing low concentrations of chloride ion, peaks in the Cu UPD were identified corresponding to Cu UPD on single Pd layers and on Pd multilayers. In this way, the dissolution of Pd in chloride-containing solution was found to occur first on the Pd multilayers, owing to a preferential dissolution at step sites. Dissolution of the remaining Pd monolayer occurs only at higher potential. Nevertheless, Pd may be completely removed from Pt(111) surfaces in the presence of chloride ion without significant disordering of the Pt(111) surface, in contrast to observations in halide-free sulfuric acid. This difference suggests a lower degree of oxide formation in the presence of chloride, preventing the roughening known to occur during oxide formation. Similar results were observed in the presence of bromide ion. Pd was stripped from the surface at lower potential in halide-containing solutions, presumably due to the formation of a Pd-halide complex.
3.3 Vacuum deposition
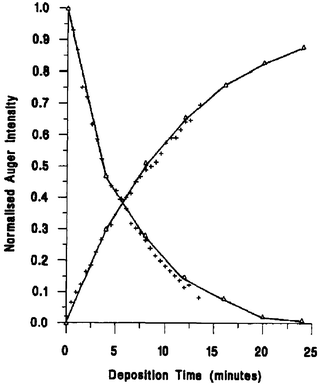 | ||
| Fig. 18 The normalized Auger signal vs. time vapor deposition plot for Pd on Pt(111). Decaying curve represents the Pt 64 eV peak, increasing curve represents Pd 330 eV peak. Crosses are experimental data, the solid line is the best fit of the Gallon model, and triangles indicate the intensity at each monolayer break. Reprinted from ref. 102, © Elsevier 1994, with permission. | ||
Pd evaporation onto Pt(100)-(1 × 1) has also been shown, using AES, to follow a Frank–van der Merwe growth mechanism.132,133 After transfer of the UHV prepared Pt/Pd surface to an electrochemical cell containing 0.1 M H2SO4 solution, a peak was observed at 0.15 V during CV with electrodes covered by submonolayers of Pd, corresponding to hydrogen adsorption and (bi)sulfate desorption on the first Pd layer. At higher Pd coverage this peak is replaced by a peak at 0.25 V, corresponding to a similar process on Pd multilayers. These voltammetric results are similar to the results obtained with Pt(100) modified by Pd through spontaneous and forced deposition128 and electrodeposition.112 A different situation was observed for evaporation of Pd onto the reconstructed Pt(100)-hex-R0.7° surface. On this surface, nucleation of the second layer of Pd occurs well before completion of the first Pd layer. This may be a result of a disordered surface, as Pd deposition lifts the reconstruction, resulting in a Pt(100)-(1 × 1) surface with a high density of Pt islands.
3.4 Monocrystalline alloys
The flame annealing method, as originally developed for preparation of Pt single crystal surfaces,1 can also be applied to the preparation of Pt/Pd alloy single crystals, hereafter denoted as PtPd(hkl).134 PtPd(111) surfaces prepared using flame annealing have been shown to form well-ordered (1 × 1) surfaces for Pd bulk concentrations ranging from 0–25% for temperatures below 900 K.26,134 In contrast, PtPd(100) and PtPd(110) surfaces have been shown to reconstruct under some circumstances.135 The PtPd(100) surface with low amounts of Pd reconstructs in UHV, resulting in a “hex” reconstruction similar to that observed for Pt(100) in UHV.136 However, for a Pd bulk concentration of 25%, no reconstruction is observed. This observation is consistent with earlier results showing that the Pd(100) surface does not reconstruct in UHV,137 and also with the observation that Pd vapor deposition onto the Pt(100)-hex-R0.7° surface lifts the reconstruction, resulting in a (1 × 1) surface.132 The presence of Pd in PtPd(110) surfaces also suppresses the (1 × 2) “missing row” reconstruction observed on pure Pt(110),138 again consistent with the observation that the Pd(110) surface does not reconstruct over a wide range of temperature in UHV.139 For the (110) surface, much lower Pd concentrations are required to prevent reconstruction than for the (100) surface, with no reconstruction observed for a bulk Pd concentration of 6%.Preparation of PtPd(111) surfaces by the flame annealing method results in an enrichment in the surface Pd content relative to the bulk value. For a bulk concentration of 25%, the surface Pd concentration, as determined by LEISS, is 44% following flame annealing and transfer to UHV. Surface segregation of Pd was shown to occur to an even greater extent on the (100) and (110) surfaces following thermal annealing.135
The electronic structure of PtPd(111) surfaces was studied in an XPS/UPS investigation by Radosavkic et al.140 The presence of Pd was found to shift the Pt 4f7/2 binding energy to lower values, in good agreement with theoretical calculations. By comparing the signal intensity for surface and subsurface Pt atoms with the intensity observed with a pure Pt(111) surface, a surface enrichment in Pd was confirmed, in agreement with LEISS results,26 while the subsurface showed an enrichment in Pt. This subsurface Pt enrichment, which prevents the formation of 3D Pd structures in the near-surface region, helps to explain the lack of hydrogen absorption observed during voltammetric experiments with alloys of up to 25% bulk Pd.135 In light of the surface Pd enrichment, it is surprising that photoelectron spectra corresponding to the valence band with adsorbed CO140 suggest, qualitatively, a surface composition similar to the bulk composition. Adsorption of CO results in a shift in the surface Pt 4f7/2 binding energy to higher values, as well as a peak splitting that has been attributed to the presence of multiple CO adsorption sites on the PtPd(111) surface.
Whereas Pd adsorption on Pt(hkl) has been shown to result in Pd island formation, the Pt/Pd bulk alloy single crystal surfaces would be expected to possess highly dispersed Pd surface atoms, based on the complete miscibility of these elements.141 Voltammetric analysis of hydrogen/anion adsorption/desorption26,134,135 and Cu UPD26 support this interpretation, as do FTIR spectra showing a single band for adsorbed CO as a result of dipole–dipole coupling between CO adsorbed on Pt and Pd sites on the PtPd(111) surface.109
3.5 Electrocatalysis
The oxidation of formic acid on Pt(100) and Pt(111) modified by forced deposition of submonolayers of Pd was compared with formic acid oxidation on clean Pt(100) and Pt(111) in a study by Llorca et al.,142 and though the activity of Pt(111) did not change greatly as a result of Pd deposition, the activity of Pt(100) did increase significantly, this latter point in good agreement with a later study using electrodeposited Pd.143 In cyclic voltammetric experiments, this increase was manifested in a ca. 200 mV shift in the formic acid oxidation peak potential on the negative-going scan (no comparison is possible on the positive going scan because Pt(100) is completely deactivated). The cyclic voltammogram of Pt(100)/Pd in sulfuric acid solution containing formic acid shows features clearly related to formic acid oxidation on Pt(100) domains, on Pd islands 1 ML in height, and on Pd islands composed of more than one layer. The oxidation potential on the 1 ML thick Pd islands was the lowest, suggesting that an optimized surface would consist of a uniform film of Pd one monolayer thick on Pt(100). However, the forced deposition method on Pt(100) does not result in such uniform films, instead showing some three-dimensional growth at coverages as low as 0.60 ML. Therefore, for forced deposition, the optimum Pd coverage was found to be 0.60 ML, but for the techniques that result in smoother adlayers (electrodeposition and evaporation) the optimum coverage, based on these results, would be expected to be approximately one monolayer. In this respect, there is some disagreement between this work and the later work by Baldauf and Kolb, who found that electrodeposited Pd multilayers were more active than Pd monolayers for formic acid oxidation on Pt(100)/Pd.143 The discrepancy may be due to the different experiments used to assess activity—chronoamperometry for the electrodeposited Pd, CV for the forced deposition. Although all Pt(hkl)/Pd surfaces studied by Baldauf and Kolb showed a higher activity than Au(hkl)/Pd, Pt(hkl), or Pd(hkl), the Pt(100)/Pd surface showed a particularly high activity, strongly dependent on the Pd coverage (Fig. 19).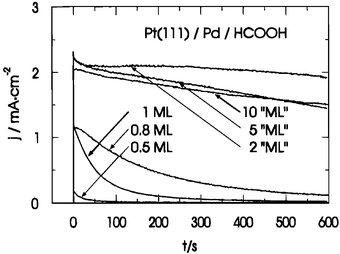 | ||
| Fig. 19 Current transients for thin Pd overlayers on Pt(111) in 0.1 M H2SO4 + 0.2 M HCOOH following a potential step from 0.07 V to 0.22 V. Reprinted with permission from ref. 143, © American Chemical Society 1996. | ||
Arenz et al. also found that Pt(111) modified by electrodeposited Pd was more active than pure Pt(111) for formic acid.109 FTIR spectra recorded during formic acid oxidation showed that, while significant amounts of CO were formed on Pt atoms, the Pd islands showed no CO formation (Fig. 20). This lack of CO poisoning of the Pd islands explains, in part, the enhanced activity for formic acid. Although CO poisoning of the Pd islands did not occur in this study, some form of Pd deactivation did occur at higher electrode potentials, most likely due to oxidation of the Pd islands.127,144 The higher degree of Pd oxidation at higher temperature leads to a decline in activity with increasing temperature, in contrast with the increase in activity with temperature typically observed for oxidation of formic acid (or other fuels) on pure Pt.109 In contrast to the results on the Pt(111)/Pd surface, FTIR spectra show that the PtPd(111) alloy surface with a surface Pd concentration of 15% produces significant amounts of CO during formic acid oxidation.109 It seems probable, based on the Pd monolayer experiments, that this CO is primarily adsorbed on Pt sites, but the strong dipole–dipole coupling prevents discrimination between CO on Pt and Pd sites. Cyclic voltammetry in a formic acid-containing solution reveals the effect of this adsorbed CO, with a significant poisoning effect observed on the positive-going scan for the alloy surfaces.
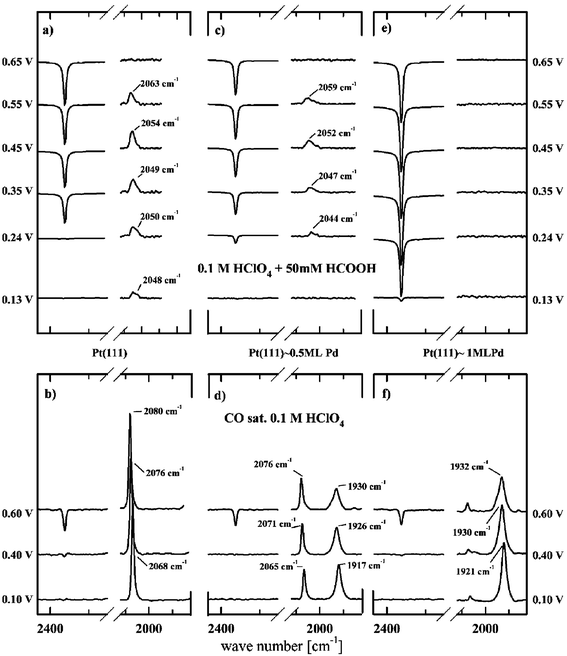 | ||
| Fig. 20 (a, c, e): FTIR spectra recorded in 0.1 M HClO4 containing 0.05 M HCOOH. Reference spectra are recorded at 0.0 V for the spectral range of 2150–2450 cm−1 (CO2 band) and at 1.0 V for the spectral range of 2150–2450 cm−1 (CO band). CO2 bands are divided by a factor of 20. (b, d, f): Sets of spectra (divided by a factor of 2) for the same surfaces in CO-saturated HClO4 solutions are shown for comparison.109 | ||
Rotating disk experiments revealed an enhanced activity of Pt(111) modified by a monolayer of Pd for H2 oxidation in acid solutions,110 while FTIR and voltammetric studies showed that similar electrodes have a decreased activity for CO oxidation in acid solutions,108,111 as well as alkaline solutions,107 relative to pure Pt(111). A voltammetric study demonstrated similarly lower CO oxidation kinetics on Pt(100) modified with submonolayers of Pd in acid and alkaline electrolytes.127 It is therefore surprising that FTIR studies showed oxidation of CO adsorbed on Pd islands at lower potential than CO adsorbed on pure Pt(100) domains.127 This apparent discrepancy was explained in terms of the higher oxophilicity of Pd relative to Pt, which results in a higher coverage of OH or adsorbed H2O at low potentials, enhancing the CO oxidation at the onset, but also creating very strong OH adsorption at high potentials, effectively poisoning the CO oxidation reaction. In HClO4 solutions, the onset of Pd–CO oxidation occurs ca. 400 mV lower on Pt(100) modified with 0.35 ML of Pd than on Pt(111) modified with 0.31 ML of Pd.108,127 This observation may have implications for the design of Pt/Pd CO-tolerant electrocatalysts for HCOOH oxidation, though as noted above, Pd–CO is not observed during formic acid oxidation on Pt(111)/Pd,109 and Pt/Pd shows little promise for use in methanol oxidation as compared with Pt/Ru.145,146 Although the relative catalytic inactivity of adsorbed Pd at higher potentials can be largely attributed to its high oxophilicity, the adsorption of chloride ions, present as a universal impurity in HClO4 solutions, may in some cases also be a limiting factor, particularly in studies that employ enhanced mass transfer through stirring or electrode rotation. The strong interaction of chloride ions with Pd blocks OH adsorption at higher potentials, an effect observed through comparison of Pt(111)/Pd in HClO4 with and without added Cl−.26,108 Similar, although weaker, site blocking in sulfuric acid is due to (bi)sulfate adsorption, which has been shown by FTIR and CO charge displacement experiments to occur at potentials as low as 0.20 V on a Pt(111) electrode modified by a submonolayer of Pd.101 The adsorption of (bi)sulfate on Pd adlayers at potentials much lower than on pure Pt(111) signals a stronger adsorption energy on Pd sites than on Pt.101 As discussed earlier for chloride adsorption, this stronger adsorption of (bi)sulfate blocks sites for OH adsorption, and therefore is one cause of the lower activity for CO oxidation that has been observed experimentally on Pt(hkl)/Pd surfaces in H2SO4 solutions.100,101,104,111,129,130
The oxygen reduction reaction (ORR) on Pt(111)/Pd has been the subject of several studies by the Lawrence Berkeley group147–149 using a rotating ring disk electrode (RRDE). In this configuration, hydrogen peroxide generated at the disk (the Pt(111)/Pd electrode) is detected at the ring. Whereas oxygen reduction on Pt(111) in sulfuric acid occurs mainly through the four electron pathway (producing water) at potentials higher than 0.2 V, on Pt(111) with a monolayer of electrodeposited Pd significant amounts of H2O2 are produced through a two electron pathway (in the potential range 0.2–0.6 V), likely due to the high coverage of (bi)sulfate anions, which limits the number of adjacent Pd sites available for O–O bond scission (Fig. 21). Oxygen reduction in perchloric acid and potassium hydroxide solutions, i.e. solutions with anions that adsorb weakly compared to (bi)sulfate, occurs primarily through the four electron pathway at potentials higher than 0.3 V (Figs. 20 and 21), further supporting this interpretation. Around 0.2 V, the H2O2 production rate in sulfuric acid is much lower than at higher potentials, presumably as a result of the decreased (bi)sulfate coverage, but higher rates of H2O2 generation are observed at potentials below 0.2 V as a result of site blockage by adsorbed hydrogen. The effect of (bi)sulfate is readily apparent in the higher rates of oxygen reduction in perchloric acid as compared with sulfuric acid. The relatively low activity of Pt(111)/Pd for oxygen reduction, as compared with Pd-free Pt(111), is clearly the result of the stronger (bi)sulfate adsorption on Pd sites than on Pt sites in sulfuric acid. In perchloric acid, the lower activity of Pt(111)/Pd relative to clean Pt(111) may be attributed to stronger OH adsorption,147 as well as adsorption of chloride impurities.149 In alkaline media, the activity of Pt(111)/Pd was shown to be maximized at a Pd coverage of 1 ML (Fig. 22),149 and at this coverage the rate of the oxygen reduction reaction is almost four times that on pure Pt(111).148,149 The activity drops rapidly at higher coverage, indicating a special activity of the Pd monolayer; this enhanced activity is probably due to an electronic effect.
 | ||
| Fig. 21 (A) Rotating ring disk electrode current–potential curves for the ORR in oxygen-saturated 0.05 M H2SO4 for the ring electrode (top) and for the disk electrode (bottom) of Pt(111) with a monolayer of Pd at various rotation rates; dashed curves are for an unmodified Pt(111) disk electrode at 1600 rpm. Recorded on a positive sweep from 0.05 V at 50 mV s−1. (B) As in (A), but in 0.1 M HClO4. Reprinted with permission from ref. 147, © American Chemical Society 2000. | ||
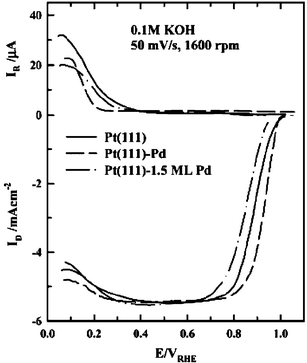 | ||
| Fig. 22 As in Fig. 20, but in 0.1 M KOH at various Pd coverages. Reprinted with permission from ref. 149, © American Chemical Society 2003. | ||
The reduction of nitrous acid and nitric oxide on Pt(hkl) modified by multilayers of Pd through the forced deposition method was studied using FTIR, in parallel with DEMS studies on rough Pd surfaces.150 Reduction of nitrous acid was determined to produce several products, including NO and N2O, and likely other species. Reduction of NO preadsorbed on Pt(hkl)/Pd, either through immersion in HNO2 solution or NO saturated solution, was shown to produce NH4+ as the only product.
4. Pt(hkl)/Me (Me = Os, Ag, Rh) surfaces
For electrocatalysis on Pt single crystal substrates, the most important noble metal adatoms, Ru and Pd, have already been discussed. However, there is a smaller body of work on other noble metal adatoms, with some electrocatalytic studies performed on Pt(hkl)/Rh, Pt(hkl)/Os, and Pt(hkl)/Ag. Preparation of Pt single crystals modified by other noble metals (Au, Re, and Ir) has also been reported, but these studies focused on UHV analysis, and no electrocatalytic studies are available in the literature.Rh surfaces have been extensively studied in UHV and electrolyte solution. Alloys of Rh with Pt and other noble metals are important catalytic materials, particularly in automotive exhaust catalysis, although the mechanism of Rh enhancement is still subject to debate. Rh shows a stronger affinity for anions than Pt,151 and, in contrast to Pt, specifically adsorbs ClO4−, with concomitant reduction to Cl−.152 Therefore, for studies where specifically adsorbed anions must be strictly avoided (e.g. atomic resolution STM153), HF electrolytes are frequently used. Rh also is a more oxophilic metal than Pt, enabling Pt/Rh surfaces to serve as bifunctional oxidation catalysts,154,155 although in this respect Rh is much less active than Ru.
Os has received little attention as a catalyst due to the difficulty in working with this metal. It is difficult to obtain in bulk form, and must usually be purchased as a powder, which can be sintered to form the bulk metal. Although the vapor pressure of Os is the lowest of the Pt-group elements, it forms a volatile oxide in contact with air, OsO4. When combined with Pt, Os has properties similar to those of Ru for the electrooxidation of CO and methanol. Recent interest in its use stems largely from the discovery that ternary and quaternary catalysts containing Os show a methanol oxidation activity even higher than that of Pt/Ru alloys.156
Ag, though the subject of many electrochemical studies, is not suitable for most electrooxidation catalysis because of its relatively low nobility. However, a favorable interaction energy between Ag and Pt stabilizes silver monolayers and bilayers on Pt single crystals, with deposition of the first UPD layer occurring at potentials roughly 0.5 V more positive than the Ag/Ag+ reversible potential in the same solution.157 Although such Pt/Ag systems are probably unsuitable for electrocatalytic applications, the study of adsorption and reaction on these bimetallic surfaces is nonetheless of fundamental interest.
4.1 Pt(hkl)/Os surfaces
Most studies of Pt(hkl)/Os have used spontaneous deposition as the primary preparation method, although some investigations of electrodeposited Os have been performed by the Tremiliosi-Filho group. Following studies of methanol oxidation on Os-modified polycrystalline Pt,59 the Urbana group studied Os deposits spontaneously formed from solutions of OsCl3 in 0.1 M H2SO4 on Pt basal single crystal surfaces using ex situ STM57,58 and in situ STM.62,160 As observed with spontaneously deposited Ru, Os is deposited on Pt(hkl) in the form of nanometer-scale islands (Fig. 23). These islands show a high degree of multilayer growth, with roughly half of the Os islands exhibiting multiple layers after a single spontaneous deposition on Pt(111),62,160 although the degree of multilayer growth is strongly sensitive to the chemical state of the dissolved Os in the deposition bath.161 During spontaneous deposition, Os islands nucleate homogeneously across the surface, insensitive to the presence of steps.62,160 The Os coverage produced by this deposition technique is high in comparison with deposition of Ru under similar conditions; however, the deposition rate is again strongly sensitive to the chemical state of the dissolved Os species, and different Os deposition baths of nominally the same composition may produce dissimilar Os deposits due to both the preparation method and, most significantly, differences between commercially produced batches of OsCl3·3H2O. Although solutions carefully prepared from the same OsCl3 batch yield reproducible results, solutions produced from different batches show different properties (such as solution color and presence of a precipitate), and produce different deposits. In all cases, aging of the solution for at least a few days is required to produce high Os deposition rates.
 | ||
| Fig. 23 EC-STM images of Os-modified Pt(111) recorded at 0.1 V in 0.1 M H2SO4 after Os was spontaneously deposited from 10−4 M OsCl3 in 0.1 M H2SO4 for 1 min (150 × 150 nm2). A chart showing the height distribution of the osmium islands (any rounded, distinguishable Os unit) accompanies the image.160 | ||
Spontaneously deposited Os on Pt(111) was further studied using ex situ XPS and in situ grazing incidence fluorescence X-ray absorption spectroscopy (GIF-XAS).161 As shown in Fig. 24, electrodes emersed from solution at potentials below 0.5 V contain only metallic Os, as determined from the Os 4f7/2 binding energy of 50.8 eV, which is only slightly shifted from the bulk Os value (50.7 eV). At potentials more positive than 0.5 V XPS indicates the presence of Os(IV) coexisting with Os(0), with the Os(0) steadily decreasing and finally disappearing at higher potential. At 0.9 V the Os 4f7/2 peak corresponding to metallic Os is replaced by a peak at slightly higher binding energy (51.1 eV), indicating the existence of an oxidation state between Os(0) and Os(IV). A similar intermediate oxidation state is observed between 1.1 and 1.2 V, corresponding to an oxidation state of Os between Os(IV) and Os(VIII). At potentials higher than 1.2 V Os is detected in the Os(VIII) state, although at these potentials most Os has been oxidatively removed from the surface, with the dissolution commencing at 0.9 V. XPS spectra recorded immediately after spontaneous deposition, i.e. before Os reduction, revealed a binding energy of 52.2 eV, corresponding to Os in a highly oxidized form.
 | ||
| Fig. 24 Potential dependence of Os oxidation states on the Pt(111) electrode after spontaneous deposition, with relative and total Os amounts determined by comparison of Os 4f and Pt 5p signals. Filled circles: total amount of Os; open circles: Os(0); squares: Os(IV); triangles: Os(VIII). Reprinted from ref. 161, © Elsevier 2003, with permission. | ||
These ex situ results were corroborated by the in situ GIF-XAS results. In the potential region below 0.5 V, the X-ray absorption energy of the white line indicates that Os is present in the metallic state. As the potential is increased above 0.5 V, the energy of the white line absorption increases, as does the white line peak height and peak width (Fig. 25). All of these parameter shifts indicate a change from metallic Os to Os oxides. As the potential is increased past 1.0 V, all GIF-XAS parameters become relatively constant, indicating that Os has reached a limiting oxidation state. Solution exchange at a potential of 1.2 V causes a decrease in the total Os signal (non-normalized edge jump height), indicating that a fraction of the detected Os is present in solution, likely in the form of OsO4.
 | ||
| Fig. 25 GIF-XAS parameters for spontaneously deposited Pt(111)/Os as functions of the electrode potential: (a) the energies of the white line peak (top), its rising (middle) and falling (bottom) sides at normalized fluorescence = 2; (b) the normalized fluorescence white line peak height; (c) the white line peak width at normalized fluorescence = 2. Reprinted from ref. 161, © Elsevier 2003, with permission. | ||
Tremiliosi-Filho and coworkers used ex situ STM to study Pt(111)/Os prepared by spontaneous deposition from H2OsCl6 solutions.158,159 Spontaneous deposition under these conditions results in Os island growth, with island nucleation insensitive to steps and defects, and predominantly monatomic island height. Oxygen coverage on Pt(100) and Pt(111) modified by Os deposition, both through spontaneous deposition and through electrolysis, was assessed using XPS.159 A higher O coverage on Pt(111) than Pt(100) was explained in terms of a higher oxidation state of Os on Pt(111), although the spectra were not analyzed in the Os 4f region to confirm this interpretation. The observation of Os oxides even for electrodes emersed at 0.05 V contrasts with earlier XPS observations of purely metallic Os below 0.5 V.161 The difference may be attributed to the lack of potential cycling treatment in the procedure used by the Tremiliosi-Filho group, as it has been shown that multiple potential cycles are necessary to completely reduce Os after spontaneous deposition.62,160,161 Also, some spontaneous deposition may occur in the time between emersion and rinsing, another probable source of the oxides detected by XPS.
In addition to the methanol oxidation studies, Pt(111)/Os and Pt(100)/Os have been investigated as catalysts for the ethanol electrooxidation reaction.158,159 On the Pt(100)/Os electrode, an optimal Os coverage of approximately 0.5 ML was determined. An optimum was also observed on Pt(111)/Os at the same deposition time (60 s spontaneous or 10 s electrolysis), although in this case the Os coverage was undetermined. In both cases, Os electrodeposits had a higher activity than spontaneously deposited Os, probably due to the higher proportion of metallic Os obtained through electrolysis.
4.2 Pt(hkl)/Ag surfaces
Although most electrochemical studies of the Pt(hkl)/Ag system have involved electrodeposition, spontaneous deposition of Ag on Pt(111) in Ag2SO4/ H2SO4 solutions has also been reported.189 Nearly complete blockage of the Pt hydrogen adsorption features indicates that high Ag coverage may be achieved using this method, an observation in contrast to results obtained with the Pt/Ru and Pt/Pd systems. Forced deposition of Ag on Pt(111) has also been reported,189 using the same H2 reduction treatment as described previously for Pd deposition.
4.3 Pt(hkl)/Rh surfaces
Electrodeposition of additional Rh layers results in the appearance of a new irreversible pair of peaks at slightly lower potential than the original hydrogen/anion adsorption/desorption peaks.190,191 These peaks are attributable to the same process, but occurring on Rh multilayers, as also demonstrated for evaporated Rh films.193 A new set of surface oxide formation/reduction peaks also appears at higher potential for Rh coverages greater than one monolayer. In all respects, the voltammetric peaks appearing on the second (and subsequent) Rh layers are very similar to those on the bulk Rh(111) electrode, in spite of the fact that the smaller Rh lattice parameter relative to Pt leads to Rh films under tensile stress. Therefore, the chemical interaction with the Pt substrate seems to be a more important factor than electronic perturbations caused by adlayer strain.190,191
FTIR spectra of Pt(111)/Rh electrodes covered by CO adsorbate also support the island-growth model of Rh deposition.99,190 CO absorption bands at 2026 and 1892 cm−1, attributed to atop and bridged CO, respectively, were observed alongside the bands corresponding to CO adsorbed on Pt sites. FTIR spectra of adsorbed NO on Pt(111) modified by small amounts of Rh reveal a peak corresponding to NO adsorbed on Pt sites at 1687 cm−1, while spectra for surfaces modified by more than one monolayer of Rh show an absorption band at 1560 cm−1 due to NO adsorbed on the second (and higher) Rh layers.190 However, no absorption bands are visible on the Pt(111) surface modified by a Rh monolayer, most likely as a result of an enhanced rate of NO dissociation on the surface with monolayer coverage.
Deposition of Rh in the form of large islands on a Pt(111) substrate was confirmed very recently using STM, as shown in Fig. 26.191 At a coverage of 0.9 ML, Rh was observed to form a mosaic of interconnected islands, with an average island diameter on the order of 20 nm. Atomic resolution images of an island revealed a hexagonal lattice with atoms separated by approximately 0.27 nm, confirming the pseudomorphic growth of Rh on Pt(111).
 | ||
| Fig. 26 STM image recorded in air of an electrodeposited Rh adlayer on Pt(111) with a coverage of ca. 0.9 ML. Reprinted from ref. 191, © Elsevier 2004, with permission. | ||
In addition to the purely electrochemical studies described above, the Pt(100)/Rh surface prepared by electrodeposition has been studied using a UHV/electrochemical transfer system by Tanaka and coworkers,194–199 who observed the disappearance of the (5 × 20) LEED pattern following Rh deposition, and its replacement by a (1 × 1) pattern with high background intensity. Rh vapor deposition on reconstructed Pt(100) has been shown to lift the reconstruction,200 but in the case of the electrodeposited Rh it is more likely that the electrochemical treatment destroyed the reconstruction; although there has been some controversy on this point,201 later work by Kolb and coworkers demonstrated that anion adsorption on “hex” reconstructed Pt(100) lifts the reconstruction,202 resulting in a high island density. Therefore, the best interpretation of the LEED observations is that lifting of the reconstruction leads to an island-rich (1 × 1) surface, and that Rh deposition occurs on this (1 × 1) surface, most probably in the form of Rh islands.203
Electrodeposition of Rh on Pt(110) has also been performed,195,198,199 and in some cases the resulting surface was subjected to an additional UHV treatment, in which cycles of sputtering and annealing were performed to produce a (1 × 2) surface.
Deposition of Rh multilayers on a Pt(100) surface prepared by flame annealing was achieved via a forced deposition from Rh2(SO4)3 solution using H2 gas as the reducing agent.130 The resulting CV in sulfuric acid media is very similar to that presented earlier for electrodeposited Rh.194 It is also strikingly similar to the CV of a disordered Rh(100) surface, suggesting that the Rh deposits prepared by both methods are poorly-ordered. FTIR spectra of the Pt(100)/Rh electrode with adsorbed CO show two CO bands, attributable to CO adsorbed on Rh in atop and bridged sites, and voltammetric stripping of the adsorbed CO in CO-free electrolyte occurs in a single peak at 0.69 V. Similar voltammetric, FTIR, and CO stripping results were observed on the Pt(110)/Rh surface.129
Reaction fronts involving NO, H2, O2, and CO have been studied in vacuo on Pt(hkl) electrodes modified by photolithographically-patterned thick (7–60 nm) evaporated Rh deposits.204–208 Such a surface geometry offers interesting possibilities for the study of catalytic reactions, but to date only gas-phase catalysis has been examined.
Thinner Rh adlayers on Pt(110)209 and Pt(100)200 produced by evaporation have also been studied in UHV, with the focus on surface alloying via annealing. Vapor deposition of Rh on Pt(110) was found to fill in the “missing row” in the (1 × 2) reconstructed surface, while on Pt(100) deposition of about 0.3 ML lifted the reconstruction, resulting in a (1 × 1) surface. Attard et al. studied the growth of Rh films on Pt(111) by evaporation,193 in a study that paralleled earlier investigations of Pd deposition on Pt(111).102 Unlike Pd, which was shown to grow in a layer-by-layer (Frank–van der Merwe) growth mode, analysis of time-dependent AES data shows that Rh vapor deposition is only quasi layer-by-layer, with second layer growth commencing before completion of the first layer during deposition at temperatures lower than 400 K. At higher temperatures, the second Rh layer is nucleated at even lower first-layer coverage. The higher tendency toward Rh clustering vis-à-vis Pd on Pt(111) has been attributed to the higher surface energy of Rh. Voltammetric features observed on the Pt(111)/Rh electrode after transfer to 0.1 M H2SO4 solution may be assigned to hydrogen desorption, coupled with anion adsorption, on the first Rh layer (peak at 0.18 V) and similar processes occurring on multilayer Rh (peak at 0.13 V), as discussed earlier for Pt(111)/Rh prepared by electrodeposition. Even at a Rh coverage of approximately 1 ML, a small butterfly peak at 0.49 V is visible, indicating that large “pure” Pt(111) domains remain. Therefore, Rh vapor deposition, like electrodeposition,99,190,191 most likely results in large Rh islands or films, rather than individual adatoms or small islands. All these features are similar to those observed on Pt(111)/Pd. Notably, a Pt(111)/Rh electrode prepared with high Rh coverage exhibits preferential dissolution of multilayer Rh during potential cycling between 0.06 and 1.06 V, resulting, after many cycles, in an electrode covered by a Rh monolayer, with the multilayer peaks almost completely eliminated.
 | ||
| Fig. 27 First voltammetric scan for the CO oxidative stripping recorded for Pt(111)/Rh in CO-free 0.1 M H2SO4 solution with different Rh coverages (indicated in the figure). Scan rate = 20 mV s−1. Reprinted from ref. 191, © Elsevier 2004, with permission. | ||
Electrocatalysis of N2O reduction on Pt(111)/Rh in 0.1 M HClO4 has also been examined recently.191 Through a comparison with pure Pt(111) and Rh(111) crystals, it was determined that N2O reduction occurs independently on the Rh and Pt phases of Pt(111) modified by submonolayer amounts of electrodeposited Rh. On the basis of the correlation of peak current densities with Rh coverage, a reduction peak at 0.15 V was ascribed to N2O reduction at Rh island edges, with a peak at 0.25 V corresponding to reduction of N2O on interior Rh atoms. The large decrease in the feature at 0.15 V with increase in Rh coverage from 0.90 to 1.03 ML suggests that relatively few Rh island edges remain at this coverage, i.e. the islands have merged together. The enhanced activity for N2O reduction at Rh island edges may be in part due to a bifunctional mechanism in which N2O is reduced on Rh sites, resulting in desorption of N2 while the remaining adsorbed O atom is reduced by H adsorbed on an adjacent Pt site. A similar mechanism was proposed to explain the high N2O reduction activity of Rh atoms on island edges on the Pt(100)/Rh surface.203
5. Modification of Ru(ijkl) by Pt
Up to this point, we have limited our discussion to the class of bimetallic electrodes produced by deposition or alloying of another noble metal with a Pt single crystal electrode. This section explores the “inverted formula”210 of Pt deposition on single crystal surfaces of other noble metals. This inverted formula, which may be described as Me(hkl)/Pt (Me = FCC) or Me(ijkl)/Pt (Me = HCP), has not been as thoroughly explored from the electrocatalytic standpoint as has been Pt(hkl)/Me, but some important theoretical and experimental observations have resulted from such studies, particularly with the Ru(ijkl)/Pt system. Notably, density functional theory (DFT) calculations211,212 and TPD experiments212,213 have demonstrated that the Pt–CO bond strength is greatly weakened when Pt exists as a monolayer supported on a Ru substrate. In order to understand this weakening, two effects must be considered: (i) the strain resulting from deposition of a pseudomorphic Pt adlayer on the Ru substrate, which has a smaller lattice constant; (ii) the chemical interaction between Pt and Ru. The Me–CO bond strength is weakened on a surface experiencing compressive strain. This can be understood in the context of the effect on the metal d-orbitals: compression leads to a greater overlap between d-orbitals and a broadening of the d-band, and as a result the d-band shifts to lower energy. Within the context of the Blyholder model,214 this lower d-electron energy leads to a decreased availability for back-donation to the CO 2π* orbital, and therefore a weaker Me–CO bond. A second effect is caused by the chemical nature of the substrate and the overlayer. A Pt overlayer on a Ru substrate donates some electron density to Ru, lowering the d-band center of Pt and again weakening the Pt–CO bond. This lowering of the Pt–CO bond strength suggests that the Ru/Pt overlayer system may possess advantages as a CO-tolerant electrocatalyst.5.1 Electrodeposition
Electrodeposition of Pt on Ru(0001) from PtCl6−2 solutions produces 3-dimensional Pt clusters, as demonstrated by RHEED and SEM.215 The average size of these clusters is approximately 3 nm.5.2 Electroless deposition
 | ||
| Fig. 28 (A) STM image (200 × 200 nm) of Pt clusters spontaneously deposited on Ru(0001) in 0.1 mM H2PtCl6 + 0.1 M H2SO4 solution. (B) Cross-section of Pt clusters at the place in image (A) indicated by the dashed line from (a) to (b). Image recorded at open circuit potential in 0.1 M H2SO4, z range 5 nm. Reprinted from ref. 216, © Elsevier 2001, with permission. | ||
5.3 Vacuum deposition
 | ||
| Fig. 29 Array of UHV STM images illustrating the thermal evolution of submonolayer Pt on Ru(0001). The various coverages (deposited at about room temperature) were: (A) 0.15 ML, (B) 0.60 ML, and (C) 0.90 ML. The image size of annealed layers is 200 × 200 nm while it is 100 × 100 nm for the layers as grown. Reprinted from ref. 221, © Elsevier 2003, with permission. | ||
5.4 Electrocatalysis
FTIR spectra of saturated CO adlayers on Ru(0001)/Pt and Ru(10![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0)/Pt surfaces in 0.1 M H2SO4 solution reveal two peaks at 2002 and 2054 cm−1 and a single peak at 2035 cm−1, respectively.218 These surfaces were prepared by spontaneous deposition, resulting in a surface characterized by nanoscale Pt islands, as described above. The presence of only one CO absorption band on the Ru(100)/Pt surface is surprising, but as the band is quite broad it most likely comprises overlapping bands due to CO adsorbed on Ru and Pt sites. On the Ru(0001)/Pt surface the peak at 2002 cm−1 may be attributed to CO adsorbed in the atop configuration on Ru sites, while the peak at 2054 cm−1 is probably due to atop CO on Pt sites. Based on the Blyholder model,214 the red shift of the CO stretching band on Pt sites relative to CO adsorbed on pure Pt(111) at the same potential (∼2070 cm−1124,224) would indicate an increased degree of back-donation to the CO 2π* orbital, and hence a stronger Pt–CO bond. As further described below, this result is clearly contradicted by TPD measurements212,213 and DFT calculations,211,212 demonstrating that the C–O stretching frequency does not correlate with the Pt–CO bond strength in a simple way. Although theoretical analysis suggests that the back-donation is the most important factor in determining the C–O stretching frequency, other effects, including the donation of electrons from the 5σ CO orbital to Pt d-states and the Pauli repulsion resulting from the overlap of CO and Pt orbitals, are also significant.225,226 These effects may explain the apparent discrepancy between Pt–CO bond strength and C–O stretching frequency, but theoretical calculations would be needed to determine the magnitude of each of these factors.
0)/Pt surfaces in 0.1 M H2SO4 solution reveal two peaks at 2002 and 2054 cm−1 and a single peak at 2035 cm−1, respectively.218 These surfaces were prepared by spontaneous deposition, resulting in a surface characterized by nanoscale Pt islands, as described above. The presence of only one CO absorption band on the Ru(100)/Pt surface is surprising, but as the band is quite broad it most likely comprises overlapping bands due to CO adsorbed on Ru and Pt sites. On the Ru(0001)/Pt surface the peak at 2002 cm−1 may be attributed to CO adsorbed in the atop configuration on Ru sites, while the peak at 2054 cm−1 is probably due to atop CO on Pt sites. Based on the Blyholder model,214 the red shift of the CO stretching band on Pt sites relative to CO adsorbed on pure Pt(111) at the same potential (∼2070 cm−1124,224) would indicate an increased degree of back-donation to the CO 2π* orbital, and hence a stronger Pt–CO bond. As further described below, this result is clearly contradicted by TPD measurements212,213 and DFT calculations,211,212 demonstrating that the C–O stretching frequency does not correlate with the Pt–CO bond strength in a simple way. Although theoretical analysis suggests that the back-donation is the most important factor in determining the C–O stretching frequency, other effects, including the donation of electrons from the 5σ CO orbital to Pt d-states and the Pauli repulsion resulting from the overlap of CO and Pt orbitals, are also significant.225,226 These effects may explain the apparent discrepancy between Pt–CO bond strength and C–O stretching frequency, but theoretical calculations would be needed to determine the magnitude of each of these factors.
Adsorption of CO on Ru(0001)/Pt prepared by Pt evaporation has been studied in UHV using TPD as well as FTIR and STM.212,213,221–223,227 Deposition of submonolayers of Pt does not change the CO adlattice on Pt-free Ru sites, which has been shown using STM to be a (√3 × √3)R30° structure at a CO coverage of 0.33 ML.213 TPD of CO also reveals essentially unchanged behavior for CO adsorbed on Ru sites, but on Pt sites the desorption occurs at lower temperature than on Pt(111) (Fig. 30), with a decrease in CO adsorption energy estimated at 0.25 eV as a result of the presence of the Ru substrate. Surface Pt/Ru alloys produced by annealing the surface have similar CO desorption characteristics, although in this case the Pt–CO bond is slightly stronger. These results are in qualitative agreement with those of Hayden et al.,75 who found CO desorption at lower temperature on Pt(110)/Ru surface alloys as compared with clean Pt(110). A somewhat larger decrease in Pt–CO binding energy, derived from TPD data as well as DFT calculations, was reported by Schlapka et al.212 In this study, the Pt coverage was varied from 1 ML to 4 or more ML. Since no dislocation lines were detected at 4 ML coverage, and since three layers of Pt atoms separate the topmost Pt layer from the Ru substrate, one may assume at this coverage that surface strain effects are unchanged from the 1 ML case, while chemical effects from the Ru substrate have completely disappeared. This assumption is confirmed by the TPD data, which show a large difference in CO desorption temperature between the 1 and 4 ML cases, but very little difference between 4 and 10 ML. In this way, the relative contributions of surface strain and Ru electronic modification were compared, with the Ru chemical contribution determined to be more significant. This result is in reasonably good agreement with the DFT calculations of Koper, who, by comparing the Pt–CO bond strength on unstrained Pt(111), strained Pt(111), and a strained Pt monolayer on Ru(0001), showed that the Ru chemical effect is somewhat larger than the strain effect, although for most other Me(111)/Pt and Me(0001)/Pt systems the chemical nature of the substrate is secondary to the strain effect.211 FTIR analysis of CO adsorbed on Ru(0001) modified by submonolayers of Pt revealed further details of the distribution of binding energies on the bimetallic surface.222 On annealed surfaces, in which Pt forms large islands, CO disappears from the Pt islands (except for the steps) at 104 K, and disappears from the steps at 140 K. At higher temperatures, CO is only present on Ru, indicating lower adsorption energy on Pt sites than on Ru sites. However, on surfaces with a low coverage of very small Pt islands, the situation is reversed: below 300 K, CO preferentially occupies the small Pt islands at the expense of Ru sites. In this way a significant dependence of binding energy on Pt island size was identified.
 | ||
| Fig. 30 Set of TPD spectra recorded after increasing CO exposure to a Ru(0001) surface covered by Pt monolayer islands (0.4 ML Pt), after room-temperature Pt deposition and subsequent annealing to 800 K (0.2 L, 0.5 L, 1.0 L, 1.5 L, 2 L, 3 L, 4 L, and 10 L exposure, pCO = 10−8 mbar, adsorption temperature = 100 K, sweep rate = 2.2 K s−1). For comparison, a saturation spectrum (4 L) obtained from the pure Ru(0001) surface is included (broken line). Reprinted from ref. 213, © Elsevier 1998, with permission. | ||
The Pt–CO bond weakening induced by the Ru substrate accelerates CO oxidation kinetics, both in vacuum, where CO oxidation by adsorbed O occurs more readily on the Ru(0001)/Pt surface than on Pt(111),213 and in solution, where electrooxidation of dissolved CO in sulfuric acid solutions commences at a lower potential on the Ru(100)/Pt surface than on polycrystalline Pt.218 On the other hand, Diemant et al. predicted that H2 electrooxidation kinetics may be worse on Ru(ijkl)/Pt surfaces,223 based on the weaker Pt–H bond observed using TPD, which implies a lower H coverage in the electrochemical environment. This lower coverage is also observable from CVs of the Ru(0001)/Pt surface.216 Of course, a higher CO tolerance would more than likely make up for any decrease in neat H2 oxidation kinetics, and Ru nanoparticles modified by adsorbed Pt were shown to compare favorably with Pt/Ru alloy nanoparticles for electrooxidation of H2/CO mixtures.217 These nanoparticle catalysts also perform well for the oxidation of methanol in the DMFC,228 while Ru(0001)/Pt surfaces exhibit faster kinetics than either pure Pt or Ru for methanol and formic acid oxidation.215 Reduction of oxygen on Pt-modified Ru single crystals and nanoparticles shows faster kinetics than on pure Ru, but the activity is lower than that of pure Pt.220
6. Conclusions
The use of well-defined noble metal model electrocatalysts has resulted in significant advancements in understanding of electrocatalytic mechanisms, benefiting both theoretical understanding and practical applications in design of surfaces with enhanced catalytic properties. These benefits are a direct result of the well-defined surface structure that may be achieved on monocrystalline surfaces, eliminating the complications of multiple crystalline phases and high defect density inherent in studies with rough and polycrystalline surfaces. An alternative strategy in the development of highly active electrocatalysts is the high-throughput combinatorial method, in which a wide variety of possible catalyst combinations are investigated in order to determine the most active composition.156 Although this is an important method in the development and screening of potential catalysts, it is limited essentially to investigation of the effects of catalyst composition, yielding no information about the effect of surface structure and the presence of specific atomic ensembles on reactivity. Therefore, further experiments on well-defined single crystal surfaces, which take the optimum compositions found by combinatorial experiments as their starting point, may yield significant additional enhancements in catalytic activity.Since Pt is the most active single-component catalyst for a great variety of surface reactions, most workers have quite reasonably approached electrocatalytic studies by starting with a Pt(hkl) surface and modifying it with other metals. However, the inverted formula, in which other noble metals are decorated by Pt, is likely to be equally fruitful in the study and development of more active electrocatalysts. Deposition of a pseudomorphic Pt monolayer on another metal produces a bimetallic surface that is similar in some respects to pure Pt, but which has slightly different electronic and geometric structure. Both aspects may be manipulated through careful selection of the substrate metal, guided by theoretical calculations. As an example of the insights that may be achieved through investigations of well-defined bimetallic surfaces, we note that modification of such Pt-overlayer systems by deposition of another metal is a promising route toward the development of new electrocatalysts. For instance, it has been demonstrated that a Pt monolayer on a Ru substrate has a much weaker Pt–CO bond strength relative to pure Pt. Furthermore, Ru islands on a Pt surface have been shown to be highly effective for oxidizing CO at the Pt/Ru edge at low potentials. By combining these two effects, i.e. deposition of a Pt monolayer on a Ru(0001) substrate with further modification by deposition of Ru islands on the Pt overlayer, a superior CO-tolerant electrocatalyst may be obtained. Such rational design, guided by fundamental in situ electrochemical and spectroscopic investigations on well-defined surfaces, as well as investigations in UHV, combinatorial experiments, and theoretical calculations, is the best approach to designing the next generation of highly active electrocatalysts.
Acknowledgements
This work is supported by the US Department of Energy, Division of Materials Sciences under Award No. DEFG02-91ER45439, through the Frederick Seitz Materials Research Laboratory at the University of Illinois at Urbana-Champaign, and by the National Science Foundation under grant NSF CHE03-4999. Additional support was provided by the Army Research Office. J. S. S. acknowledges support from a National Science Foundation Graduate Research Fellowship.References
- J. Clavilier, R. Faure, G. Guinet and R. Durand, J. Electroanal. Chem., 1980, 107, 205 CrossRef CAS.
- J. Clavilier, in Interfacial Electrochemistry: Theory, Experiment, and Applications, ed. A. Wieckowski, Marcel Dekker, New York, 1999, p. 231 Search PubMed.
- A. T. Hubbard, Acc. Chem. Res., 1980, 13, 177 CrossRef CAS.
- A. T. Hubbard, J. Vac. Sci. Technol., 1980, 17, 49 CrossRef CAS.
- F. G. Will, J. Electrochem. Soc., 1965, 112, 451 CAS.
- E. Budevski, G. Staikov and W. J. Lorenz, Electrochim. Acta, 2000, 45, 2559 CrossRef CAS.
- W. J. Li, J. A. Virtanen and R. M. Penner, Appl. Phys. Lett., 1992, 60, 1181 CrossRef CAS.
- D. Hofmann, W. Schindler and J. Kirschner, Appl. Phys. Lett., 1998, 73, 3279 CrossRef.
- H. J. Mamin, P. H. Guethner and D. Rugar, Phys. Rev. Lett., 1990, 65, 2418 CrossRef CAS.
- R. Schuster, V. Kirchner, X. H. Xia, A. M. Bittner and G. Ertl, Phys. Rev. Lett., 1998, 80, 5599 CrossRef CAS.
- D. M. Kolb, R. Ullmann and T. Will, Science, 1997, 275, 1097 CrossRef CAS.
- J. Meier, K. A. Friedrich and U. Stimming, Faraday Discuss., 2002, 121, 365 RSC.
- M. M. P. Janssen and J. Moolhuysen, Electrochim. Acta, 1976, 21, 869 CrossRef CAS.
- S. R. Brankovic, J. X. Wang and R. R. Adzic, Surf. Sci., 2001, 474, L173 CrossRef CAS.
- J. Clavilier, M. J. Llorca, J. M. Feliu and A. Aldaz, J. Electroanal. Chem., 1991, 310, 429 CrossRef CAS.
- K. Bromann, C. Felix, H. Brune, W. Harbich, R. Monot, J. Buttet and K. Kern, Science, 1996, 274, 956 CrossRef CAS.
- S. Kramer, R. R. Fuierer and C. B. Gorman, Chem. Rev., 2003, 103, 4367 CrossRef.
- E. Bauer, Z. Kristallogr., 1958, 110, 372.
- F. C. Frank and J. H. van der Merwe, Proc. R. Soc. London, 1949, A198, 216 CAS.
- M. Volmer and A. Weber, Z. Phys. Chem., 1926, 119, 277 Search PubMed.
- J. N. Stranski and L. Krastanov, Sitzber. Akad. Wiss. Wien, 1938, 146, 796 Search PubMed.
- C. T. Campbell, Annu. Rev. Phys. Chem., 1990, 41, 775 CrossRef CAS.
- H. Niehus, W. Heiland and E. Taglauer, Surf. Sci. Rep., 1993, 17, 213 CrossRef CAS.
- H. A. Gasteiger, P. N. Ross and E. J. Cairns, Surf. Sci., 1993, 293, 67 CrossRef CAS.
- J. C. Davies, B. E. Hayden and D. J. Pegg, Electrochim. Acta, 1998, 44, 1181 CrossRef CAS.
- T. J. Schmidt, N. M. Markovic, V. Stamenkovic, P. N. Ross, G. A. Attard and D. J. Watson, Langmuir, 2002, 18, 6969 CrossRef CAS.
- O. J. Adlhart and K. O. Heuer, Fuel Cell Catalysis: Final Report, U.S. Army Electronics Research and Development Laboratory, Contract No. DA36-039-SC-90691, 1963 Search PubMed.
- O. A. Petrii, B. I. Podlovchenko, A. N. Frumkin and H. Lal, J. Electroanal. Chem., 1965, 10, 253 CrossRef CAS.
- H. Binder, A. Kohling and G. Sandstede, in Hydrocarbon Fuel Cell Technology, ed. B. S. Baker, Academic Press, New York, 1965, p. 91 Search PubMed.
- L. W. Niedrach, D. W. McKee, J. Paynter and I. Danzig, Electrochem. Tech., 1967, 5, 318 Search PubMed.
- D. Kardash, C. Korzeniewski and N. Markovic, J. Electroanal. Chem., 2001, 500, 518 CrossRef CAS.
- M. Watanabe and S. Motoo, J. Electroanal. Chem., 1975, 60, 267 CrossRef CAS.
- T. Iwasita, F. C. Nart and W. Vielstich, Ber. Bunsen-Ges. Phys. Chem., 1990, 94, 1030 CAS.
- M. Krausa and W. Vielstich, J. Electroanal. Chem., 1994, 379, 307 CrossRef CAS.
- Y. Y. Tong, H. S. Kim, P. K. Babu, P. Waszczuk, A. Wieckowski and E. Oldfield, J. Am. Chem. Soc., 2002, 124, 468 CrossRef CAS.
- C. Lu and R. I. Masel, J. Phys. Chem. B, 2001, 105, 9793 CrossRef CAS.
- C. Lu, C. Rice, R. I. Masel, P. K. Babu, P. Waszczuk, H. S. Kim, E. Oldfield and A. Wieckowski, J. Phys. Chem. B, 2002, 106, 9581 CrossRef CAS.
- H. S. Wang, C. Wingender, H. Baltruschat, M. Lopez and M. T. Reetz, J. Electroanal. Chem., 2001, 509, 163 CrossRef CAS.
- L. Gao, H. L. Huang and C. Korzeniewski, Electrochim. Acta, 2004, 49, 1281 CrossRef CAS.
- W. Chrzanowski and A. Wieckowski, Langmuir, 1997, 13, 5974 CrossRef CAS.
- W. Chrzanowski and A. Wieckowski, in Interfacial Electrochemistry: Theory, Experiment, and Applications, ed. A. Wieckowski, Marcel Dekker, New York, 1999, p. 937 Search PubMed.
- W. Chrzanowski and A. Wieckowski, Langmuir, 1998, 14, 1967 CrossRef CAS.
- Catalysis and Electrocatalysis at Nanoparticle Surfaces, ed. A. Wieckowski, E. Savinova and C. Vayenas, Marcel Dekker, New York, 2003 Search PubMed.
- K. A. Friedrich, K. P. Geyzers, U. Linke, U. Stimming and J. Stumper, J. Electroanal. Chem., 1996, 402, 123 CrossRef CAS.
- G. Tremiliosi, H. Kim, W. Chrzanowski, A. Wieckowski, B. Grzybowska and P. Kulesza, J. Electroanal. Chem., 1999, 467, 143 CrossRef CAS.
- M. Cappadonia, J. Schmidberger, W. Schwegle and U. Stimming, Proc.-Electrochem. Soc., 1996, 96–8, 269 Search PubMed.
- K. A. Friedrich, K. P. Geyzers, F. Henglein, A. Marmann, U. Stimming, W. Unkauf and R. Vogel, Proc.-Electrochem. Soc., 1996, 96–8, 119 Search PubMed.
- S. Cramm, K. A. Friedrich, K. P. Geyzers, U. Stimming and R. Vogel, Fresenius’ J. Anal. Chem., 1997, 358, 189 CrossRef CAS.
- K. A. Friedrich, K. P. Geyzers, A. Marmann, U. Stimming and R. Vogel, Z. Phys. Chem., 1999, 208, 137 Search PubMed.
- K. A. Friedrich, K. P. Geyzers, A. J. Dickinson and U. Stimming, J. Electroanal. Chem, 2002, 524, 261 CrossRef.
- H. A. Gasteiger, N. Markovic, P. N. Ross and E. J. Cairns, J. Phys. Chem., 1993, 97, 12020 CrossRef CAS.
- W. F. Lin, M. S. Zei, M. Eiswirth, G. Ertl, T. Iwasita and W. Vielstich, J. Phys. Chem. B, 1999, 103, 6968 CrossRef CAS.
- W. Chrzanowski, H. Kim and A. Wieckowski, Catal. Lett., 1998, 50, 69 CrossRef CAS.
- H. Massong, H. S. Wang, G. Samjeske and H. Baltruschat, Electrochim. Acta, 2000, 46, 701 CrossRef CAS.
- G. Samjeske, X. Y. Xiao and H. Baltruschat, Langmuir, 2002, 18, 4659 CrossRef CAS.
- E. Herrero, J. M. Feliu and A. Wieckowski, Langmuir, 1999, 15, 4944 CrossRef.
- A. Crown, I. R. Moraes and A. Wieckowski, J. Electroanal. Chem., 2001, 500, 333 CrossRef CAS.
- A. Crown and A. Wieckowski, Phys. Chem. Chem. Phys., 2001, 3, 3290 RSC.
- A. Crown, H. Kim, G. Q. Lu, I. R. de Moraes, C. Rice and A. Wieckowski, J. New Mater. Electrochem. Syst., 2000, 3, 275 Search PubMed.
- A. Crown, C. Johnston and A. Wieckowski, Surf. Sci., 2002, 506, L268 CrossRef CAS.
- V. D. Colle, M. J. Giz and G. Tremiliosi, J. Braz. Chem. Soc., 2003, 14, 601 Search PubMed.
- S. Strbac, C. Johnston, G. Q. Lu, A. Crown and A. Wieckowski, Surf. Sci. Search PubMed , in press.
- P. Waszczuk, J. Solla-Gullon, H. S. Kim, Y. Y. Tong, V. Montiel, A. Aldaz and A. Wieckowski, J. Catal., 2001, 203, 1 CrossRef CAS.
- H. Kim, I. R. de Moraes, G. Tremiliosi, R. Haasch and A. Wieckowski, Surf. Sci., 2001, 474, L203 CrossRef CAS.
- C. Vericat, M. Wakisaka, R. Haasch, P. S. Bagus and A. Wieckowski, J. Solid State Electrochem., 2004, 8, 794 Search PubMed.
- W. E. O’Grady, P. L. Hagans, K. I. Pandya and D. L. Maricle, Langmuir, 2001, 17, 3047 CrossRef CAS.
- R. Viswanathan, G. Y. Hou, R. X. Liu, S. R. Bare, F. Modica, G. Mickelson, C. U. Segre, N. Leyarovska and E. S. Smotkin, J. Phys. Chem. B, 2002, 106, 3458 CrossRef CAS.
- P. K. Babu, H. S. Kim, E. Oldfield and A. Wieckowski, J. Phys. Chem. B, 2003, 107, 7595 CrossRef CAS.
- J. McBreen and S. Mukerjee, J. Electrochem. Soc., 1995, 142, 3399 CAS.
- T. Iwasita, H. Hoster, A. John-Anacker, W. F. Lin and W. Vielstich, Langmuir, 2000, 16, 522 CrossRef CAS.
- W. F. Lin, J. M. Jin, P. A. Christensen and K. Scott, Electrochim. Acta, 2003, 48, 3815 CrossRef CAS.
- J. C. Davies, B. E. Hayden, D. J. Pegg and M. E. Rendall, Surf. Sci., 2002, 496, 110 CrossRef CAS.
- T. D. Jarvi, T. H. Madden and E. M. Stuve, Electrochem. Solid State Lett., 1999, 2, 224 CrossRef CAS.
- H. Hoster, T. Iwasita, H. Baumgartner and W. Vielstich, Phys. Chem. Chem. Phys., 2001, 3, 337 RSC.
- J. C. Davies, B. E. Hayden and D. J. Pegg, Surf. Sci., 2000, 467, 118 CrossRef CAS.
- A. Lamouri, Y. Gofer, Y. Luo, G. S. Chottiner and D. A. Scherson, J. Phys. Chem. B, 2001, 105, 6172 CrossRef CAS.
- N. Yee, G. S. Chottiner and D. A. Scherson, J. Phys. Chem. B, 2004, 108, 5847 CrossRef CAS.
- E. Herrero, K. Franaszczuk and A. Wieckowski, J. Electroanal. Chem., 1993, 361, 269 CrossRef CAS.
- W. Chrzanowski, H. Kim, G. Tremiliosi-Filho, A. Wieckowski, B. Grzybowska and P. Kulesza, J. New Mater. Electrochem. Syst., 1998, 1, 31 Search PubMed.
- G. Q. Lu, P. Waszczuk and A. Wieckowski, J. Electroanal. Chem., 2002, 532, 49 CrossRef CAS.
- G.-Q. Lu, J. O. White and A. Wieckowski, Surf. Sci., 2004, 564, 131 CrossRef CAS.
- M. T. M. Koper, J. J. Lukkien, A. P. J. Jansen and R. A. van Santen, J. Phys. Chem. B, 1999, 103, 5522 CrossRef CAS.
- M. T. M. Koper, N. P. Lebedeva and C. G. M. Hermse, Faraday Discuss., 2002, 121, 301 RSC.
- J. S. Spendelow, G. Q. Lu, P. J. A. Kenis and A. Wieckowski, J. Electroanal. Chem., 2004, 568, 215 CrossRef CAS.
- N. P. Lebedeva, M. T. M. Koper, J. M. Feliu and R. A. van Santen, J. Electroanal. Chem., 2002, 524, 242 CrossRef.
- J. F. Rodriguez, M. E. Bothwell, G. J. Cali and M. P. Soriaga, J. Am. Chem. Soc., 1990, 112, 7392 CrossRef CAS.
- M. P. Soriaga, Y.-G. Kim and J. E. Soto, in Interfacial Electrochemistry: Theory, Experiment, and Applications, ed. A. Wieckowski, Marcel Dekker, New York, 1999, p. 249 Search PubMed.
- T. Solomun, J. Electroanal. Chem., 1988, 255, 163 CrossRef CAS.
- A. Cuesta, L. A. Kibler and D. M. Kolb, J. Electroanal. Chem., 1999, 466, 165 CrossRef CAS.
- H. Naohara, S. Ye and K. Uosaki, J. Phys. Chem. B, 1998, 102, 4366 CrossRef CAS.
- H. Naohara, S. Ye and K. Uosaki, J. Electroanal. Chem., 1999, 473, 2 CrossRef CAS.
- M. Baldauf and D. M. Kolb, Electrochim. Acta, 1993, 38, 2145 CrossRef CAS.
- L. A. Kibler, M. Kleinert, R. Randler and D. M. Kolb, Surf. Sci., 1999, 443, 19 CrossRef CAS.
- A. Capon and R. Parsons, J. Electroanal. Chem., 1975, 65, 285 CrossRef CAS.
- A. H. Taylor, R. D. Pearce and S. B. Brummer, J. Chem. Soc., Faraday Trans., 1971, 67, 801 Search PubMed.
- A. Capon and R. Parsons, J. Electroanal. Chem., 1973, 44, 1 CrossRef CAS.
- B. Beden, A. Bewick and C. Lamy, J. Electroanal. Chem., 1983, 148, 147 CrossRef CAS.
- G. A. Attard and A. Bannister, J. Electroanal. Chem., 1991, 300, 467 CrossRef CAS.
- J. Inukai and M. Ito, J. Electroanal. Chem., 1993, 358, 307 CrossRef CAS.
- B. Alvarez, V. Climent, A. Rodes and J. M. Feliu, Phys. Chem. Chem. Phys., 2001, 3, 3269 RSC.
- B. Alvarez, V. Climent, A. Rodes and J. M. Feliu, J. Electroanal. Chem., 2001, 497, 125 CrossRef CAS.
- G. A. Attard, R. Price and A. Alakl, Electrochim. Acta, 1994, 39, 1525 CrossRef CAS.
- B. Alvarez, A. Rodes, J. M. Perez and J. M. Feliu, J. Phys. Chem. B, 2003, 107, 2018 CrossRef CAS.
- R. Hoyer, L. A. Kibler and D. M. Kolb, Electrochim. Acta, 2003, 49, 63 CrossRef CAS.
- M. J. Ball, C. A. Lucas, N. M. Markovic, V. Stamenkovic and P. N. Ross, Surf. Sci., 2002, 518, 201 CrossRef CAS.
- A. Gil, A. Clotet, J. M. Ricart, F. Illas, B. Alvarez, A. Rodes and J. M. Feliu, J. Phys. Chem. B, 2001, 105, 7263 CrossRef CAS.
- M. Arenz, V. Stamenkovic, T. J. Schmidt, K. Wandelt, P. N. Ross and N. M. Markovic, Surf. Sci., 2002, 506, 287 CrossRef CAS.
- M. Arenz, V. Stamenkovic, T. J. Schmidt, K. Wandelt, P. N. Ross and N. M. Markovic, Surf. Sci., 2003, 523, 199 CrossRef CAS.
- M. Arenz, V. Stamenkovic, T. J. Schmidt, K. Wandelt, P. N. Ross and N. M. Markovic, Phys. Chem. Chem. Phys., 2003, 5, 4242 RSC.
- N. M. Markovic, C. A. Lucas, V. Climent, V. Stamenkovic and P. N. Ross, Surf. Sci., 2000, 465, 103 CrossRef CAS.
- C. A. Lucas, N. M. Markovic, M. Ball, V. Stamenkovic, V. Climent and P. N. Ross, Surf. Sci., 2001, 479, 241 CrossRef CAS.
- M. J. Ball, C. A. Lucas, N. M. Markovic, V. Stamenkovic and P. N. Ross, Surf. Sci., 2003, 540, 295 CrossRef CAS.
- T. M. Chang and E. A. Carter, Surf. Sci., 1994, 318, 187 CrossRef CAS.
- T. M. Chang and E. A. Carter, J. Phys. Chem., 1995, 99, 7637 CrossRef CAS.
- M. Han, P. Mrozek and A. Wieckowski, Phys. Rev. B: Condens. Matter, 1993, 48, 8329 CrossRef CAS.
- M. I. Rojas, M. G. Del Popolo and E. P. M. Leiva, Langmuir, 2000, 16, 9539 CrossRef CAS.
- L. A. Kibler, M. Kleinert and D. M. Kolb, Surf. Sci., 2000, 461, 155 CrossRef CAS.
- L. A. Kibler, M. Kleinert, V. Lazarescu and D. M. Kolb, Surf. Sci., 2002, 498, 175 CrossRef CAS.
- B. Alvarez, J. M. Feliu and J. Clavilier, Electrochem. Commun., 2002, 4, 379 CrossRef CAS.
- V. Climent, R. Gomez and J. M. Feliu, Electrochim. Acta, 1999, 45, 629 CrossRef CAS.
- N. M. Markovic, B. N. Grgur and P. N. Ross, J. Phys. Chem. B, 1997, 101, 5405 CrossRef CAS.
- C. A. Lucas, N. M. Markovic and P. N. Ross, Surf. Sci., 1999, 425, L381 CrossRef CAS.
- Y. V. Tolmachev, A. Menzel, A. V. Tkachuk, Y. S. Chu and H. D. You, Electrochem. Solid State Lett., 2004, 7, E23 CrossRef CAS.
- I. Villegas and M. J. Weaver, J. Chem. Phys., 1994, 101, 1648 CrossRef CAS.
- I. Villegas, X. P. Gao and M. J. Weaver, Electrochim. Acta, 1995, 40, 1267 CrossRef CAS.
- D. Zurawski, M. Wasberg and A. Wieckowski, J. Phys. Chem., 1990, 94, 2076 CrossRef CAS.
- M. Arenz, V. Stamenkovic, P. N. Ross and N. M. Markovic, Electrochem. Commun., 2003, 5, 809 CrossRef CAS.
- M. J. Llorca, J. M. Feliu, A. Aldaz and J. Clavilier, J. Electroanal. Chem., 1993, 351, 299 CrossRef CAS.
- R. Gomez, A. Rodes, J. M. Perez, J. M. Feliu and A. Aldaz, Surf. Sci., 1995, 327, 202 CrossRef CAS.
- R. Gomez, A. Rodes, J. M. Perez, J. M. Feliu and A. Aldaz, Surf. Sci., 1995, 344, 85 CrossRef CAS.
- A. AlAkl and G. A. Attard, J. Phys. Chem. B, 1997, 101, 4597 CrossRef CAS.
- G. A. Attard and R. Price, Surf. Sci., 1995, 335, 63 CrossRef CAS.
- G. A. Attard and R. Price, Surf. Sci., 1996, 345, 236 CrossRef CAS.
- D. J. Watson and G. A. Attard, Electrochim. Acta, 2001, 46, 3157 CrossRef CAS.
- D. J. Watson and G. A. Attard, Surf. Sci., 2002, 515, 87 CrossRef CAS.
- M. A. Van Hove, R. J. Koestner, P. C. Stair, J. P. Biberian, L. L. Kesmodel, I. Bartos and G. A. Somorjai, Surf. Sci., 1981, 103, 218 CrossRef CAS.
- G. W. Simmons, Y.-N. Wang, J. Marcos and K. Klier, J. Phys. Chem., 1991, 95, 4522 CrossRef CAS.
- D. L. Adams, H. B. Nielsen, M. A. Van Hove and A. Ignatiev, Surf. Sci., 1981, 104, 47 CrossRef CAS.
- M. Wolf, A. Goschnick, J. Lobodacackovic, M. Grunze, W. N. Unertl and J. H. Block, Surf. Sci., 1987, 182, 489 CrossRef CAS.
- D. Radosavkic, N. Barrett, R. Belkhou, N. Marsot and C. Guillot, Surf. Sci., 2002, 516, 56 CrossRef CAS.
- M. Hansen and K. Anderko, Constitution of Binary Alloys., 2nd edn. McGraw-Hill, New York, 1986 Search PubMed.
- M. J. Llorca, J. M. Feliu, A. Aldaz and J. Clavilier, J. Electroanal. Chem., 1994, 376, 151 CrossRef CAS.
- M. Baldauf and D. M. Kolb, J. Phys. Chem., 1996, 100, 11375 CrossRef CAS.
- M. S. McGovern, PhD Thesis, University of Illinois at Urbana-Champaign, 2004.
- A. Hamnett and B. J. Kennedy, Electrochim. Acta, 1988, 33, 1613 CrossRef CAS.
- F. Kadirgan, B. Beden, J. M. Leger and C. Lamy, J. Electroanal. Chem., 1981, 125, 89 CrossRef CAS.
- V. Climent, N. M. Markovic and P. N. Ross, J. Phys. Chem. B, 2000, 104, 3116 CrossRef CAS.
- T. J. Schmidt, V. Stamenkovic, M. Arenz, N. M. Markovic and P. N. Ross, Electrochim. Acta, 2002, 47, 3765 CrossRef CAS.
- M. Arenz, T. J. Schmidt, K. Wandelt, P. N. Ross and N. M. Markovic, J. Phys. Chem. B, 2003, 107, 9813 CrossRef CAS.
- B. Alvarez, A. Rodes, J. M. Perez, J. M. Feliu, J. L. Rodriguez and E. Pastor, Langmuir, 2000, 16, 4695 CrossRef CAS.
- P. Zelenay, G. Horanyi, C. K. Rhee and A. Wieckowski, J. Electroanal. Chem., 1991, 300, 499 CrossRef CAS.
- C. K. Rhee, M. Wasberg, P. Zelenay and A. Wieckowski, Catal. Lett., 1991, 10, 149 CAS.
- S. L. Yau, Y. G. Kim and K. Itaya, J. Am. Chem. Soc., 1996, 118, 7795 CrossRef CAS.
- D. F. A. Koch, D. A. J. Rand and R. Woods, J. Electroanal. Chem., 1976, 70, 73 CrossRef CAS.
- N. R. de Tacconi, J. M. Leger, B. Beden and C. Lamy, J. Electroanal. Chem., 1982, 134, 117 CrossRef CAS.
- E. Reddington, A. Sapienza, B. Gurau, R. Viswanathan, S. Sarangapani, E. S. Smotkin and T. E. Mallouk, Science, 1998, 280, 1735 CrossRef CAS.
- F. El Omar, R. Durand and R. Faure, J. Electroanal. Chem., 1984, 160, 385 CrossRef.
- V. P. Santos and G. Tremiliosi-Filho, J. Electroanal. Chem., 2003, 554, 395 CrossRef.
- V. P. Santos, V. Del Colle, R. M. Bezerra and G. Tremiliosi, Electrochim. Acta, 2004, 49, 1221 CrossRef.
- C. Johnston, S. Strbac and A. Wieckowski, in preparation.
- C. K. Rhee, M. Wakisaka, Y. V. Tolmachev, C. M. Johnston, R. Haasch, K. Attenkofer, G. Q. Lu, H. You and A. Wieckowski, J. Electroanal. Chem., 2003, 554, 367 CrossRef.
- A. T. Hubbard, J. L. Stickney, S. D. Rosasco, M. P. Soriaga and D. Song, J. Electroanal. Chem., 1983, 150, 165 CrossRef CAS.
- J. L. Stickney, S. D. Rosasco, D. Song, M. P. Soriaga and A. T. Hubbard, Surf. Sci., 1983, 130, 326 CrossRef CAS.
- A. Wieckowski, B. C. Schardt, S. D. Rosasco, J. L. Stickney and A. T. Hubbard, Surf. Sci., 1984, 146, 115 CrossRef CAS.
- A. Wieckowski, S. D. Rosasco, B. C. Schardt, J. L. Stickney and A. T. Hubbard, Inorg. Chem., 1984, 23, 565 CrossRef CAS.
- T. Solomun, B. C. Schardt, S. D. Rosasco, A. Wieckowski, J. L. Stickney and A. T. Hubbard, J. Electroanal. Chem., 1984, 176, 309 CrossRef CAS.
- J. L. Stickney, S. D. Rosasco, B. C. Schardt and A. T. Hubbard, J. Phys. Chem., 1984, 88, 251 CrossRef CAS.
- D. G. Frank, T. Golden, O. M. R. Chyan and A. T. Hubbard, Appl. Surf. Sci., 1991, 48–9, 166 CrossRef.
- D. G. Frank, O. M. R. Chyan, T. Golden and A. T. Hubbard, J. Phys. Chem., 1994, 98, 1895 CrossRef CAS.
- P. Zelenay, M. Gamboaaldeco, G. Horanyi and A. Wieckowski, J. Electroanal. Chem., 1993, 357, 307 CrossRef CAS.
- B. P. Costa, J. Canullo, D. V. Moll, R. C. Salvarezza, M. C. Giordano and A. J. Arvia, J. Electroanal. Chem., 1988, 244, 261 CrossRef CAS.
- R. Durand, R. Faure, D. Aberdam and S. Traore, Electrochim. Acta, 1989, 34, 1653 CrossRef CAS.
- D. Aberdam, C. Salem, R. Durand and R. Faure, Surf. Sci., 1990, 239, 71 CrossRef CAS.
- C. M. Vitus and B. C. Schardt, Abstr. Pap. Am. Chem. Soc., 1990, 200, 173.
- N. C. Gibson, P. M. Saville and D. A. Harrington, J. Electroanal. Chem., 1991, 318, 271 CrossRef CAS.
- M. Labayen, E. Herrero, J. M. Feliu and D. A. Harrington, J. Electroanal. Chem., 2000, 488, 32 CrossRef CAS.
- M. Labayen, D. A. Harrington, M. Saidy and K. A. R. Mitchell, Surf. Sci., 2001, 490, 256 CrossRef CAS.
- N. Kimizuka and K. Itaya, Faraday Discuss., 1992, 117 Search PubMed.
- N. Shinotsuka, K. Sashikata and K. Itaya, Surf. Sci., 1995, 335, 75 CrossRef CAS.
- J. F. Rodriguez, D. L. Taylor and H. D. Abruna, Electrochim. Acta, 1993, 38, 235 CrossRef CAS.
- D. L. Taylor and H. D. Abruna, J. Electrochem. Soc., 1993, 140, 3402 CAS.
- I. Oda, H. Ogasawara and M. Ito, Langmuir, 1996, 12, 1094 CrossRef CAS.
- A. M. Bittner, J. Electroanal. Chem., 1997, 431, 51 CrossRef CAS.
- J. X. Wang, N. S. Marinkovic, R. R. Adzic and B. M. Ocko, Surf. Sci., 1998, 398, L291 CrossRef CAS.
- N. S. Marinkovic, J. X. Wang, J. S. Marinkovic and R. R. Adzic, J. Phys. Chem. B, 1999, 103, 139 CrossRef CAS.
- T. Langkau and H. Baltruschat, Electrochim. Acta, 2002, 47, 1595 CrossRef CAS.
- G. A. Attard and A. Al-Akl, J. Electroanal. Chem., 2003, 554, 439 CrossRef.
- E. Herrero, L. J. Buller and H. D. Abruna, Chem. Rev., 2001, 101, 1897 CrossRef CAS.
- J. Clavilier, L. H. Klein, A. Vaskevich and A. A. ElShafei, J. Chem. Soc., Faraday Trans., 1996, 92, 3777 RSC.
- R. Gomez and J. M. Feliu, Electrochim. Acta, 1998, 44, 1191 CrossRef CAS.
- R. Gomez, F. J. G. de Dios and J. M. Feliu, Electrochim. Acta, 2004, 49, 1195 CrossRef CAS.
- G. Kokkinidis, J. Electroanal. Chem., 1986, 201, 217 CrossRef CAS.
- G. A. Attard, R. Price and A. Alakl, Surf. Sci., 1995, 335, 52 CrossRef CAS.
- M. Taniguchi, E. K. Kuzembaev and K. Tanaka, Surf. Sci., 1993, 290, L711 CrossRef CAS.
- A. Sasahara, H. Tamura and K. Tanaka, Catal. Lett., 1994, 28, 161 CAS.
- H. Tamura and K. Tanaka, Langmuir, 1994, 10, 4530 CrossRef CAS.
- K. Tanaka, B. E. Nieuwenhuys and H. Tamura, J. Chin. Chem. Soc., 1995, 42, 303 CAS.
- A. Sasahara, H. Tamura and K. Tanaka, J. Phys. Chem., 1996, 100, 15229 CrossRef CAS.
- K. Tanaka and A. Sasahara, J. Mol. Catal. A, 2000, 155, 13 CrossRef CAS.
- H. T. Wu and T. T. Tsong, Surf. Sci., 1994, 318, 358 CrossRef CAS.
- D. M. Kolb, Prog. Surf. Sci., 1996, 51, 109 CrossRef CAS.
- M. S. Zei, N. Batina and D. M. Kolb, Surf. Sci., 1994, 306, L519 CrossRef CAS.
- F. J. G. de Dios, R. Gomez and J. M. Feliu, Electrochem. Comm., 2001, 3, 659 CrossRef.
- E. Schutz, N. Hartmann, Y. Kevrekidis and R. Imbihl, Faraday Discuss., 1996, 47 Search PubMed.
- F. Esch, S. Gunther, E. Schutz, A. Schaak, I. G. Kevrekidis, M. Marsi, M. Kiskinova and R. Imbihl, Catal. Lett., 1998, 52, 85 CrossRef CAS.
- E. Schutz, N. Hartmann, Y. Kevrekidis and R. Imbihl, Catal. Lett., 1998, 54, 181 CrossRef CAS.
- F. Esch, S. Gunther, E. Schutz, A. Schaak, I. G. Kevrekidis, M. Marsi, M. Kiskinova and R. Imbihl, Surf. Sci., 1999, 443, 245 CrossRef CAS.
- S. Y. Shvartsman, E. Schutz, R. Imbihl and I. G. Kevrekidis, Catal. Today, 2001, 70, 301 CrossRef CAS.
- Y. L. He, J. K. Zuo, G. C. Wang and J. J. Low, Surf. Sci., 1991, 255, 269 CrossRef CAS.
- S. T. Kuk and A. Wieckowski, J. Power Sources Search PubMed , in press.
- T. E. Shubina and M. T. M. Koper, Electrochim. Acta, 2002, 47, 3621 CrossRef CAS.
- A. Schlapka, M. Lischka, A. Gross, U. Kasberger and P. Jakob, Phys. Rev. Lett., 2003, 91 Search PubMed.
- F. B. de Mongeot, M. Scherer, B. Gleich, E. Kopatzki and R. J. Behm, Surf. Sci., 1998, 411, 249 CrossRef CAS.
- G. Blyholder, J. Phys. Chem., 1964, 68, 2772 CrossRef CAS.
- M. S. Zei, T. Lei and G. Ertl, Z. Phys. Chem. (Munich), 2003, 217, 447 CrossRef CAS.
- S. R. Brankovic, J. McBreen and R. R. Adzic, J. Electroanal. Chem., 2001, 503, 99 CrossRef CAS.
- S. R. Brankovic, J. X. Wang and R. R. Adzic, J. Serb. Chem. Soc., 2001, 66, 887 CAS.
- S. R. Brankovic, N. S. Marinkovic, J. X. Wang and R. R. Adzic, J. Electroanal. Chem., 2002, 532, 57 CrossRef CAS.
- S. R. Brankovic, J. X. Wang, Y. Zhu, R. Sabatini, J. McBreen and R. R. Adzic, J. Electroanal. Chem., 2002, 524, 231 CrossRef.
- H. Inoue, S. R. Brankovic, J. X. Wang and R. R. Adzic, Electrochim. Acta, 2002, 47, 3777 CrossRef CAS.
- U. Kasberger and P. Jakob, Surf. Sci., 2003, 540, 76 CrossRef CAS.
- A. Schlapka, U. Kasberger, D. Menzel and P. Jakob, Surf. Sci., 2002, 502, 129 CrossRef.
- T. Diemant, T. Hager, H. E. Hoster, H. Rauscher and R. J. Behm, Surf. Sci., 2003, 541, 137 CrossRef CAS.
- K. Yoshimi, M.-B. Song and M. Ito, Surf. Sci., 1996, 368, 389 CrossRef CAS.
- P. S. Bagus and W. Muller, Chem. Phys. Lett., 1985, 115, 540 CrossRef CAS.
- F. Illas, S. Zurita, J. Rubio and A. M. Marquez, Phys. Rev. B: Condens. Matter, 1995, 52, 12372 CrossRef CAS.
- R. J. Behm, Acta Phys. Pol., A, 1998, 93, 259 Search PubMed.
- A. S. Arico, V. Baglio, E. Modica, A. Di Blasi and V. Antonucci, Electrochem. Commun., 2004, 6, 164 CrossRef CAS.
| This journal is © the Owner Societies 2004 |