Principles of sequence-recognition in aromatic polyimides†
Howard M.
Colquhoun
*,
Zhixue
Zhu
*,
Christine J.
Cardin
and
Yu
Gan
School of Chemistry, University of Reading, Whiteknights, Reading, UK RG6 6AD. E-mail: h.m.colquhoun@rdg.ac.uk; z.x.zhu@rdg.ac.uk
First published on 8th November 2004
Abstract
Pyrene-based molecular tweezers show sequence-specific binding to aromatic polyimides through sterically-controlled donor–acceptor π-stacking and hydrogen bonding; 1H NMR spectra of tweezer-complexes with polyimides having different sequence-restrictions show conclusively that the detection of long range sequence-information results from multiple tweezer-binding at adjacent imide residues.
The most fundamental mechanisms of biology depend on supramolecular recognition of monomer sequence-information in linear copolymers. This is most evident in the expression of nucleic acid sequences during protein synthesis,1 but is also crucially important in gene-regulation by proteins,2 and in the operation of restriction enzymes which cleave DNA at sequence-specified linkages.3 Recognition of monomer sequences in synthetic polymer systems is by contrast relatively unknown,4 though NMR spectroscopic evidence was very recently obtained for sequence-selectivity in the binding of a molecular tweezer (1a) to aromatic co-polyimides.5 We have now identified the specific interactions associated with this type of binding by crystallographic analysis of a tweezer-complex with a model di-imide. In addition, an analysis of tweezer-complexation with sequence-restricted polyimides shows conclusively that long-range sequence effects apparent in the 1H NMR spectra arise from multiple tweezer-binding to adjacent imide residues.
The X-ray structure‡ (Fig. 1) of a 1 ∶ 1 complex between tweezer 1b and the model di-imide 2 (Ka = 6 × 103 M−1 in 6 ∶ 1 v/v CHCl3/hexafluoropropan-2-ol) shows that tweezer-binding occurs via (i) a double π-stacking arrangement of its electron-rich pyrenyl arms with the electron-poor pyromellitimide unit (mean atom to pyrene-plane distances 3.38, 3.39 Å), (ii) a pair of hydrogen bonds (yellow) between the tweezer amide-NH protons and a pyromellitimide carbonyl oxygen (N⋯O = 3.13, 3.22 Å), and (iii) di-imide chain folding through ca. 180° (as predicted by computational modelling),5 bringing one of its two 4-chlorophenylenesulfone residues into π-stacking contact with a pyrenyl tweezer-arm (mean C to pyrene-plane distance = 3.66 Å; Cl⋯pyrene = 3.59 Å). The second ether-sulfone unit does not fold around the tweezer in the same way but adopts a more extended, non-interacting conformation.
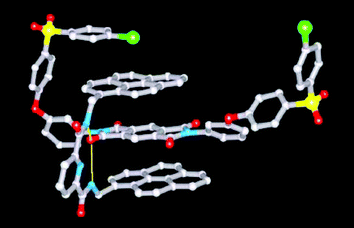 | ||
| Fig. 1 X-Ray structure of the 1 ∶ 1 complex between tweezer 1b and the model di-imide 2. Hydrogen atoms are omitted for clarity. | ||
Development of fine structure in the 1H NMR resonances of tweezer-bound imide residues in high molar mass polyimides has tentatively been ascribed to the amplification of long-range polymer sequence information by the tweezer.5 Given fast-exchange conditions, a possible mechanism for this involves tweezer-binding at one or both neighbouring imide residues, thereby enhancing the aromatic ring-current shielding effects produced by a tweezer bound at the “observed” site. This would enable differentiation between sequences where the tweezer can bind (i) at neither of the two sites adjacent to the observed binding site, (ii) at one adjacent site or (iii) at both adjacent sites.
We have now subjected this adjacent-binding model to a rigorous experimental test, making use of two novel polyimides with analogous structures but different sequence-restrictions. These polymers (6 and 7) were synthesised at high molar mass (Mn = 143,000 and 112,000 daltons respectively) by polycondensation of pyromellitic dianhydride with (i) a 1 ∶ 1 mixture of the symmetrical diamines 3 and 4, and (ii) the unsymmetrical diamine 5 alone. Designating a methylated ring in 6 or 7 as “H” (hindered), a non-methylated ring as “U” (unhindered), and a di-imide residue as “I”, then both polymers will contain the three different imide-centred sequences UIU, UIH (or HIU) and HIH, one of which (HIH) also exists as a pair of non-interconverting conformational isomers (Fig. 2).6 For both polymers, the sequences UIU, UIH/HIU and HIH should appear in relative abundances 1 ∶ 2 ∶ 1 (on the basis of a random distribution of U and H residues). As shown in Fig. 3 [spectra (a) and (d)], these abundances are clearly reflected in the imide-resonances (δ 8.4–8.6) observed by 1H NMR spectroscopy.
Spectra (b) and (e) show the effects of adding just 1.25 mol% (relative to imide) of tweezer 1a. Crucially only one of the three imide resonances shows a complexation-shift and is thus assigned to the unhindered sequence UIU. This complexation-shift correlates well with that observed for the homopolyimide derived from the unhindered diamine 3 (see ESI). The imide resonance at lowest field is unshifted and can be confidently assigned to the doubly hindered sequence HIH, leaving the central resonance assignable as HIU/UIH – its twofold degeneracy agreeing with the relative intensity of this resonance. Increasing the proportion of tweezer to 5 mol% leads to a further upfield shift of the UIU resonance, but although this remains a sharp singlet for polymer 7, it broadens and separates into an apparent 1 ∶ 2 ∶ 1 triplet for copolymer 6 (see ESI for more detailed spectroscopic data). As outlined below, these observations clearly demonstrate that higher-order sequence information relating to the environment of UIU is being expressed in the 1 ∶ 1 copolymer 6, but, as a result of inbuilt sequence-restrictions, not in polymer 7.

 | ||
| Fig. 3 1H NMR spectra of polyimides 6 and 7; (a)/(d) pure polymer, (b)/(e) polymer plus tweezer 1a (1.25 mol% relative to imide residues), and (c)/(f) polymer plus tweezer 1a (5 mol% relative to imide). | ||
Designating the residues from monomers 3, 4, and 5 as U–U, H–H and H–U respectively, the symmetrical nature of 3 and 4 means that in copolymer 6 the strongly-binding triplet UIU can occur only within the following restricted range of sequences:
HIU–UIU–UIH (provides no adjacent UIU binding site)
HIU–UIU–UIU (provides one adjacent UIU binding site)
UIU–UIU–UIH (provides one adjacent UIU binding site)
UIU–UIU–UIU (provides two adjacent UIU binding sites)
The emergence of a 1 ∶ 2 ∶ 1 imide-resonance-pattern for UIU in copolymer 6 is thus entirely consistent with the idea that supramolecular tweezer-binding induces resolution of long-range sequence-information in the 1H NMR spectra of polyimides. In polymer 7 however, the use of an unsymmetrical diamine places different restrictions on sequences higher than triplets so that the only allowed UIU-centred three-triplet sequences are now:
HIH–UIU–HIH (provides no adjacent UIU binding site)
HIH–UIU–HIU (provides no adjacent UIU binding site)
UIH–UIU–HIH (provides no adjacent UIU binding site)
UIH–UIU–HIU (provides no adjacent UIU binding site)
There are thus again three different sequence-environments for UIU in polymer 7, but the adjacent-binding theory now predicts that the imide resonance arising from this sequence will show no higher-order splitting in the presence of the tweezer, since adjacent tweezer-binding is sequence-forbidden. This prediction is clearly borne out by the spectra shown in Fig. 3 (d–f). Even more remarkably, for polymer 7 at higher tweezer ∶ imide ratios (> 1 ∶ 1), additional development of fine structure is observed in the imide resonances of the non-binding (HIH) and weakly-binding (HIU) sequences, as shown in Fig. 4. Enumeration of the possible HIU-centred sequences in polymer 7 (below) shows equal populations of sequences in which strong tweezer-binding can and cannot occur at an adjacent imide residue, so clearly accounting for the splitting of the HIH resonance into an apparent 1 ∶ 1 doublet.
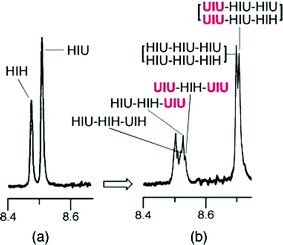 | ||
| Fig. 4 Partial 1H NMR spectra of polyimide 7 in the presence of the tweezer 1a; (a) 5 mol% tweezer relative to imide (cf.Fig. 3f), and (b) 100 mol% tweezer relative to imide. Assignments refer to the protons of the central di-imide residue (I) in each sequence shown. The HIH-centred sequence-assignments shown in spectrum (b) refer to the syn-isomer of this sequence and are repeated for the corresponding resonances at lower field assigned to the anti-isomer. | ||
UIU–HIU–HIU (provides one adjacent UIU binding site)
UIU–HIU–HIH (provides one adjacent UIU binding site)
HIU–HIU–HIU (provides no adjacent UIU binding site)
HIU–HIU–HIH (provides no adjacent UIU binding site)
A similar analysis (below) of the possible HIH-centred sequences in polymer 7 shows that HIH can occur in three distinct environments, with relative populations 1 ∶ 2 ∶ 1:
UIU–HIH–UIU (provides two adjacent UIU binding sites)
UIU–HIH–UIH (provides one adjacent UIU binding site)
HIU–HIH–UIU (provides one adjacent UIU binding site)
HIU–HIH–UIH (provides no adjacent UIU binding site)
Although an apparent 1 ∶ 2 ∶ 1 triplet would therefore be predicted, a pair of such “triplets” is in fact observed (Fig. 4). This is however readily accounted for on the basis that, as noted above and shown in Fig. 2, the sequence HIH exists as a pair of non-interconverting conformational isomers. The syn isomer, with both methyl groups on the same side of the imide unit, can in principle interact weakly, via its unhindered face, with a single pyrene unit of the tweezer. In contrast, each face of the anti isomer is blocked by a methyl group and so an even weaker interaction with the tweezer would be expected. A small upfield shift for the syn- and an even smaller shift for the anti-HIH resonance would thus be predicted, accounting for the splitting of this resonance at high tweezer concentrations (Fig. 4).
A control experiment (ESI, Fig. S5) using the homopolyimide from diamine 4 and pyromellitic anhydride, in which every imide unit is of the type “HIH”, shows exactly the same 1 ∶ 1 splitting of the imide resonance, the same very small interaction-shifts but no fine-structure development.
Finally, a corresponding analysis of the environments of HIU and HIU in copolymer 6 predicts that at high tweezer ∶ imide ratios the corresponding resonances should both split to give simple doublets. As shown in the ESI (Fig. S2) this is indeed the pattern of resonances observed.
It is clear that: (i) the aromatic ring-current shift induced by a tweezer-type molecule such as 1a represents a powerful tool for identifying polyimide chain-sequences by 1H NMR, (ii) in a polymer “triplet” sequence, only small variations in the steric requirements of the flanking monomer residues are needed to achieve high selectivity in supramolecular recognition of the central residue, (iii) more extended polyimide sequences (triplets of triplets) can be assigned in detail by 1H NMR, through the effects of singly- or doubly-adjacent tweezer-binding, and (iv) these effects result from strong tweezer-binding to unhindered di-imide residues, through a combination of electronically-complementary, π–π stacking (involving polymer chain-folding) and N–H⋯O hydrogen bonding between the amide groups of the tweezer and a carbonyl oxygen of the imide residue.
We thank the University of Reading Research Endowment Trust for funding, and the EPSRC X-ray Crystallography Service for data collection facilities.
Notes and references
- M. Nirenberg, Trends Biochem. Sci., 2004, 29, 46 CrossRef CAS.
- C. M. Falcon and K. S. Matthews, Biochemistry, 2000, 39, 11074 CrossRef CAS.
- J. M. Bujnicki, Curr. Protein Pept. Sci., 2003, 4, 327 Search PubMed.
- Site-specific, but not sequence-specific, binding of macrocyclic receptors to synthetic polymers during polyrotaxane formation has been described,8–10 and polyrotaxane formation involving sequence-dependent π-stacking of aromatic/aliphatic homopolymers with a bipyridinium-based macrocycle has also been reported11,12.
- H. M. Colquhoun and Z. Zhu, Angew. Chem., Int. Ed., 2004, 43, 5040 CrossRef CAS.
- K. D. Shimizu, T. M. Dewey and J. Rebek, J. Am. Chem. Soc., 1994, 116, 5145 CrossRef CAS . This paper reports a rotational barrier ΔG‡ of ca. 30 kcal mol−1 for a bis(di-ortho)-substituted N,N′-diarylpyromellitimide.
- H. M. Colquhoun, Z. X. Zhu and D. J. Williams, Org. Lett., 2003, 5, 4353 CrossRef CAS.
- C. G. Gong, T. E. Glass and H. W. Gibson, Macromolecules, 1998, 31, 208 CrossRef.
- S. Choi, J. W. Lee, Y. H. Ko and K. Kim, Macromolecules, 2002, 35, 3526 CrossRef CAS.
- D. Tuncel and J. H. G. Steinke, Macromolecules, 2004, 27, 288 CrossRef.
- G. J. Owen and P. Hodge, Chem. Commun., 1997, 11 RSC.
- P. Hodge, P. Monvisade, G. J. Owen, F. Heatley and Y. Pang, New J. Chem., 2000, 24, 703 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: synthesis and characterisation data for all new materials; binding constant data; 1H NMR data for tweezer–polymer and tweezer–model complexation. See http://www.rsc.org/suppdata/cc/b4/b412801j/ |
| ‡ Previous work7 has shown that tweezers 1a and 1b adopt essentially identical binding-geometries with pyromellitimides. In the present study, crystal quality led to [1b + 2] being chosen for X-ray analysis. Single crystals of the complex were grown by vapour diffusion of an equimolar solution of the two components in chloroform/hexafluoropropan-2-ol (6/1 v/v) with diethyl ether as non-solvent. [1b + 2]: C87H53Cl2N5O12S2·3C3H2F6O·CHCl3Mr = 2118.9, triclinic, P-1, a = 10.9503(4), b = 17.6840(10), c = 23.9264(12) Å, α = 104.520(2), β = 96.031(3), γ = 91.682(3)°. V = 4452.9(4) Å3, T = 120 K, Z = 2, Dc 1.58 g cm−3, μ(Mo-Kα) = 0.32 mm−1, F (000) = 2152. Independent measured reflections 9168. R1 = 0.139, wR2 = 0.391 for 6742 independent observed reflections [2θ ≤ 20.8°, I > 2σ(I)]. Average I/σ(I) 15.5. CCDC 248234. See http://www.rsc.org/suppdata/cc/b4/b412801j/ for crystallographic data in .cif or other electronic format. |
| This journal is © The Royal Society of Chemistry 2004 |