A convergent, versatile route to two synthetic conjugate anti-toxin malaria vaccines
Peter H.
Seeberger†
*,
Regina L.
Soucy
,
Yong-Uk
Kwon†
,
Daniel A.
Snyder
and
Takuya
Kanemitsu
Department of Chemistry: Massachusetts Institute of Technology, Cambridge, MA 02139, USA. E-mail: seeberger@org.chem.ethz.ch; Fax: +41) 1-633123; Tel: (+41) 1-6332103
First published on 9th July 2004
Abstract
The synthesis of two glycosylphosphatidyl inositol (GPI) glycans that constitute the malaria toxin and promising anti-toxin vaccine constructs using a scalable route is described.
Malaria infects 5–10% of humanity, and kills up to three million people each year, mostly children in Africa.1 Current malaria treatments are often impractical in many endemic areas, and drug resistance is a growing problem. At the same time, there is still no effective malaria vaccine.2 Conjugate carbohydrate vaccines have shown great utility as public health tools in preventing the infection of children by Haemophilus influenzae type b and Streptococcus pneumoniae.3 We have previously demonstrated the efficacy of anti-toxin vaccination in a mouse model of malaria.4 Here, we report the development of a general and practical synthesis strategy for access to defined malaria toxin structures, and its application to the synthesis of a second-generation vaccine.
The malaria parasite, Plasmodium falciparum, expresses a large amount of glycosylphosphatidylinositol (GPI) in protein anchored and free form on the cell surface.5 Mounting evidence suggests that the proinflammatory-cytokine cascade triggered by this GPI is responsible for much of malaria's morbidity and mortality.6 Vaccination with synthetic GPI produces anti-GPI antibodies, which neutralize this toxin and result in host survival.4 Based on the GPI toxin 1, our initial vaccine candidate 2a was designed and conjugated to a carrier protein (Fig. 1).4,7 However, as the native toxin is linked to the cell membrane via an inositol phosphate diester, we reasoned that presenting the antigen in the proper orientation, as in 3a/3b, could result in better vaccination.
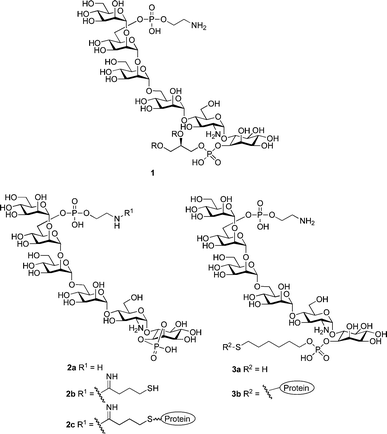 | ||
| Fig. 1 Malarial GPI (1) and model vaccine constructs (2c and 3b). | ||
To obtain ready access to large quantities of 2a and 3a, we devised a convergent, modular synthesis, involving a minimum of late-stage modification, and using robust chemistry throughout. Assembly proceeded via a key 4+2 glycosylation, which allowed for the same tetrasaccharide building block to be used in generation of both 2a and 3a. In addition to the two inositol-containing disaccharides 12 and 13, three mannose synthons 4, 10 and 11 were used (Scheme 1). After the completion of the hexamers, the phosphate diester functions were installed using H-phosphonates 19 and 20 prior to global deprotection and conjugation (Scheme 2).
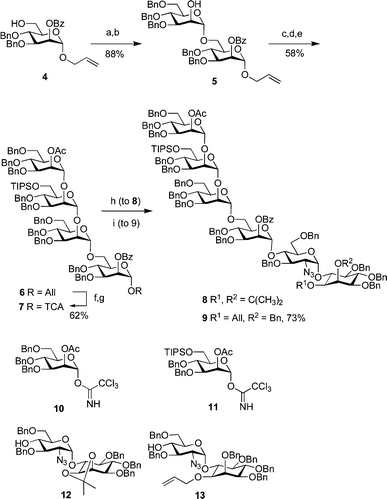 | ||
| Scheme 1 (a) 10, TMSOTf; (b) AcCl, MeOH; (c) 11, TMSOTf; (d) Mg(OMe)2, MeOH; (e) 10, TMSOTf; (f) PdCl2, NaOAc, HOAc, H2O; (g) Cl3CCN, DBU; (h) 12, TMSOTf; (i) 13, TMSOTf. | ||
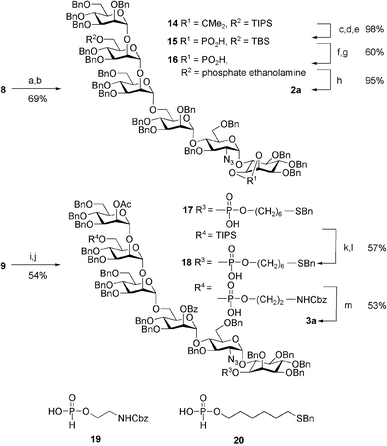 | ||
| Scheme 2 (a) MeOH, NaOMe; (b) BnBr, NaH; (c)TsOH, MeOH; (d) TBSCl, Im.; (e) Cl2PO2Me, Py.; (f) TBAF; (g) 1. 19, PivCl, pyridine; 2. I2; (h) Pd(OH)2, H2; (i) Sc(OTf)3, H2O; (j) 1. 20, PivCl, pyridine; 2. I2; (k) PdCl2, NaOAc, HOAc, H2O; (l) 1. 19, PivCl, pyridine; 2. I2; (m) 1. NaOMe, MeOH; 2. Na, NH3. | ||
This synthesis built on previous efforts towards GPI structures by our4,7 and other laboratories.8 Solution-phase methods are considerably less rapid when compared to our automated assembly but allow for ready scale-up, an important consideration in preparation for preclinical and clinical trials.
The production of the key tetramannose trichloroacetimidate building-block 7 started from C2-benzoyl mannose 4 (see Scheme 1). Glycosylation of 4 with 10, followed immediately by selective removal of the 2-O-acetate in the presence of the benzoate ester using acetyl chloride in methanol, provided disaccharide alcohol 5. Repetition of this maneuver, first using donor 11 and then 10 again, provided fully-protected tetrasaccharide 6. Removal of the anomeric allyl group with PdCl2 in wet acetic acid, was followed by the formation of glycosyl trichloroacetimidate 7 using Cl3CCN/DBU. Demonstrating the scalability of this chemistry, central tetrasaccharide 7 was produced readily on a 20 g scale. Coupling of 7 with 12 afforded hexamer 8en route to 2a. The union of 7 and 13 provided 9 to be elaborated into 3a. It should be noted that the 2-O-benzoate resulted in a significantly improved glycosylating agent when compared to a tetrasaccharide containing the 2-O-benzyl ether used previously: 85% yield as opposed to 39%.
The ester functions of hexamer 8 were first replaced with benzyl ethers to fashion 14 (see Scheme 2). Elaboration of 14 as reported previously furnished 15.7 Desilylation and phosphorylation using H-phosphonate 19 provided fully-protected intermediate 16. Global deprotection in one step using Pd(OH)2 was followed by reaction of the primary amine with 2-iminothiolane,9 to generate thiol 2b, ready for coupling to maleimide-activated BSA and formation of model vaccine 2c.4
Removal of the allyl ether from hexasaccharide 9 (see Scheme 2), using PdCl2 in wet acetic acid was followed by phosphorylation with 20 to give 17. The TIPS ether was cleaved using Sc(OTf)3, and the ethanolaminephosphate linker was installed using H-phosphonate 19, yielding fully-protected 18. Global deprotection was accomplished by the removal of ester groups using sodium methoxide in methanol and subsequent Birch reaction using sodium in ammonia to afford 3a, ready for conjugation to maleimide-functionalized BSA, giving the new model vaccine 3b.
The products of these syntheses (2b and 3a) were attached to BSA both as a model for attachment to the antigenic proteins desired for vaccination, and to produce useful substrates for ELISA tests for anti-GPI IGs in both naturally immune and vaccinated individuals.10 Work is currently underway to determine, via rodent trials, the best carrier protein and adjuvants for the vaccines. Also, we are engaged in synthetic studies producing a variety of substructures of the GPIs, to be used in determining the minimum antigen structure necessary to produce good immune response. The method presented here has 14 steps and provides 3a in 6.4% overall yield.
In conclusion, we have demonstrated the development of a practical synthesis of malarial GPI structures, and applied these methods to the generation of conjugate anti-toxin malaria vaccines from fully synthetic oligosaccharides, resulting in more efficient access both to previously tested and second-generation vaccines.
This research was supported by GlaxoSmithKline (Scholar Award to P.H.S.), the Alfred P. Sloan Foundation (Fellowship to P.H.S.) and Merck & Co (Academic Development Award to P.H.S). Y.-U. Kwon thanks Korea Science and Engineering Foundation (KOSEF) for financial support (Postdoctoral Fellowship).
Notes and references
- WHO, Division of Control of Tropical Diseases, World Health Stat. Q., 1992, 45, 257 Search PubMed.
- M. F. Good, Nat. Rev. Immunol., 2001, 1, 117 CrossRef CAS.
- B. Kuberan and R. J. Linhardt, Curr. Org. Chem., 2000, 4, 653 CAS.
- L. Schofield, M. C. Hewitt, K. Evans, M. Siomos and P. H. Seeberger, Nature, 2002, 418, 785 CrossRef CAS.
- A. Guha-Niyogi, D. R. Sullivan and S. J. Turco, Glycobiol., 2001, 11, 45R CrossRef CAS.
- (a) L. Schofield and F. Hackett, J. Exp. Med., 1993, 177, 145 CrossRef CAS; (b) S. D. Tachado and L. Schofield, Biochem. Biophys. Res. Commun., 1994, 205, 984 CrossRef CAS; (c) L. Schofield, S. Novakovic, P. Gerold, R. T. Schwarz, M. J. McConville and S. D. Tachado, J. Immunol., 1996, 156, 1886 Search PubMed; (d) S. D. Tachado, P. Gerold, R. T. Schwarz, M. J. McConville and L. Schofield, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 4022 CrossRef CAS.
- M. C. Hewitt, D. A. Snyder and P. H. Seeberger, J. Am. Chem. Soc., 2002, 124, 13434 CrossRef CAS.
- (a) C. Murakata and T. Ogawa, Carb. Res., 1992, 235, 95 Search PubMed; (b) U. E. Udodong, R. Madsen, C. Roberts and B. Fraser-Reid, J. Am. Chem. Soc., 1993, 115, 7886 CrossRef CAS; (c) T. G. Mayer, B. Kratzer and R. R. Schmidt, Angew. Chem. Int. Ed. Engl., 1994, 33, 2177 CrossRef; (d) A. S. Campbell and B. Fraser-Reid, J. Am. Chem. Soc., 1995, 117, 10387 CrossRef CAS; (e) D. K. Baeschlin, A. R. Chaperon, L. G. Green, M. G. Hahn, S. J. Ince and S. V. Ley, Chem. Eur. J., 2000, 6, 172 CrossRef CAS; (f) K. Pekari, D. Tailler, R. Weingart and R. R. Schmidt, J. Org. Chem., 2001, 66, 7432 CrossRef CAS; (g) K. Pekari and R. R. Schmidt, J. Org. Chem., 2003, 68, 1295 CrossRef CAS.
- (a) D. R. Tolan and R. R. Traut, J. Biol. Chem., 1981, 256, 10129 CAS; (b) C. A. Alagon and T. P. King, Biochemistry, 1980, 19, 4341 CrossRef.
- C. Evans, P. H. Seeberger, L. Schofield, unpublished results.
Footnote |
| † New Address: Laboratory for Organic Chemistry, ETH Hönggerberg, HCI F315, Wolfgang-Pauli-Str. 10, CH-8093 Zürich, Switzerland |
| This journal is © The Royal Society of Chemistry 2004 |