Potassium hexacyanoferrate(II)—a new cyanating agent for the palladium-catalyzed cyanation of aryl halides
Thomas
Schareina
,
Alexander
Zapf
and
Matthias
Beller
*
Leibniz-Institut für Organische Katalyse an der Universität Rostock e.V., Buchbinderstr. 5–6, 18055 Rostock, Germany. E-mail: matthias.beller@ifok.uni-rostock.de
First published on 12th May 2004
Abstract
A new advantageous cyanating agent, potassium hexacyanoferrate(II), is described for the palladium-catalyzed cyanation of aryl halides. All cyanide ions on the iron(II) center can be transferred to the aryl halide using palladium(II) acetate and dppf as the catalyst. Under optimized reaction conditions good yields of benzonitriles and unprecedented catalyst productivities are observed.
Substituted benzonitriles represent important building blocks for fine chemical synthesis. They are integral parts of dyes, natural products, herbicides, agrochemicals, and pharmaceuticals. On a larger scale aryl cyanides are produced mainly via ammoxidation of the corresponding toluenes. In addition, still the Rosenmund–von Braun reaction [reaction of aryl halides with copper(I) cyanide] and the Sandmeyer reaction [reaction of diazonium salts with copper(I) cyanide] are used to a considerable extent, although very recently also a copper-catalyzed variant has been described.1 Compared to these classic methods transition metal-catalyzed cyanations of aryl halides are somewhat underdeveloped.2
In the past, both nickel3 and palladium complexes4 have been used as catalysts for the cyanation of aryl halides. Mechanistic studies revealed that the general problem of these reactions is the deactivation of the transition metal catalyst by formation of too stable cyano complexes.4a,5 Recently, we have been able to show that catalyst deactivation can be prevented by defined control of the concentration of dissolved cyanide ions.6 Typically this is achieved by the use of nonpolar solvents such as toluene or xylene, that allow only low concentrations of solubilized cyanide salts (generally KCN or NaCN). Another way to control the Pd/cyanide ratio is the slow dosage of liquid or soluble cyanide sources. Here, we demonstrated that trimethylsilylcyanide7 or acetone cyanohydrin6 are suitable reagents for this purpose, which lead to an increase in catalyst efficiency.
Despite the recent progress in palladium-catalyzed cyanations of aryl triflates, iodides, bromides, and even chlorides all known protocols have disadvantages and offer significant potential for improvements. On the one hand catalyst productivities are low (in general TON 20–400) compared to other palladium-catalyzed coupling reactions. In addition, ortho-substituted aryl halides react often sluggishly and give low product yields. Importantly, the known cyanide sources have also severe drawbacks which prevent wider application of this methodology: alkali cyanides are highly poisonous, zinc cyanide leads to stoichiometric amounts of heavy metal salt wastes, and TMSCN is sensitive to moisture and can easily liberate hydrogen cyanide, which is also true for acetone cyanohydrin. In order to overcome these problems, we were looking for alternative cyanide sources, which are less poisonous and can easily be handled without special precautions.
Here, we describe for the first time the use of potassium hexacyanoferrate(II) (K4[Fe(CN)6]) as cyanating agent for the general synthesis of benzonitriles (Scheme 1).
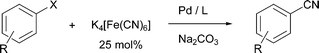 | ||
| Scheme 1 Cyanation of aryl halides using potassium hexacyanoferrate(II) as cyanide source (X = Br, Cl). | ||
Table 1 shows selected results that were obtained while investigating the cyanation of bromobenzene with K4[Fe(CN)6].† As starting point a previously developed catalyst system consisting of palladium(II) acetate and 1,5-bis(diphenylphosphino)pentane (dpppe) in the presence of sodium carbonate was used. While nonpolar solvents such as toluene and dioxane are not suited for this reaction (Table 1, entries 1 and 2), we were surprised to discover that in DMAc or NMP benzonitrile is formed in significant yield (49 and 56%, respectively) (Table 1, entries 3 and 4). Applying alkali cyanides the latter solvents are not useful for the cyanation of aryl bromides or chlorides due to the increased cyanide concentration in solution, which leads to catalyst deactivation.
| Entry | Solvent | Cat. conc. (mol%) | Ligand [conc. (mol%)] | T (°C) | Conv. (%) | Yield (%) | TON |
|---|---|---|---|---|---|---|---|
| a 2,2′-Bis(diphenylphosphino)diphenyl ether. b 9,9-Dimethyl-4,5-bis(diphenylphosphino)xanthene. c 2,2′-Bis{bis[3,5-bis(trifluoromethyl)phenyl]phosphinomethyl}-1,1′-binaphthyl. d 17 mol% K4[Fe(CN)6]. | |||||||
| 1 | Toluene | 1 | dpppe [2] | 120 | 12 | 6 | 6 |
| 2 | Dioxane | 1 | dpppe [2] | 120 | 14 | 9 | 9 |
| 3 | DMAc | 1 | dpppe [2] | 120 | 57 | 49 | 49 |
| 4 | NMP | 1 | dpppe [2] | 120 | 68 | 56 | 56 |
| 5 | NMP | 1 | PPh3 [4] | 120 | 64 | 54 | 54 |
| 6 | NMP | 1 | PCy3 [4] | 120 | 84 | 70 | 70 |
| 7 | NMP | 1 | Bpephosa [2] | 120 | 73 | 59 | 59 |
| 8 | NMP | 1 | dppf [2] | 120 | 100 | 81 | 81 |
| 9 | NMP | 1 | Xantphosb [2] | 120 | 29 | 18 | 18 |
| 10 | NMP | 1 | Iphosc [2] | 120 | 12 | 1 | 1 |
| 11 | NMP | 0.1 | dppf [0.2] | 120 | 100 | >99 | 1000 |
| 12d | NMP | 0.1 | dppf [0.2] | 120 | 96 | 92 | 920 |
| 13 | NMP | 0.01 | dppf [0.02] | 140 | 100 | 97 | 9700 |
| 14 | NMP | 1 | dppf [2] | 100 | 59 | 43 | 43 |
Testing different ligands in the presence of K4[Fe(CN)6] revealed, that both monodentate and chelating phosphines are suitable for the cyanation of bromobenzene. 1,1′-Bis(diphenylphosphino)ferrocene (dppf) gave full conversion and the best yield (81%; Table 1, entry 8), but tricyclohexylphosphine was almost as efficient (70%; Table 1, entry 6). Advantageously, even simple PPh3 gave a moderate yield of benzonitrile (54%; Table 1, entry 5). Using 1 mol% of palladium catalyst reductive dehalogenation is observed as side-reaction to a minor amount. Surprisingly, with a decreased amount of catalyst (0.1 mol%) an even higher selectivity and yield of benzonitrile is obtained (>99%; Table 1, entry 11). We explain this behavior by a decrease of agglomeration of molecular palladium complexes to “palladium black”, which gives less selective reactions. A similar effect has been described by de Vries et al. for Heck olefinations of aryl iodides.8 Even higher turnover numbers can be realized when the reaction temperature is increased by 20 K. In the presence of 0.01 mol% palladium 97% of benzonitrile is formed resulting in a TON of almost 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 (Table 1, entry 13). This is the highest turnover number which we are aware for a palladium-catalyzed cyanation of aryl halides. Generally, 0.25 equiv. of hexacyanoferrate(II) are applied as cyanating agent in our reaction. This corresponds to 1.5 equivalents of cyanide with respect to aryl halide. However, lowering the amount of the cyanide source to 0.17 equiv. still gave 92% of benzonitrile (Table 1, entry 12). This demonstrates that all the six cyanide ions bound to the iron(II) center are used in the cyanation reaction. Interestingly, potassium hexacyanoferrate(III) in contrast is not suitable as the cyanide source (<5% of benzonitrile), presumably because of its oxidizing properties. Apart from Pd(OAc)2 other palladium sources, e.g. Pd2(dba)3, performed equally well in the model reaction. An important parameter is the reaction temperature, which should not be <120 °C (Table 1, entry 14). Apparently at lower temperature cyanide is not transferred from the iron center.
000 (Table 1, entry 13). This is the highest turnover number which we are aware for a palladium-catalyzed cyanation of aryl halides. Generally, 0.25 equiv. of hexacyanoferrate(II) are applied as cyanating agent in our reaction. This corresponds to 1.5 equivalents of cyanide with respect to aryl halide. However, lowering the amount of the cyanide source to 0.17 equiv. still gave 92% of benzonitrile (Table 1, entry 12). This demonstrates that all the six cyanide ions bound to the iron(II) center are used in the cyanation reaction. Interestingly, potassium hexacyanoferrate(III) in contrast is not suitable as the cyanide source (<5% of benzonitrile), presumably because of its oxidizing properties. Apart from Pd(OAc)2 other palladium sources, e.g. Pd2(dba)3, performed equally well in the model reaction. An important parameter is the reaction temperature, which should not be <120 °C (Table 1, entry 14). Apparently at lower temperature cyanide is not transferred from the iron center.
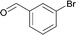
The new protocol was then applied to the cyanation of various aryl halides (Table 2). In general, using 0.1 mol% catalyst at 120 °C good to excellent yields of the desired benzonitriles are obtained. In the case of electron deficient aryl bromides, e.g. for 4-bromoacetophenone (Table 2, entries 1 and 2), even 0.01 mol% palladium are sufficient for high yields (87%). Methyl 4-bromobenzoate (78%) and 4-(trifluoromethyl)bromobenzene (94%) work similarly well, while 3-bromobenzaldehyde gives 66% of 3-cyanobenzaldehyde (Table 2, entries 3–5).
| Entry | Aryl halide | T (°C) | Cat. conc. (mol%) | Conv. (%) | Yield (%) | TON |
|---|---|---|---|---|---|---|
|
1
2 |
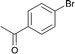
|
120
120 |
0.1
0.01 |
100
100 |
86
87 |
860
8700 |
| 3 |
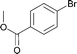
|
100 | 0.1 | 100 | 78 | 780 |
| 4 |
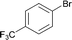
|
120 | 0.1 | 98 | 94 | 940 |
| 5 |
|
120 | 0.1 | 96 | 66 | 660 |
|
6
7 |
|
120
140 |
0.1
0.5 |
60
93 |
51
68 |
510
136 |
| 8 |
|
120 | 0.1 | 100 | 95 | 950 |
|
9
10 |
|
120
160 |
0.25
0.25 |
48
88 |
41
75 |
164
300 |
| 11 |
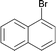
|
120 | 0.1 | 100 | 90 | 900 |
| 12 |
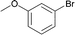
|
120 | 0.1 | 92 | 88 | 880 |
| 13 |
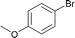
|
120 | 0.1 | 100 | 95 | 950 |
| 14 |
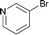
|
140 | 0.1 | 97 | 86 | 860 |
| 15 |
|
140 | 0.1 | 95 | 87 | 870 |
| 16 |
|
140 | 0.1 | 91 | 78 | 780 |
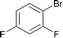
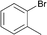
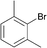
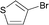
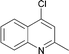
Interestingly, also a number of ortho-substituted aryl bromides work well in the procedure. In the case of 2,4-difluorobromobenzene an increase in the catalyst concentration (to 0.5 mol%) and reaction temperature (to 140 °C) leads to an improved yield of 68% vs. 51% (Table 2, entries 6 and 7). 2-Bromotoluene gives 95% of 2-cyanotoluene under standard conditions (Table 2, entry 8). Even 2,6-dimethylbromobenzene is converted to 2-cyano-m-xylene in 75% yield in the presence of 0.25 mol% catalyst (Table 2, entries 9 and 10). 1-Bromonaphthalene yields 90% 1-cyanonaphthalene (Table 2, entry 11). Electronically deactivated m- and p-bromoanisole can be cyanated with very good yield (88–95%; Table 2, entries 12 and 13). N- and S-heterocycles such as 3-bromopyridine, 3-bromothiophene, and 4-chloroquinaldine give the desired nitriles in 78–87% yield (Table 2, entries 14–16).
In summary, we have developed a new general protocol for the cyanation of aryl halides. For the first time potassium hexacyanoferrate(II) has been used as cyanide source. The advantages of this “coupling reagent” are obvious: in contrast to other cyanating agents potassium hexacyanoferrate(II) is less poisonous‡ and can be handled without special precaution. Due to the slow release of cyanide ions a significantly improved catalyst productivity compared to previously known procedures is achieved.
The authors thank Dr W. Baumann, Dr C. Fischer, Mrs S. Buchholz and Ms K. Reincke for analytical support. Generous support from the state Mecklenburg-Vorpommern and the Fonds der Chemischen Industrie are gratefully acknowledged. OMG (now Umicore) is thanked for gifts of palladium compounds.
Notes and references
- J. Zanon, A. Klapars and S. L. Buchwald, J. Am. Chem. Soc., 2003, 125, 2890 CrossRef CAS.
- (a) G. P. Ellis and T. M. Romney-Alexander, Chem. Rev., 1987, 87, 779 CrossRef CAS; (b) M. Sundermeier, A. Zapf and M. Beller, Eur. J. Inorg. Chem., 2003, 3513 CrossRef CAS.
- (a) L. Cassar, M. Foà, F. Montanari and G. P. Marinelli, J. Organomet. Chem., 1979, 173, 335 CrossRef CAS; (b) Y. Sakakibara, F. Okuda, A. Shimoyabashi, K. Kirino, M. Sakai, N. Uchino and K. Takagi, Bull. Chem. Soc. Jpn., 1988, 61, 1985 CAS; (c) Y. Sakakibara, Y. Ido, K. Sasaki, M. Sakai and N. Uchino, Bull. Chem. Soc. Jpn., 1993, 66, 2776 CAS.
- (a) K. Takagi, T. Okamoto, Y. Sakakibara and S. Oka, Chem. Lett., 1973, 471 CAS; (b) A. Sekiya and N. Ishikawa, Chem. Lett., 1975, 277 CAS; (c) K. Takagi, T. Okamoto, Y. Sakakibara, A. Ohno, S. Oka and N. Hayama, Bull. Chem. Soc. Jpn., 1975, 48, 3298 CAS; (d) K. Takagi, T. Okamoto, Y. Sakakibara, A. Ohno, S. Oka and N. Hayama, Bull. Chem. Soc. Jpn., 1976, 49, 3177 CAS; (e) J. R. Dalton and S. L. Regen, J. Org. Chem., 1979, 44, 4443 CrossRef CAS; (f) Y. Akita, M. Shimazaki and A. Ohta, Synthesis, 1981, 974 CrossRef CAS; (g) N. Chatani and T. Hanafusa, J. Org. Chem., 1986, 51, 4714 CrossRef CAS; (h) K. Takagi, K. Sasaki and Y. Sakakibara, Bull. Chem. Soc. Jpn., 1991, 64, 1118 CAS; (i) Y. Anderson and B. Långström, J. Chem. Soc., Perkin Trans. 1, 1994, 1395 RSC; (j) B. A. Anderson, E. C. Bell, F. O. Ginah, N. K. Harn, L. M. Pagh and J. P. Wepsiec, J. Org. Chem., 1998, 63, 8224 CrossRef CAS; (k) T. Okano, J. Kiji and Y. Toyooka, Chem. Lett., 1998, 425 CAS; (l) P. E. Maligres, M. S. Waters, F. Fleitz and D. Askin, Tetrahedron Lett., 1999, 40, 8193 CrossRef CAS; (m) F. Jin and P. N. Confalone, Tetrahedron Lett., 2000, 41, 3271 CrossRef CAS; (n) M. Sundermeier, A. Zapf, M. Beller and J. Sans, Tetrahedron Lett., 2001, 42, 6707 CrossRef CAS; (o) J. Ramnauth, N. Bhardwaj, P. Renton, S. Rakhit and S. Maddaford, Synlett, 2003, 2237 CAS.
- M. Sundermeier, A. Zapf, S. Mutyala, W. Baumann, J. Sans, S. Weiss and M. Beller, Chem. Eur. J., 2003, 9, 1828 CrossRef CAS.
- M. Sundermeier, A. Zapf and M. Beller, Angew. Chem., 2003, 115, 1700 ( Angew. Chem., Int. Ed. , 2003 , 42 , 1661 ) CrossRef.
- M. Sundermeier, S. Mutyala, A. Zapf, A. Spannenberg and M. Beller, J. Organomet. Chem., 2003, 684, 50 CrossRef CAS.
- A. H. M. de Vries, J. M. C. A. Mulders, J. H. M. Mommers, H. J. W. Henderickx and J. G. de Vries, Org. Lett., 2003, 5, 3285 CrossRef CAS.
Footnotes |
| † General procedure: Under inert conditions 2.0 mmol Na2CO3, 0.5 mmol K4[Fe(CN)6] {K4[Fe(CN)6]·3H2O is ground to a fine powder and dried in vacuum (ca. 2 mbar) at 80 °C over night}, Pd(OAc)2 and ligand are placed in a pressure tube. Then 2.0 mmol aryl halide and 2.0 mL solvent are added. Stock solutions of Pd(OAc)2 and ligand were used for Pd contents smaller than 0.5%. The pressure tube is sealed and heated at 120 °C for 16 h. After cooling to room temperature 3.0 mL dichloromethane and 0.4 mL diethylene glycol di-n-butyl ether (internal standard) are added and the mixture is analyzed by GC. For isolating the products the reaction mixture is washed with water and the organic phase is dried over Na2SO4. After evaporation of the solvents the residue is subjected to column chromatography (silica, hexane–ethyl acetate). All prepared benzonitriles are known compounds and identified by comparison (GC/MS) with authentic samples. |
| ‡ KCN is extremely toxic [LDL0 (oral, human) = 2.86 mg kg−1] and develops HCN on contact with acidic water. K4[Fe(CN)6] is nontoxic and used in food industry for metal precipitation in wine. Also it has been used as anti-agglutinating auxiliary for NaCl (table salt) (cf. Roempp Lexikon Chemie – Version 2.0, Georg Thieme Verlag, Stuttgart/New York, 1999). It is soluble in water without decomposition. |
| This journal is © The Royal Society of Chemistry 2004 |