The first general method for α-trifluoromethylation of carboxylic acids using BrF3†
Aviv
Hagooly
and
Shlomo
Rozen
*
School of Chemistry, Raymond and Beverly Sackler Faculty of Exact Sciences, Tel-Aviv University, Tel-Aviv 69978, Israel. E-mail: rozens@post.tau.ac.il
First published on 9th February 2004
Abstract
2-Carbomethoxy-1,1-bis(methylsulfide)-1-alkenes, easily made from carboxylic acids, CS2 and MeI, were treated with BrF3 producing eventually the desired α-trifluoromethyl carboxylate derivatives – RCH(CF3)COOR′ – in good yields.
The α-position to the carboxylate moiety is unique when organic acids associated with biological activity are the issue. The importance of the CF3 group has been outlined in numerous cases1 and recently, Olah, Prakash and others have achieved remarkable results using Me3SiCF3 as a tool for introducing the trifluoromethyl group into electrophilic centers [e.g. R2CO → R2C(CF3)OH].2
Still, only a few, highly specific α-trifluoromethyl carboxylates have been described so far. Recent examples include constructing α-alkoxy-α-trifluoromethyl acids3 and a procedure for electrophilic trifluoromethylation of the strongly nucleophilic carbon of β-ketocarboxylates.4 Attempting α-alkylation of β,β,β- trifluoropropionates was proven impractical since a facile defluorination takes place even at −78 °C: (CF3CH2COOR + B−
→ CF2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCOOR).5 Clearly, a general method for introducing this important group into the α-position of a given carboxylic acid is needed. We describe here a method, based on the use of BrF3, which closes this gap.
CHCOOR).5 Clearly, a general method for introducing this important group into the α-position of a given carboxylic acid is needed. We describe here a method, based on the use of BrF3, which closes this gap.
Bromine trifluoride has been rarely used in organic chemistry when not heavily halogenated molecules are in question. It plays a pivotal role in the synthesis of some modern anaesthetics such as sevoflurane6 and recently in constructing the CF2![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 7 and CF38 groups. In most of these procedures the soft acidic bromine atom of the BrF3 complexifies itself with soft basic nitrogen or sulfur atoms, placing the naked nucleophilic fluorides in the immediate vicinity of the electrophilic carbon α to the heteroatom. The formation of the CF bonds is thus facilitated and the reaction is usually completed within a few seconds. This greatly helps to keep undesirable radical side reactions to a minimum.
7 and CF38 groups. In most of these procedures the soft acidic bromine atom of the BrF3 complexifies itself with soft basic nitrogen or sulfur atoms, placing the naked nucleophilic fluorides in the immediate vicinity of the electrophilic carbon α to the heteroatom. The formation of the CF bonds is thus facilitated and the reaction is usually completed within a few seconds. This greatly helps to keep undesirable radical side reactions to a minimum.
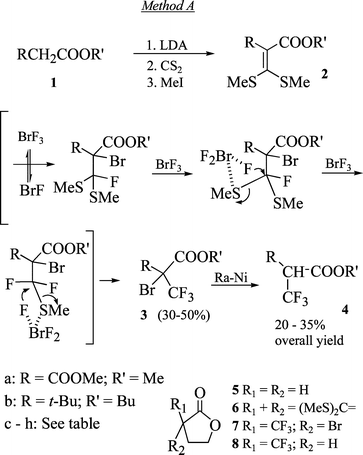
One of the best methods to place a sulfur atom near the α-position of an ester group of type 1 is to react its corresponding enolate with CS2 followed by MeI.9 In order to substitute both sulfur atoms of the resulting 2-carbomethoxy-1,1-bis(methylsulfide)-1-alkene 2c–h, five molar equivalents of BrF3
(method A
–
Scheme 1) had to be used to form 2-bromo-2-trifluoromethyl carboxylates 3c–h. The bromine atom could then be removed by Raney nickel and the desired α-trifluoromethyl esters 4c–h were obtained. The presence of the bromine atom suggests that the first step of the reaction is a nucleophilic attack of the olefinic center on the bromine atom in either BrF3 or BrF, which is always present in the reagent (a known equilibrium since BrF3 always contains some bromine). A second, and if supplied also a third molecule, of BrF3 attacks the sulfur atoms resulting in CF bond formation with the nearby electrophilic carbon.8a Although the presence of an aromatic ring is usually prohibitive since it is easily brominated by the reagent10 the reaction is not restricted only to straight chain acids. Butyrolactone 5 was converted to the corresponding bis(methylsulfide) derivative 611 and reacted with BrF3. Since the reaction is fast and is performed at 0 °C, the lactone ring was not affected and 2-bromo-2-trifluoromethylbutyrolactone 7 was obtained. Treatment with Raney nickel produced the desired 2-trifluoromethylbutyrolactone 8.12 Similarly, dimethyl malonate 1a afforded the known dimethyl 2-trifluoromethylmalonate 4a.13 Strong steric hindrance to the carbon α to the carboxylate moiety as in butyl neopentanoate 1b is responsible for low yields of the corresponding 1,1-bis(methyl sulfide)
2b
(20%), but the reactions with BrF3 and Raney nickel proceed as expected resulting in butyl 2-trifluoromethyl-α-t-butyl acetate 4b. The main disadvantage of this route, however, is the use of a large excess of BrF3, which prompts radical reactions responsible in most cases for the low overall yield of 20–35%.
 | ||
| Scheme 1 | ||
The reaction was considerably improved and the yields were more than doubled when a somewhat different route (method B – Scheme 2) was developed. When only 2.5 molar equivalents of BrF3 were reacted for less than a minute with the disulfides 2, mixtures of more than 85% of methyl 2-bromo-2-[difluoro(methylsulfide)methyl]alkanoates 9, the respective sulfoxides 10, and traces of the sulfones 11 were obtained. These mixtures were not resolved but treated ‘as is’ with HOF·CH3CN at room temperature, transferring within a few minutes14 all sulfur-containing compounds to the corresponding 11 which contain the good leaving sulfone group. These were reacted with Bu4NF,15 eliminating both bromine and sulfone groups to give the target α-trifluoromethylalkanoates 4 in overall yields of up to 70% based on the starting esters. It should be mentioned here that this method is also very suitable for introducing the important isotope 18F into the CF3 group for positron emitting tomography (PET) purposes.
 | ||
| Scheme 2 | ||
The scope of this reaction was investigated and is summarized in Table 1. The straight chain methyl heptanoate 1c, methyl undecanoate 1d and methyl tetradecanoate 1e were α-trifluoromethylated to produce 4c,164d17 and 4e, respectively, in 65 – 70% overall yield. Both cyclic derivatives 1f and 1g reacted rapidly to form the unknown methyl 3-cyclopentyl-2-trifluoromethylpropanoate 4f and methyl 4-cyclohexyl-2-trifluoromethylbutanoate 4g. Bromine trifluoride is known to substitute chlorine atoms as demonstrated by the synthesis of the anaesthetic sevoflurane, but again the complexation and the fast reaction with the sulfur atoms in the reaction of 2h leave the chlorine intact and methyl 5-chloro-2-trifluoromethylpentanoate 4h was eventually obtained. It is known that unprotected alcohols are quickly oxidized by BrF3 to acyl fluorides,18 but when protected, either as ethers or pivaloyl esters (e.g.1i or 1j), the reaction proceeds as expected and ethyl 4-ethoxy-2-trifluoromethylbutanoate 4i and ethyl 6-pivalooxy-2-trifluoromethylhexanoate 4j were formed. The reason for choosing a pivaloyl ester as a protecting group is its tolerance toward strong bases, which are required for the activation of the α-position in the 1
→
2 transformation. This is also the reason why ketones must first be protected as ketals (e.g.1k
→
1l), but after the formation of 2l this protecting group could be removed. The ketone 2k was thus reacted with BrF3 with no complications to produce methyl 7-oxo-2-trifluoromethyloctanoate 4k.
|
|
||
|---|---|---|
| Compound | Ra | Overall yield of 4b (%) |
| a For spectral characterization of some representative compound 9s, 10s, and all 11s see the ESI. b All α-trifluoromethyl esters of type 4 are oils. They are fully characterized by IR, 1H, 13C, 19F NMR, HRMS and microanalysis. c These compounds are ethyl esters. | ||
| c | CH3(CH2)4 | 6516 |
| d | CH3(CH2)8 | 7017 |
| e | CH3(CH2)11 | 65 |
| f |

|
65 |
| g |

|
65 |
| h | Cl(CH2)3 | 60 |
| i c | EtO(CH2)2 | 55 |
| j c | t-BuCOO(CH2)4 | 60 |
| k | CH3CO(CH2)4 | 50 |
| l |

|
|

In conclusion, we have demonstrated for the first time a general method for constructing various types of α-trifluoromethyl carboxylic acids suitable also for incorporation of the positron emitting isotope 18F into such molecules.
We thank the USA-Israel Binational Science Foundation (BSF), Jerusalem, Israel for financial support.
References
- See, for example: B. R. Langlois and T. Billard, Synthesis, 2003, 185 Search PubMed; G. Li, Y. Chen, J. R. Missert, A. Rungta, T. J. Dougherty, Z. D. Grossman and R. K. Pandey, J. Chem. Soc., Perkin Trans. 1, 1999, 1785 CrossRef CAS; J. W. Hilborn, Z. H. Lu, A. R. Jurgens, Q. K. Fang, P. Byers, S. A. Wald and C. H. Senanayake, Tetrahedron Lett., 2001, 42, 8919 RSC.
- G. K. S. Prakash, M. Mandal and G. A. Olah, Org. Lett., 2001, 3, 2847 CrossRef CAS; G. K. S. Prakash, M. Mandal and G. A. Olah, Angew. Chem., Int. Ed., 2001, 40, 589 CrossRef CAS; S. Caron, N. M. Do, P. Arpin and A. Larivee, Synthesis, 2003, 1693 CAS.
- R. Fernandez, E. M. Zamora, C. Pareja, J. Vazquez, E. Diez, A. Monge and J. M. Lassaletta, Angew. Chem., Int. Ed., 1998, 37, 3428 CrossRef CAS.
- J. A. Ma and D. Cahard, J. Org. Chem., 2003, 68, 8726 CrossRef CAS.
- T. Yokozawa, T. Nakai and N. Ishikawa, Tetrahedron Lett., 1984, 25, 3987 CrossRef CAS.
- D. F. Halpern and M. L. Robin, US Pat., 4,996,371, 1991; K. Ramig, Synthesis, 2002, 2627 Search PubMed.
- S. Rozen, E. Mishani and A. Bar-Haim, J. Org. Chem., 1994, 59, 2918 CrossRef CAS; S. Rozen and I. Ben-David, J. Org. Chem., 2001, 66, 496 CrossRef CAS; R. Sasson, A. Hagooly and S. Rozen, Org. Lett., 2003, 5, 769 CrossRef CAS; A. Hagooly, R. Sasson and S. Rozen, J. Org. Chem., 2003, 68, 8287 CrossRef CAS.
- (a) A. Hagooly, I. Ben-David and S. Rozen, J. Org. Chem., 2002, 67, 8430 CrossRef CAS; (b) S. Rozen, D. Rechavi and A. Hagooly, J. Fluorine Chem., 2001, 111, 161 CrossRef CAS; (c) I. Ben-David, D. Rechavi, E. Mishani and S. Rozen, J. Fluorine Chem., 1999, 97, 75 CrossRef CAS.
- D. A. Konen, P. E. Pfeffer and L. S. Silbert, Tetrahedron, 1976, 32, 2507 CrossRef CAS.
- S. Rozen and O. Lerman, J. Org. Chem., 1993, 58, 239 CrossRef CAS.
- R. K. Dieter, J. Org. Chem., 1981, 46, 5031 CrossRef CAS.
- N. Reineke, N. A. Zaidi, M. Mitra, D. O'Hagan, A. S. Batsanov, J. A. K. Howard and D. Y. Naumov, J. Chem. Soc., Perkin Trans. 1, 1996, 147 RSC.
- N. Ishikawa and T. Yokozawa, Bull. Chem. Soc. Jpn., 1983, 56, 724 CAS.
- S. Rozen and Y. Bareket, J. Org. Chem., 1997, 62, 1457 CrossRef CAS ; other oxygen transfer agents such as DMDO could also be used, but HOF·CH3CN was found to be the most efficient one.
- E. Anselmi, J. C. Blazejewski and C. Wakselman, Tetrahedron Lett., 1998, 39, 9651 CrossRef CAS.
- K. Tomoya, Jap. Pat., 02,238,896, 1990(Chem. Abstr., 1991, 114, 120301t).
- K. Iseka, Y. Kuroki, T. Nagai and Y. Kobayashi, Chem. Pharm. Bull., 1996, 44, 477.
- S. Rozen and I. Ben-David, J. Fluorine Chem., 1996, 76, 145 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: complete experimental details and instructions of how to work and handle BrF3 and HOF·CH3CN. 1H NMR, 13C NMR, 19F NMR, IR and microanalysis data for all compounds. See http://www.rsc.org/suppdata/cc/b3/b315705a/ |
| This journal is © The Royal Society of Chemistry 2004 |