Borrowing hydrogen: a catalytic route to C–C bond formation from alcohols†
Michael G.
Edwards
a,
Rodolphe F. R.
Jazzar
a,
Belinda M.
Paine
a,
Duncan J.
Shermer
a,
Michael K.
Whittlesey
*a,
Jonathan M. J.
Williams
*a and
Dean D.
Edney
b
aDepartment of Chemistry, University of Bath, Claverton Down, Bath, UK BA2 7AY. E-mail: chsmkw@bath.ac.uk; chsjmjw@bath.ac.uk
bGlaxoSmithKline Research & Development, Old Powder Mills, Tonbridge, Kent, UK TN11 9AN
First published on 18th November 2003
Abstract
Ruthenium complexes have been shown to perform efficient transfer hydrogenation reactions between alcohols and alkenes; in combination with an in situ Wittig reaction, indirect formation of C–C bonds has been achieved from alcohols.
The concept of catalytic electronic activation as a route to new C–C bond forming reactions is an attractive one since it allows the use of alcohols as substrates.1 One model for this involves the indirect Wittig reaction of a temporarily oxidized alcohol (Scheme 1) to yield an alkene, which is then reduced to give the final longer chain alkane.2 The key to the catalytic cycle is the borrowing of hydrogen—dehydrogenation of alcohol to aldehyde releases H2, which would be stored by the catalytic metal fragment and then returned in the final hydrogenation step. The development of catalytic systems is therefore likely to involve metal complexes in which H2 dissociation and re-coordination is facile, preferably without the requirement for forcing conditions.3
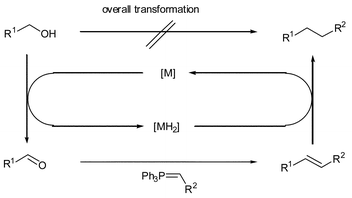 | ||
| Scheme 1 Catalytic electronic activation: indirect Wittig reaction upon alcohols. | ||
Our previous attempts at achieving this reaction using an iridium-based system required heating at 150 °C for 72 h.2 Herein we report on a ruthenium-catalysed approach requiring milder reaction conditions. The use of a ruthenium N-heterocyclic carbene complex allows the reaction to be carried out at significantly lower temperatures and reaction times. We have recently reported the facile C–H bond activation of the N-heterocyclic carbene 1,3-bis(2,4,6-trimethylphenyl)imidazol-2-ylidene (IMes) in Ru(IMes)(PPh3)2(CO)H2 (1) at room temperature in the presence of a sacrificial alkene (Scheme 2).4 The C–H cleavage product 2 readily reforms the starting dihydride upon reaction with H2. The rapid hydrogenation of alkenes5 and the reversibility of the pathway illustrated in Scheme 2 prompted us to investigate the suitability of alcohols as hydrogen donors in this process.6
 | ||
| Scheme 2 Reversible dehydrogenation/hydrogenation pathway of complex 1. | ||
It was subsequently demonstrated that vinyltrimethylsilane (3) could be completely hydrogenated by complex 1 at 70 °C to provide ethyltrimethylsilane (4) with isopropanol acting as the hydrogen donor (Scheme 3). Indeed, the crossover transfer hydrogenation7 reaction between one equivalent of alcohol and one equivalent of alkene could also be readily achieved (Scheme 3). (±)-Phenethyl alcohol (6) and tert-butyl cinnamate (5) were converted solely into acetophenone (8) and tert-butyl dihydrocinnamate (7) using 5 mol% of complex 1.
 | ||
| Scheme 3 Transfer hydrogenation reactions with complex 1. | ||
The ability to effect a crossover transfer hydrogenation is critical to the success of the indirect Wittig reaction identified in Scheme 1. In the absence of crossover hydrogenation the cycle is unable to proceed once the initial catalyst has been exhausted. Following the success of these transfer hydrogenation reactions the catalytic activity of complex 1 in the indirect Wittig process was examined (Scheme 4, Table 1). Table 1 shows the performance of complex 1 (5 mol% loading) for reaction of benzyl alcohol (9) with the ester ylide (10) in toluene solution at 80 °C. In all cases 5 mol% of vinyltrimethylsilane (3) was added to accomplish the initial dehydrogenation of the catalyst required for the reaction to proceed. The reaction catalysed by complex 1 afforded 90% of the dihydrocinnamate product (11) after 24 hours (entry 1). The complex Ru(PPh3)3(CO)H2 also proved to be successful in the indirect Wittig reaction, although it proved to be inferior to complex 1 over the same reaction period (entry 2).8
 | ||
| Scheme 4 Ruthenium catalysed indirect Wittig reactions with benzyl ester ylide 10. | ||
| Entry | Precursor (5 mol%) | Ligand (mol%) | Conversion (%)b |
|---|---|---|---|
| a The reactions were carried out on a 0.5 mmol scale in toluene (1.5 mL) at 80 °C for 24 hours using 1.1 equivalents of ylide 10. b Determined by 1H NMR spectroscopy. c The precursor and IMes were heated at 70 °C for 1.5 hours in toluene before addition of the remaining reagents. | |||
| 1 | 1 | — | 90 |
| 2 | Ru(PPh3)3(CO)H2 | — | 80 |
| 3 | Ru(PPh3)3(CO)H2 | IMes (5) | 87 |
| 4c | Ru(PPh3)3(CO)H2 | IMes (5) | 86 |
The highest levels of activity of complex 1 are associated with isolated material; however in situ generation of the catalyst proved to be successful (entries 3 and 4). Thus, whilst the carbene complex 1 is the most successful catalyst, commercially available complex Ru(PPh3)3(CO)H2 was also reasonably effective.
We further demonstrated that the indirect Wittig reaction catalysed by 1 could be successfully achieved in high yield by the use of alternative phosphorane ester ylides with other alcohol substrates (Scheme 5). Using optimised reaction conditions (1 mol% 1, 1.00 M concentration, 80 °C) the indirect Wittig adducts 11, 12, 13, 15 and 17 were obtained in good to excellent isolated yields following column chromatography (70–84%). These results demonstrate that complex 1 displays high catalytic activity for C–C bond formation via this route at moderately low temperatures (80 °C). In contrast, the previously reported2 iridium catalysed reactions afforded indirect Wittig adducts in lower yield, 47–71%, even under extremely forcing reaction conditions (150 °C, 72 hours) and at considerably higher catalyst loadings (5 mol%). In addition, the results indicate that the presence of an N-heterocyclic carbene ligand is beneficial for improved reactivity in comparison with other complexes.
 | ||
Scheme 5 Synthesis of indirect Wittig reaction adducts (1 mol%
1, 1.1 equiv. Ph3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCO2R, 2 mol% H2CCHSiMe3, PhMe, 1.00 M, 80 °C); (a) 24 hour reaction time; (b) 48 hour reaction time. CHCO2R, 2 mol% H2CCHSiMe3, PhMe, 1.00 M, 80 °C); (a) 24 hour reaction time; (b) 48 hour reaction time. | ||
In conclusion, we have demonstrated that ruthenium complexes act as catalysts for the formation of C–C bonds from alcohol substrates via an intriguing indirect Wittig reaction.
We thank the EPSRC, GlaxoSmithKline and the University of Bath for financial support and Johnson Matthey plc for the loan of ruthenium trichloride.
Notes and references
- P. J. Black, W. Harris and J. M. J. Williams, Angew. Chem., Int. Ed., 2001, 40, 4475 CrossRef CAS.
- M. G. Edwards and J. M. J. Williams, Angew. Chem., Int. Ed., 2002, 41, 4740 CrossRef CAS.
- J. E. Bäckvall, J. Organomet. Chem., 2002, 652, 105 CrossRef CAS.
- R. F. R. Jazzar, S. A. Macgregor, M. F. Mahon, S. P. Richards and M. K. Whittlesey, J. Am. Chem. Soc., 2002, 124, 4944 CrossRef CAS.
- Several hydrogenation reactions using hydrogen gas have been reported for NHC complexes. See for example: H. M. Lee, D. C. Smith Jr., Z. He, E. D. Stevens, C. S. Yi and S. P. Nolan, Organometallics, 2001, 20, 794 Search PubMed; H. M. Lee, T. Jiang, E. D. Stevens and S. P. Nolan, Organometallics, 2001, 20, 1255 CrossRef CAS; M. T. Powell, D.-R. Hou, M. C. Perry, X. Cui and K. Burgess, J. Am. Chem. Soc., 2001, 123, 8878 CrossRef CAS; L. D. Vázquez-Serrano, B. T. Owens and J. M. Buriak, Chem. Commun., 2002, 2518 CrossRef CAS; M. C. Perry, X. Cui, M. T. Powell, D.-R. Hou, J. H. Riebenspies and K. Burgess, J. Am. Chem. Soc., 2003, 125, 113 RSC.
- Transfer hydrogenation reactions have been reported with a range of transition metal NHC complexes: A. C. Hillier, H. M. Lee, E. D. Stevens and S. P. Nolan, Organometallics, 2001, 20, 4246 Search PubMed; J. Louie, C. W. Bielawski and R. H. Grubbs, J. Am. Chem. Soc., 2001, 123, 11312 CrossRef CAS; M. A. Albrecht, J. R. Miecznikowski, A. Samuel, J. W. Faller and R. H. Crabtree, Organometallics, 2002, 21, 3596 CrossRef CAS; A. A. Danopoulos, S. Winston and W. B. Motherwell, Chem. Commun., 2002, 1376 CrossRef CAS; M. A. Albrecht, R. H. Crabtree, J. A. Mata and E. Peris, Chem. Commun., 2002, 32 RSC; M. Poyatos, J. A. Mata, E. Falomir, R. H. Crabtree and E. Peris, Organometallics, 2003, 22, 1110 RSC; M. Poyatos, E. Mas-Marzá, J. A. Mata, M. Sanaú and E. Peris, Eur. J. Inorg. Chem., 2003, 1215 CrossRef CAS.
- The literature contains very few reports of stoichiometric crossover transfer hydrogenation reactions between alcohols and alkenes. A few examples of crossover transfer hydrogenation using a small excess of alkene acceptor have been reported. See for example: M. E. Krafft and B. Zorc, J. Org. Chem., 1986, 51, 5482 Search PubMed.
- Ru(PPh3)3(CO)H2 has been reported as catalysing hydrogenation of alkenes, aldehydes and ketones, although very forcing conditions are required for substrates with CO groups. F. Kakiuchi, S. Sekine, Y. Tanaka, A. Kamatani, M. Sonoda, N. Chatani and S. Murai, Bull. Chem. Soc. Jpn., 1995, 68, 62 Search PubMed; R. A. Sanchez-Delgado and O. L. de Ochoa, J. Organomet. Chem., 1980, 202, 427 CAS; R. A. Sanchez-Delgado, A. Andriollo, O. L. de Ochoa, T. Suarez and N. Valencia, J. Organomet. Chem., 1981, 209, 77 CrossRef CAS; G. Speier and L. Marko, J. Organomet. Chem., 1981, 210, 253 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: Experimental procedures and characterization data for compounds 1, 11, 12, 13, 15 and 17 along with general experimental procedures. See http://www.rsc.org/suppdata/cc/b3/b312162c/ |
| This journal is © The Royal Society of Chemistry 2004 |