Carbon–carbon bond cleavage by cytochrome P450BioI (CYP107H1)†
Max J.
Cryle
and
James J.
De Voss
*
Department of Chemistry, University of Queensland, St. Lucia, Brisbane, Australia 4072. E-mail: j.devoss@uq.edu.au; Fax: +61 7 3365 4299; Tel: +61 7 3365 3825
First published on 17th November 2003
Abstract
Cytochrome P450BioI (CYP107H1) is believed to supply pimelic acid equivalents for biotin biosynthesis in Bacillus subtilis: we report here that the mechanistic pathway adopted by this multifunctional P450 for the in-chain cleavage of fatty acids is via consecutive formation of alcohol and threo-diol intermediates, with the likely absolute configuration of the intermediates also reported.
The cytochromes P450 (P450s) comprise a superfamily of oxidative hemoproteins that catalyse an impressive array of oxidative transformations. Typically, this involves oxidation at an unactivated carbon centre to yield the corresponding alcohol.1 However, the oxidative potential of these enzymes is perhaps best illustrated by their ability to catalyse the cleavage of C–C bonds.2 We report here that the mechanistic pathway adopted by the multifunctional P450BioI for the in-chain cleavage of fatty acids is via alcohol and threo-diol intermediates. This represents the first prokaryotic P450 capable of catalysing C–C bond cleavage in an unfunctionalised substrate.
A number of mechanistically different reaction types can be distinguished for P450 catalysed C–C bond scission, but each type is represented by only a few examples.2 Additionally, almost all of the described reactions are restricted to eukaryotes; CYP51, which occurs in both eukaryotes and prokaryotes, is the only prokaryotic example apart from P450BioI.3 Most P450 C–C bond scissions occur α to an existing functional group,2,4–8 but perhaps more impressive are those catalysed by multifunctional P450s which oxidatively cleave an unactivated alkyl moiety. One such reaction that has been fully described is the transformation of cholesterol into pregnenolone and 4-methylpentanal catalysed by the eukaryotic P450scc.2‡ In this, two hydroxylation reactions lead specifically to 20(R),22(R)-dihydroxycholesterol and the C–C bond between the two oxygenated carbons is then oxidatively cleaved. A similar process occurs in the metabolism of the antimicrobial agent olanexidine, but it is unclear whether a single P450 is responsible for all of the oxidative steps.9
We have recently reported the over-expression and characterisation of a Bacillus subtilus enzyme, P450BioI, that is involved in biotin biosynthesis.10 Analysis of mutant phenotypes had suggested that it played a role in the formation of pimelic acid (heptanedioic acid) which provides the majority of the carbon backbone of biotin.11 Expression of P450BioI in E. coli resulted in the isolation of the enzyme alone and in complex with an acyl carrier protein acylated with C14 to C18 fatty acids (acyl ACP).10 It was demonstrated that turnover of this complex resulted in the formation of a pimeloyl ACP, via cleavage of the alkyl chain of the acyl moiety. Free fatty acids also serve as substrates for a catalytically active P450BioI system.10,12,13 Initially, it was postulated that ω-functionalisation of the fatty acid may occur, providing an alternative route for the production of pimelic acid via subsequent chain shortening reactions.13 However, we have recently demonstrated that the major positions of free fatty acid oxidation are exclusively non-terminal.12 For example, tetradecanoic acid (1) is oxidised to mainly 11-, 12- and 13-hydroxytetradecanoic acids along with a small amount of pimelic acid. The lack of specificity observed in the oxidation of the free fatty acids is unsurprising, given that it appears likely the natural substrate is an ACP thioester of a fatty acid.10 However, we believed that as pimelic acid was indeed produced from free fatty acids, the route by which this was formed would likely be the same as that by which the pimeloyl ACP was produced. We thus set out to determine the pathway by which P450BioI catalysed the C–C bond cleavage of fatty acids to produce pimelic acid using tetradecanoic acid 1 as the illustrative substrate.
P450scc provides the only delineated example of oxidative bond fission of an unactivated alkyl chain.‡ However, it was clear that other pathways might be followed as direct P450 catalysed C–C bond cleavage has also been observed in simple alcohols7 and ketones.2 Thus, we planned to synthesise a range of possible intermediates and determine the relative level of pimelic acid production, reasoning that more advanced intermediates would lead to greater pimelic acid production.
Therefore, compounds 2–6 were synthesised and incubated with a catalytically active P450BioI system consisting of purified E. coli flavodoxin reductase, cindoxin and P450BioI10 and the level of pimelic acid production quantified by GC analysis.
The results (Fig. 1) show clear differences in the levels of pimelic acid production and delineate the pathway by which P450BioI cleaves the fatty acid chain. The C7 alcohol 2 is a better substrate than 1 but the C7 oxo fatty acid 4 does not improve pimelic acid production. Neither the C8 alcohol 3 nor the C8 oxo fatty acid 5 is processed at all. The erythro 7,8-diol (rac6a) is a poorer substrate than the threo diastereomer (rac6b) which leads to an approximately 15-fold increase in C–C bond cleavage relative to 1. These results are consistent with an oxidation pathway that is initiated by C7 hydroxylation (Scheme 1). Subsequent C8 oxidation forms the threo diol that then undergoes oxidative cleavage. The threo diol would be predicted to arise from the enzyme acting upon one face of the extended fatty acid chain.
 | ||
| Fig. 1 Pimelic acid production by P450BioI from racemic substrates (substrate concentration = 1 mM). | ||
Unambiguous identification of the products of C–C bond cleavage was necessary, as a mechanism involving diol cleavage would predict formation of two aldehyde fragments. The increased turnover of threo-7,8-dihydroxytetradecanoic acid (rac6b) revealed the presence of heptanal and 7-oxoheptanoic acid in comparable amounts at short reaction times. Both compounds, either as synthetic standards or produced via P450 catalysed bond scission, were converted to the corresponding acids over time (>70% after 24 h) under enzyme turnover conditions via aerial oxidation, independent of NADPH. All of these results are consistent with C–C bond cleavage via a route corresponding to that seen for P450scc.2
The absolute configurations of the preferred enzymic substrates were investigated by the synthesis and evaluation of scalemic samples of the 7-hydroxy (2) and threo 7,8-dihydroxytetradecanoic acids (6b). The former was available in ∼70% ee as determined by enantioselective HPLC (Chiracel OD), with the key step in the syntheses being a CBS borane reduction of the appropriate acetylenic ketone.14 The enantiomerically pure threo diols (R,R and S,S6b) were synthesised from chiral, non-racemic tartaric acid, utilizing methodology developed by Seebach for differentiating either end of the starting diacid.15 Incubation of these substrates with a catalytically active system indicated that the S2 and the corresponding R,R diol were preferentially processed by P450BioI (Fig. 2). The enantioselectivity of the enzyme was low, with less favored R2 and S,S6b still being good substrates for the enzyme. A plausible explanation for this lack of selectivity stems from the fact that the enantiomers of 6b differ only in the location of a carboxyl or a methyl group five methylene groups removed from the diol moiety. In the putative natural substrate, an acyl ACP, the carrier protein would presumably dictate the binding orientation. However, with the free fatty acids, two binding orientations are possible that present the same apparent stereochemistry of the diol at the active site differing only in the location of a distal methyl or carboxylate.
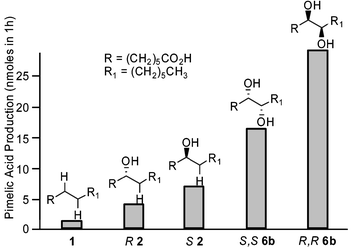 | ||
| Fig. 2 Pimelic acid production by P450BioI from scalemic substrates (substrate concentration = 1 mM). | ||
 | ||
| Scheme 1 | ||
Accurate dissociation constants for the enzyme–substrate complexes were difficult to determine spectrophotometrically, probably due to the instability of the uncomplexed enzyme.10 However, it was clear from the estimated association constants10 that the hydroxylated fatty acids bound significantly (at least 4 fold) more tightly to the enzyme than the starting fatty acid, but both alcohol and diol appeared to bind equally well. This is analogous to the situation seen with P450scc in which the hydroxylated intermediates were bound equally, but more tightly than cholesterol.2 This is presumed to increase the efficiency of the reaction: the intermediates are not released or displaced by the initial substrate but continue on to product. The fact that the C7 alcohol and the 7,8-diol both facilitate and direct subsequent oxidation reactions is clearly reflected in the analysis of the enzymic turnovers. While the 11- and 12-hydroxytetradecanoic acids are the major products isolated from P450BioI catalyzed hydroxylation of 1, they are not substrates for further oxidation.122 does not accumulate in enzymic oxidations of 1, nor does 6 appear in turnover of 1 or 2. Finally, the only alternative oxidation pathway apart from C–C bond cleavage seen for 2 is conversion into the corresponding ketone 4. Interestingly, this pathway was much more significant for R2 than S2 (>10 fold, quantified by GC), which produces less pimelic acid. This suggests that the orientation of the substrate in the active site is determined by the stereochemistry at C7 and this in turn directs subsequent oxidation reactions.
In conclusion, we have identified the pathway by which the multifunctional, prokaryotic P450BioI catalyses the cleavage of a C–C bond in a fatty acid (Scheme 1). By analogy we predict cleavage of an acyl ACP, its natural substrate, will occur in the same way. The mechanism involves the consecutive formation of an alcohol and a vicinal diol and subsequent C–C bond cleavage. This is analogous to that seen with P450scc and P450BioI is thus only the second P450 identified as being capable of such bond fission, and the first from a prokaryote.
MJC is grateful for an Australian Postgraduate Research Award.
Notes and references
- P. R. Ortiz de Montellano and J. J. De Voss, Nat. Prod. Rep., 2002, 19, 477 RSC.
- P. R. Ortiz de Montellano, in Cytochrome P450: Structure, Mechanism, and Biochemistry, Second Edition, ed. P. R. Ortiz de Montellano, Plenum, New York, 1995 Search PubMed.
- M. J. Cryle, J. E. Stok and J. J. De Voss, Aust. J. Chem., 2003, 56, 749 CrossRef CAS.
- H. Fukuda, T. Fujii, E. Sukita, M. Tazaki, S. Nagahama and T. Ogawa, Biochem. Biophys. Res. Commun., 1994, 201, 516 CrossRef CAS.
- T. Hakamatsuka, M. F. Hashim, Y. Ebizuka and U. Sankawa, Tetrahedron, 1991, 47, 5969 CrossRef CAS.
- J. R. Reed, D. Vanderwel, S. Choi, G. Pomonis, R. C. Reitz and G. J. Blomquist, Proc. Natl. Acad. Sci. USA, 1994, 91, 10000 CAS.
- V. Stanjek, M. Miksch, P. Lueer, U. Matern and W. Boland, Angew. Chem., Int. Ed., 1999, 38, 400 CrossRef CAS.
- W. Yin, G. A. Doss, R. A. Stearns, A. G. Chaudhary, C. E. Hop, R. B. Franklin and S. Kumar, Drug Metab. Dispos., 2003, 31, 215 Search PubMed.
- K. Umehara, S. Kudo, Y. Hirao, S. Morita, T. Ohtani, M. Uchida and G. Miyamoto, Drug Metab. Dispos., 2000, 28, 1417 Search PubMed.
- J. E. Stok and J. J. De Voss, Arch. Biochem. Biophys., 2000, 384, 351 CrossRef CAS.
- S. Bower, J. B. Perkins, R. R. Yocum, C. L. Howitt, P. Rahaim and J. Pero, J. Bacteriol., 1996, 178, 4122 CAS.
- M. J. Cryle, N. J. Matovic and J. J. De Voss, Org. Lett., 2003, 5, 3341 CrossRef CAS.
- A. J. Green, S. L. Rivers, M. Cheesman, G. A. Reid, L. G. Quaroni, I. D. G. Macdonald, S. K. Chapman and A. W. Munro, J. Biol. Inorg. Chem., 2001, 6, 523 Search PubMed.
- K. A. Parker and M. W. Ledeboer, J. Org. Chem., 1996, 61, 3214 CrossRef CAS.
- E. Hungerbuehler and D. Seebach, Helv. Chim. Acta, 1981, 64, 687 CrossRef.
Footnotes |
| † Electronic Supplementary Information (ESI) available: 2, R2, S2, 4, SS6b, RR6b characterisation and synthesis. Enzymatic oxidation and analysis protocols. See http://www.rsc.org/suppdata/cc/b3/b311652b/ |
| ‡ Whilst C–C bond cleavage is observed with CYP19 and CYP51 in steroid biosynthesis, both proceed via aldehyde intermediates which are inaccessible to P450BioI. |
| This journal is © The Royal Society of Chemistry 2004 |