First synthesis of an amythiamicin pyridine cluster†
Mark C. Bagley*a, James W. Dalea, Robert L. Jenkinsa and Justin Bowerb
aSchool of Chemistry, Cardiff University, PO Box 912, Cardiff, UK CF10 3TB. E-mail: bagleymc@cf.ac.uk; Fax: +44 29 20874030; Tel: +44 29 20874029
bVernalis, Granta Park, Abington, Cambridge, UK CB1 6GB
First published on 25th November 2003
Abstract
The pyridine-containing central domain of the amythiamicin group of thiopeptide antibiotics is prepared in protected form in 9 steps, 93% ee and 18% overall yield from (S)-2-[1-(tert-butoxycarbonylamino)-2-methylpropyl]thiazole-4-carboxylic acid by Michael addition–cyclodehydration of a 2-(2-thiazolyl)enamine and 1-(2-thiazolyl)propynone.
The amythiamicins1 are members of the thiopeptide class of antibiotics, a family of sulfur-containing highly modified cyclic peptides isolated for their effectiveness against methicillin resistant Staphylococcus aureus (MRSA).2 Structurally, amythiamicin A is a macrocyclic thiopeptide that contains a heterocyclic ensemble of heavily modified amino acids centred about a polythiazolylpyridine core. Although no route to amythiamicin A has appeared in the literature, the synthesis of other thiopeptide antibiotics and their heterocyclic components has attracted considerable international interest, resulting in the total synthesis of micrococcin P13 and promothiocin A.4 At this time the configuration of three of the five stereogenic centres present in amythiamicin is not known but, as a working hypothesis, it was assumed that they were derived from the more common L-amino acids based on degradation studies on the structurally related antibiotic GE2270A (MDL 62,879).5
As part of our interest in the discovery of new heteroannulation methods for the synthesis of pyridines,6,7 we set out to establish a rapid route to the heterocyclic core of the amythiamicins by Michael addition–cyclodehydration of enamine 2 and alkynone 3 that would proceed with total control of regiochemistry and generate the pyridine heterocycle in the correct oxidation state (Scheme 1). Central to our strategy was the need to minimise subsequent manipulation following the heteroannulation reaction, facilitated by the use of 1-(2-thiazolyl)propynone 3, anticipated to be highly reactive and to require an unusual Bohlmann–Rahtz procedure in order for efficient cyclization to the target pyridine 1. Although a simpler 1-(2-thiazolyl)propynone has been used before in a Bohlmann–Rahtz reaction with an enamine this required a five-fold excess of reagent, limiting its use as a method to access pyridines substituted at C-6.4 We sought to validate a more efficient and rapid route to these pyridines and, in so doing, facilitate the synthesis of the amythiamicin central heterocyclic domain with suitable functionality for elaboration of the natural product.
 | ||
| Scheme 1 Synthetic approach to the central amythiamicin domain 1. | ||
The first component, 1-(2-thiazolyl)propyn-1-one 3, was prepared in five steps (Scheme 2) from 2,2-diethoxyacetamide (4) by thionation with phosphorus pentasulfide, Hantzsch thiazole synthesis with ethyl bromopyruvate in ethanol in the presence of 4 Å molecular sieves, followed by acid catalysed acetal deprotection to give aldehyde 7 that could be stored at room temperature. Addition of ethynylmagnesium bromide in THF with aqueous work up and subsequent oxidation of propargylic alcohol 8 with manganese(IV) oxide gave propynone 3. As anticipated, this substrate proved unstable at room temperature and so was generated from aldehyde 7 as required. In order to establish new heteroannulation conditions suitable for this substrate, propynone 3 was reacted with ethyl β-aminocrotonate by a variety of different methods (Table 1) to give model pyridine 10 substituted at C-6 in accordance with the target amythiamicin domain. When dienone 9, generated by Michael addition in ethanol at 50 °C, was heated to 150 °C (entry 1) cyclodehydration to pyridine 10 was incomplete and so alternative conditions were sought. Acid catalysed one-step procedures failed to give satisfactory yields (entries 2, 3), as did tandem oxidation–heteroannulation8 of propargylic alcohol 8 (entry 4) and so we investigated a new cyclodehydration method, hitherto reported only as an aside in the development of one-step acid-catalysed Bohlmann–Rahtz procedures.6 Dienone 9, generated by Michael addition in ethanol, was stirred at 50 °C in toluene–acetic acid to give pyridine 10 in > 98% yield (entry 5).
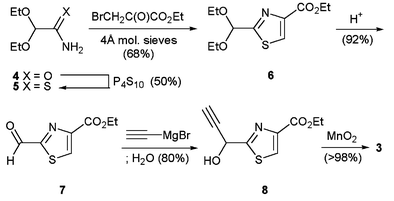 | ||
| Scheme 2 Synthesis of 1-(2-thiazolyl)propyn-1-one 3. | ||

With successful conditions established in a model system for rapid introduction of the C-6 substituent, efforts were directed towards establishing the group at C-3 with similar rapidity. Hantzsch thiazole 11 was reduced with lithium aluminium hydride to give alcohol 12, which was protected as 2-(trimethylsilyl)ethoxy-methyl (SEM) ether 13 (Scheme 3). Directed 5-lithiation with n-butyllithium and reaction with chlorotrimethylsilane provided thiazole 14 as a protected pyridine C-3 building block for the synthesis of amythiamicin.
 | ||
| Scheme 3 Synthesis of thiazole building block 14. | ||
N-Protected L-valine was transformed to valine-derived thiazole 15 by the modified Hantzsch procedure of Meyers,9 followed by hydrolysis. Reaction with ethyl chloroformate under basic conditions, aminolysis of the mixed anhydride with aqueous ammonia and thionation of amide 16 using Lawesson's reagent (LR) gave thioamide 17 (Scheme 4). Hantzsch thiazole synthesis with ethyl bromopyruvate under basic conditions followed by hydroxythiazoline dehydration, using a mixture of trifluoroacetic anhydride and 2,6-lutidine, gave chiral bis-thiazole 18 in 96% ee [HPLC on Chiralpak AD]. Hydrolysis with lithium hydroxide, formation of Weinreb amide 20 and reaction with the lithio-derivative of 2-methylthiazole 14 gave Claisen condensation product 21. Protodesilylation using tetrabutylammonium fluoride in THF followed by microwave irradiation of thiazole 22 and ammonium acetate in toluene at 120 °C provided enamine 2 for submission to the key Bohlmann–Rahtz reaction. Utilizing conditions successful for the synthesis of model pyridine 10, Michael addition of enamine 2 and propynone 3 in ethanol followed by acetic acid-catalysed cyclodehydration at 70 °C, gave the amythiamicin heterocyclic domain 1 with total regiocontrol in 85% yield and 93% ee. This work represents the first synthesis of this heterocyclic cluster, in protected form, generated in only 9 steps and 18% overall yield from 15, and constitutes a rapid route to the amythiamicin antibiotic family.
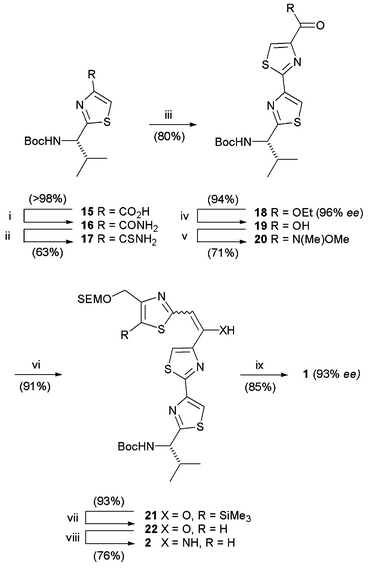 | ||
| Scheme 4 Reagents and conditions: (i) EtO2CCl, THF, Et3N then aq. NH3; (ii) LR; (iii) NaHCO3, ethyl bromopyruvate then trifluoroacetic anhydride, 2,6-lutidine; (iv) LiOH, H2O, MeOH; (v) EtO2CCl, THF, Et3N then HN(Me)OMe·HCl; (vi) 14, n-BuLi; H2O; (vii) TBAF, THF, RT, 1 h; (viii) NH4OAc, μwave, 120 °C (100 W), PhMe, 30 min; (ix) 3, EtOH, 60 °C; toluene–AcOH, 70 °C. | ||
Notes and references
- K. Shimanaka, N. Kinoshita, H. Iinuma, M. Hamada and T. Takeuchi, J. Antibiot., 1994, 47, 668 CAS; K. Shimanaka, Y. Takahashi, H. Iinuma, H. Naganawa and T. Takeuchi, J. Antibiot., 1994, 47, 1145 CAS; K. Shimanaka, Y. Takahashi, H. Iinuma, H. Naganawa and T. Takeuchi, J. Antibiot., 1994, 47, 1153 CAS.
- K. Shimanaka, H. Iinuma, M. Hamada, S. Ikeno, K. S. Tsuchiya, M. Arita and M. Hori, J. Antibiot., 1995, 48, 182 CAS.
- M. A. Ciufolini and Y.-C. Shen, Org. Lett., 1999, 1, 1843 CrossRef CAS.
- C. J. Moody and M. C. Bagley, Chem. Commun., 1998, 2049 RSC; M. C. Bagley, K. E. Bashford, C. L. Hesketh and C. J. Moody, J. Am. Chem. Soc., 2000, 122, 3301–3313 CrossRef CAS.
- P. Tavecchia, P. Gentili, M. Kurz, C. Sottani, R. Bonfichi, E. Selva, S. Lociuro, E. Restelli and R. Ciabatti, Tetrahedron, 1995, 51, 4867 CrossRef CAS.
- M. C. Bagley, C. Brace, J. W. Dale, M. Ohnesorge, N. G. Phillips, X. Xiong and J. Bower, J. Chem. Soc., Perkin Trans. 1, 2002, 1663 RSC.
- M. C. Bagley, J. W. Dale, D. D. Hughes, M. Ohnesorge, N. G. Phillips and J. Bower, Synlett, 2001, 1523 CrossRef CAS; M. C. Bagley, R. Lunn and X. Xiong, Tetrahedron Lett., 2002, 43, 8331 CrossRef CAS; M. C. Bagley, J. W. Dale and J. Bower, Chem. Commun., 2002, 1682 RSC.
- M. C. Bagley, D. D. Hughes, H. M. Sabo, P. H. Taylor and X. Xiong, Synlett, 2003, 1443 CrossRef CAS.
- E. Aguilar and A. I. Meyers, Tetrahedron Lett., 1994, 35, 2473 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: experimental details. See http://www.rsc.org/suppdata/cc/b3/b310944e/ |
| This journal is © The Royal Society of Chemistry 2004 |