
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multifunctional siRNA/ferrocene/cyclodextrin nanoparticles for enhanced chemodynamic cancer therapy†
Gowtham
Raj
a,
D. S.
Vasudev
a,
Sarah
Christopher
a,
Anupama
Babulal
a,
P.
Harsha
a,
Soumakanya
Ram
a,
Mehul
Tiwari
a,
Markus
Sauer
b and
Reji
Varghese
 *a
*a
aSchool of Chemistry, Indian Institute of Science Education and Research (IISER) Thiruvananthapuram, Trivandrum-695551, Kerala, India. E-mail: reji@iisertvm.ac.in
bDepartment of Biotechnology and Biophysics, Biocenter, Julius Maximilian University of Würzburg, Würzburg, Germany
First published on 17th January 2024
Abstract
The therapeutic outcome of chemodynamic therapy (CDT) is greatly hindered by the presence of oxidative damage repair proteins (MTH1) inside cancer cells. These oxidative damage repair proteins detoxify the action of radicals generated by Fenton or Fenton-like reactions. Hence, it is extremely important to develop a simple strategy for the downregulation of MTH1 protein inside cancer cells along with the delivery of metal ions into cancer cells. A one-pot host–guest supramolecular approach for the codelivery of MTH1 siRNA and metal ions into a cancer cell is reported. Our approach involves the fabrication of an inclusion complex between cationic β-cyclodextrin and a ferrocene prodrug, which spontaneously undergoes amphiphilicity-driven self-assembly to form spherical nanoparticles (NPs) having a positively charged surface. The cationic surface of the NPs was then explored for the loading of MTH1 siRNA through electrostatic interactions. Using HeLa cells as a representative example, efficient uptake of the NPs, delivery of MTH1 siRNA and the enhanced CDT of the nanoformulation are demonstrated. This work highlights the potential of the supramolecular approach as a simple yet efficient method for the delivery of siRNA across the cell membrane for enhanced chemodynamic therapy.
Introduction
Chemodynamic therapy (CDT) is one of the widely used techniques for the treatment of cancer.1–4 It involves the delivery of a transition metal/metal-based system into a cancer cell, which upon reaction with hydrogen peroxide (H2O2) overexpressed inside the cancer cell produces highly toxic reactive oxygen species (ROS). These reactions are generally known as Fenton or Fenton-like reactions. Excessive generation of radicals inside the cancer cell induces oxidative damage, which leads to cell death. Unlike other therapeutic approaches such as radiotherapy, photothermal therapy (PTT) and photodynamic therapy (PDT), where sophisticated instruments are typically required, CDT is straightforward and explores mainly endogenous chemicals inside the cancer cells as a trigger for their activity. This makes CDT a superior approach for cancer therapy over other conventional strategies.5–8One of the important challenges associated with CDT for cancer therapy is the presence of oxidative damage-repairing proteins inside the cells.9,10 They potentially neutralize the ROS generated by Fenton/Fenton-like reactions by various defense mechanisms, thereby significantly reducing the efficacy of CDT. For example, the MTH1 protein, a nucleotide pool sanitizing enzyme, can hydrolyze the oxidized nucleotides formed by ROS, thus protecting the cancer cells from oxidative damage.11–14 This suggests that suppressing MTH1 protein synthesis inside cancer cells along with the delivery of a Fenton active metal catalyst would be a promising strategy to improve the efficiency of CDT. Very recently, Tan et al. have shown that MTH1 protein expression inside cancer cells can be significantly downregulated by using small interfering RNAs (siRNAs), and showed a dramatic enhancement in the therapeutic efficacy of PDT.11 Motivated by this report, we envisioned that the co-delivery of siRNA for the inhibition of MTH1 protein expression and a Fenton reagent would be an ideal strategy for an efficient CDT, which has not yet been attempted. Delivery of siRNAs into cancer cells was typically achieved by using cationic nanoparticles (NPs) as the delivery vehicle in a noncovalent fashion.15–19 Hence, we want to explore a supramolecular approach for the co-delivery of a Fenton reagent and siRNA for the inhibition of MTH1 protein synthesis into cancer cells for enhanced CDT.20
Herein, we demonstrate the host–guest interaction between cationic β-cyclodextrin (β-CD+, host) and a ferrocene prodrug (1, guest) as a nanoplatform for the co-delivery of MTH1 siRNA and a Fenton reagent (Fe2+) into a cancer cell. β-Cyclodextrin was chosen as the host molecule in our design because of (i) its high affinity to form a host–guest complex with Fenton active ferrocene derivatives, (ii) water solubility and (iii) excellent biocompatibility. The supramolecular host–guest interaction between ferrocene and β-CD is well established and has been extensively applied for the design of diverse supramolecular functional nanomaterials.21–26 Our design strategy relies on the initial host–guest interaction between cationic β-CD+ and the prodrug 1 in aqueous medium for the formation of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 amphiphilic β-CD+/1 complex, which then spontaneously undergoes amphiphilicity-driven self-assembly for the formation of cationic spherical nanoparticles (β-CD+/1 NPs). The most important structural feature of β-CD+/1 NPs is the protrusion of cationic β-CD+ on the surface of the NPs due to its hydrophilic nature. The positively charged outer surface of the NPs permits the integration of MTH1 siRNA (siRNA) onto their surface via electrostatic interactions to form β-CD+/1/siRNA NPs. The internalization of β-CD+/1/siRNA NPs into cancer cells via an endocytotic pathway followed by their lysosomal escape and distribution in the cytosol is demonstrated. The disassembly of the NPs in the cytosol and the subsequent delivery of siRNA in the cytosol is shown, which efficiently downregulates the expression of the MTH1 protein and causes an increase of CDT action. Simultaneously, the prodrug 1 undergoes the Fenton reaction with the overexpressed H2O2 inside the cancer cells to produce ˙OH and p-quinone methide (an antioxidant scavenger). The latter is a potential scavenger of glutathione (GSH),27 which is also beneficial for the enhancement of CDT activity. The combined effect of the delivery of siRNA, generation of ˙OH through the Fenton reaction and depletion of GSH by the production of an antioxidant scavenger leads to a dramatic increase in the overall efficacy of CDT (Scheme 1).
:1 amphiphilic β-CD+/1 complex, which then spontaneously undergoes amphiphilicity-driven self-assembly for the formation of cationic spherical nanoparticles (β-CD+/1 NPs). The most important structural feature of β-CD+/1 NPs is the protrusion of cationic β-CD+ on the surface of the NPs due to its hydrophilic nature. The positively charged outer surface of the NPs permits the integration of MTH1 siRNA (siRNA) onto their surface via electrostatic interactions to form β-CD+/1/siRNA NPs. The internalization of β-CD+/1/siRNA NPs into cancer cells via an endocytotic pathway followed by their lysosomal escape and distribution in the cytosol is demonstrated. The disassembly of the NPs in the cytosol and the subsequent delivery of siRNA in the cytosol is shown, which efficiently downregulates the expression of the MTH1 protein and causes an increase of CDT action. Simultaneously, the prodrug 1 undergoes the Fenton reaction with the overexpressed H2O2 inside the cancer cells to produce ˙OH and p-quinone methide (an antioxidant scavenger). The latter is a potential scavenger of glutathione (GSH),27 which is also beneficial for the enhancement of CDT activity. The combined effect of the delivery of siRNA, generation of ˙OH through the Fenton reaction and depletion of GSH by the production of an antioxidant scavenger leads to a dramatic increase in the overall efficacy of CDT (Scheme 1).
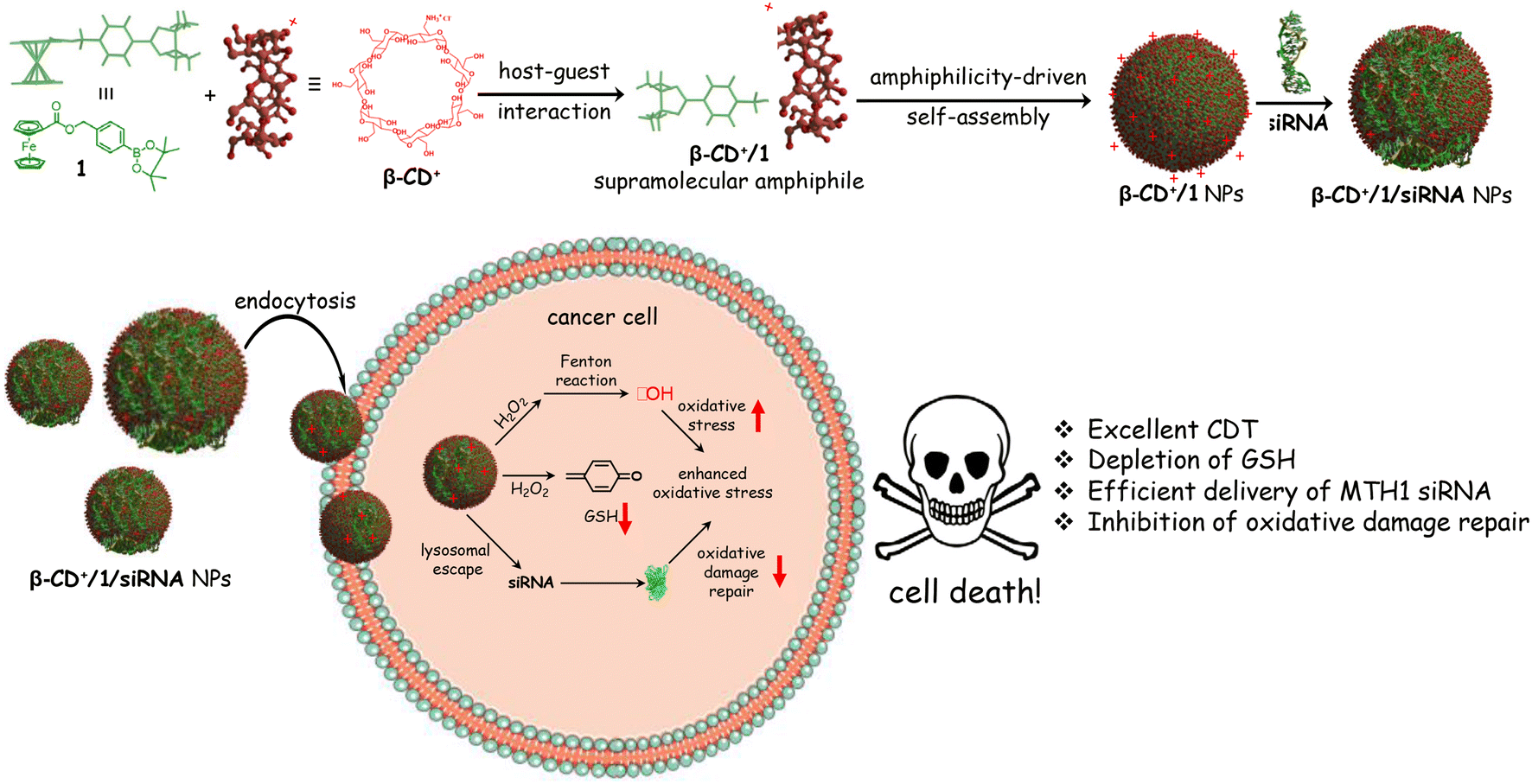 | ||
| Scheme 1 Chemical structure of 1, β-CD+ and their host–guest complex formation followed by the formation of β-CD+/1 NPs. Loading of siRNA onto the surface of β-CD+/1 NPs to form β-CD+/1/siRNA NPs is also shown. The cellular internalization of β-CD+/1/siRNA NPs and the different responses of the NPs inside the cancer cellular environment are depicted schematically. | ||
Results and discussion
The synthesis of the cationic host β-CD+ and the guest prodrug 1 was achieved through multistep organic reactions.28,29 Details of the syntheses and the characterization of β-CD+, 1 and all the intermediates are provided in the ESI.† The supramolecular synthesis of the amphiphilic host–guest complex of β-CD+ and 1 (β-CD+/1) in water:THF (99:1) was achieved by annealing a 1:5 molar ratio of β-CD+ (1 μM in Milli-Q water) and 1 (5 μM in THF) at 90 °C for 5 minutes followed by slow cooling to room temperature over a period of 1 h. Excess 1 present in the solution was then removed by repeated centrifugation at 4000 rpm for 3 minutes, and the supernatant obtained after several rounds of centrifugation was used for further experiments. The supramolecular complex β-CD+/1 spontaneously undergoes amphiphilicity-driven self-assembly to form β-CD+/1 NPs.
We then characterized the complex formation between β-CD+ and 1 and the spontaneous self-assembly of the β-CD+/1 complex into the NPs by using various spectroscopic and microscopic tools. The host β-CD+ is water soluble, whereas the guest prodrug 1 is nearly water insoluble. Accordingly, the absorption spectra of 1 in THF:water (THF:water = 5:95, 5 μM) exhibited a very weak absorption band of the ferrocene unit of 1 at 265 nm due to its very poor water solubility (Fig. S1†). Interestingly, the emergence of characteristic absorption of 1 at 265 nm was clearly observed with the addition of β-CD+. The intensity of the band was found to increase with the increase of β-CD+ concentration and a strong absorption band of 1 at 265 nm was observed at a β-CD+ concentration of 5 μM (Fig. 1a). These results indicate the initial formation of the inclusion complex between β-CD+ and 1 (β-CD+/1) as reported,30,31 which spontaneously undergoes amphiphilicity-driven self-assembly to β-CD+/1 NPs. The emergence of an absorption peak of 1 with the addition of β-CD+ can be attributed to the solubilization of 1 in water with the spontaneous formation of β-CD+/1 NPs. Tapping-mode atomic force microscopic (AFM) analyses revealed the formation of spherical NPs for β-CD+/1 aggregates with an average diameter of ∼80 nm (Fig. S2†). Zeta potential measurement of β-CD+/1 NPs showed a value of +40.54 mV, which clearly reveals that the surface charge of the NPs is positive (Fig. 1b). All these data collectively confirm the initial formation of the 1:1 supramolecular complex β-CD+/1, which subsequently undergoes amphiphilicity-driven self-assembly for the formation of β-CD+/1 spherical NPs with cationic β-CD+ exposed to the outer surface of the NPs.
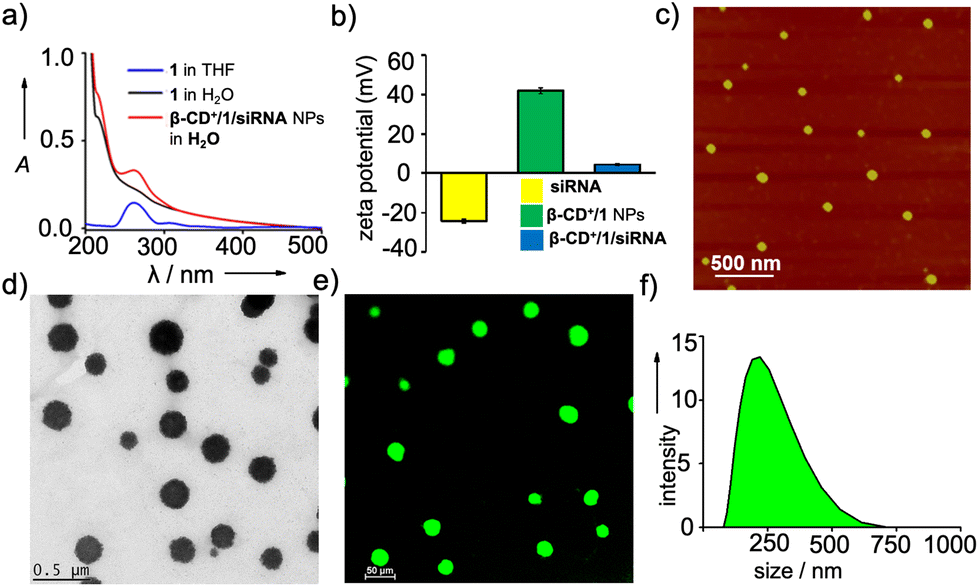 | ||
| Fig. 1 (a) Comparison of the absorption spectra of 1 (in THF and water) and β-CD+/1 NPs (water). (b) Comparison of the zeta potential values of siRNA, β-CD+/1 NPs and β-CD+/1/siRNA NPs. (c) AFM and (d) TEM images of β-CD+/1/siRNA NPs. (e) CLSM image of β-CD+/1/siRNA-FAM NPs and (f) DLS size distribution of β-CD+/1/siRNA NPs. | ||
After establishing the formation of β-CD+/1 NPs, we have explored the loading of siRNA onto the cationic surface of the NPs through electrostatic interactions (Table 1). For this purpose, siRNA (20 nM) was mixed with β-CD+/1 NPs (1 μM each) and annealed at 25 °C for 30 minutes, followed by slow cooling to room temperature. Zeta potential measurements showed values of +4.89, −24.72 and +40.54 mV for β-CD+/1/siRNA, siRNA and β-CD+/1 NPs, respectively (Fig. 1b). The decrease in the zeta potential value of β-CD+/1 NPs from +40.54 mV to +4.89 mV for β-CD+/1/siRNA clearly indicates the successful loading of negatively charged siRNA onto the positively charged surface of β-CD+/1 NPs to form β-CD+/1/siRNA NPs. Furthermore, no significant change in the zeta potential value of +4.5 mV for β-CD+/1/siRNA was observed over a time period of 6 h (Fig. S4†). This shows the structural stability of the electrostatic interaction between negatively charged siRNA and positively charged β-CD+/1 with respect to time. Also, the structural stability of β-CD+/1/siRNA NPs in serum was studied using zeta potential analyses. For this, β-CD+/1/siRNA NPs were treated with 10% heat-inactivated serum for 6 h, and the corresponding zeta potential analyses showed a value of +4.1 mV. This shows that even after 6 h of incubation, degradation of β-CD+/1/siRNA NPs and release of siRNA have not occurred (Fig. S4†). The structural stability of β-CD+/1/siRNA NPs is extremely important as it potentially prevents the premature release of siRNA. Tapping-mode AFM (Fig. 1c) and transmission electron microscopic (TEM) (Fig. 1d) analyses revealed the formation of spherical NPs for β-CD+/1/siRNA. Moreover, confocal laser scanning microscopy (CLSM) analyses of 6-carboxyfluorescein (FAM) labelled siRNA (siRNA-FAM) showed the formation of green fluorescent spherical NPs for β-CD+/1/siRNA-FAM (Fig. 1e). These results suggest that the morphology of β-CD+/1 NPs remains intact even after the loading of siRNA. It is to be noted that the average diameter of β-CD+/1/siRNA NPs obtained from the AFM analyses is ∼80 nm, indicating that the loading of siRNA does not cause significant change to the diameter of β-CD+/1 NPs, as expected. However, it must be pointed out that our strategy cannot offer any control over the diameter of the NPs as it is evident from TEM (Fig. 1d), CLSM (Fig. 1e) and DLS (Fig. 1f); all of them show large size distributions for the NPs.
| RNA | Sequence (5′ → 3′) |
|---|---|
| siRNA | GAC GAC AGC UAC UGG UUU CTT (sense) |
| GAA ACC AGU AGC UGU CGU CTT (antisense) | |
| siRNA-FAM | FAM-GAC GAC AGC UAC UGG UUU CTT (sense) |
| GAA ACC AGU AGC UGU CGU CTT (antisense) |
We next investigated the peroxidase-like catalytic activity of β-CD+/1 NPs. The catalytic activity of β-CD+/1 NPs is due to the presence of ferrocene-derived prodrug 1, which can efficiently react with H2O2 to produce toxic ˙OH. The X-ray photoelectron spectroscopic (XPS) analyses of 1 confirmed the oxidation state of iron (Fe) as +2. The peaks observed at the binding energies of 707.3 and 720 eV correspond to Fe2+, which is responsible for the peroxidase-like catalytic activity of β-CD+/1 NPs (Fig. S5†). The catalytic activity of β-CD+/1 NPs was monitored by using the methylene blue (MB) degradation assay. β-CD+/1 NPs in the presence of H2O2 (10 mM) produce ˙OH, which can degrade MB, causing a decrease in the characteristic absorption peak of MB at 660 nm. As shown in Fig. 2a, excellent degradation of MB was observed for the β-CD+/1 NP (1 mM)-treated MB (15 μM) sample in the presence of H2O2 (10 mM) when compared to the corresponding β-CD+/1 NP-untreated MB samples. These observations are in accordance with our design strategy that the production of ˙OH occurs via the β-CD+/1 NP catalyzed Fenton reaction.
 | ||
| Fig. 2 (a) Comparison of the absorption spectra of MB (black trace), MB in the presence of H2O2 (red trace) and MB in the presence of H2O2 and β-CD+/1 NPs (blue trace). (b) DTNB assay for GSH depletion analyses: absorption spectral changes of DTNB in the presence of GSH (blue trace), β-CD+/1 NPs (green trace) and β-CD+/1 and GSH (yellow trace). (c) Colorimetric detection of Fe3+ released from β-CD+/1 NPs upon reaction with H2O2. Visual color changes of the NP solution upon formation of a red coloured [Fe(bipy)3]2+ complex in the presence and in the absence of H2O2 are also shown. (d) A plot of percentage of Fe3+ released by β-CD+/1 NPs in the presence and in the absence of H2O2. | ||
In addition to the generation of ˙OH, prodrug 1 of β-CD+/1 NPs also reacts with H2O2 to produce p-quinone methide, which can undergo 1,6-conjugate nucleophilic addition with GSH to form alkylated products.27 Hence, p-quinone methide is an efficient GSH scavenger, which is highly beneficial for the efficiency of CDT.27 Initially, the formation of p-quinone methide during the reaction between β-CD+/1 and H2O2 was confirmed by LR-MS analyses, which showed an m/z of 106.14 corresponding to the mass of p-quinone methide (Fig. S6†). The depletion of GSH concentration by the reaction with p-quinone methide was then monitored by using 5,5′-dithiobis[2-nitrobenzoic acid] (DTNB) as the indicator.32,33 DTNB has its characteristic absorption band centered at 312 nm and exhibits a red shift to 412 nm upon reaction with GSH, which is due to the formation of 2-nitro-5-thiobenzoic acid.34 In order to study GSH depletion by the NPs, GSH (25 μM) was incubated with β-CD+/1 NPs (25 μM), H2O2 (100 μM) and DTNB (10 μM) for 0.5 h, and the absorption spectral changes were then monitored. As a control experiment, the absorption spectrum of the reaction mixture containing GSH and DTNB without β-CD+/1 NPs and H2O2 was recorded. As expected, a red shift from 312 to 412 nm was observed for GSH treated with DTNB, indicating the reaction between GSH and DTNB to form 2-nitro-5-thiobenzoic acid, whereas no change in the absorption spectrum was observed for GSH treated DTNB in the presence of β-CD+/1 NPs and H2O2 (Fig. 2b). This clearly discloses that p-quinone methide formed by the reaction between β-CD+/1 NPs and H2O2 efficiently consumes GSH, and as a consequence of this reaction, no significant amount of GSH was available in the reaction medium for the subsequent reaction with DTNB. These results clearly confirm the GSH scavenging capability of β-CD+/1 NPs in the presence of H2O2.
The reaction of β-CD+/1 NPs with H2O2 also releases free Fe3+ ions in the solution, and the formation of free Fe3+ in the solution was subsequently characterized by using the standard bipyridine assay.27 This was achieved by reducing the released Fe3+ to Fe2+ by using sodium dithionate (Na2S2O6), which was then reacted with 2,2′-bipyridine (bipy) to form a red coloured [Fe(bipy)3]2+ complex (Fig. 2c). Accordingly, strong red coloured complex formation was observed for β-CD+/1 NPs (100 μM) in the presence of H2O2 (9.8 mM), Na2S2O6 (1 M) and bipy (300 μM). The release efficiency of Fe3+ was found to be 31.6% for the H2O2-treated β-CD+/1 NPs, whereas only negligible release of Fe3+ (2.6%) was observed for the H2O2-untreated β-CD+/1 NPs (Fig. 2d).27
After the characterization of the functional behaviours of the NPs, we studied the in vitro toxicity of the NPs inside the cancer cells. For the cellular imaging, FAM functionalized siRNA (siRNA-FAM) was used. For this purpose, initially, β-CD+/1/siRNA-FAM NPs (β-CD+, 1: 10 μM each and siRNA-FAM: 20 nM) were synthesized following the earlier protocol. HeLa cells were taken as a representative cell line in our study and incubated with siRNA-FAM and β-CD+/1/siRNA-FAM NPs for 6 h. As shown in Fig. 3a, no internalization was observed for free siRNA-FAM, whereas efficient internalization was observed for β-CD+/1/siRNA-FAM NPs as evident from the green fluorescence associated with the HeLa cells. Internalization of NPs was then quantified using fluorescence activated cell sorting (FACS) studies (Fig. 3b). A high mean fluorescence intensity (MFI) shift was observed in the β-CD+/1/siRNA-FAM NP-treated HeLa cells (575), when compared to the MFI shift of 116 in the siRNA-FAM-treated HeLa cells. Time-dependent CLSM analyses showed that internalization of β-CD+/1/siRNA-FAM NPs into the HeLa cells occurs within 3 h of incubation. In contrast, no internalization was observed for siRNA2-treated cells even after 6 h of incubation, as expected (Fig. S7†). In order to get a better insight into the kinetics of siRNA release from β-CD+/1/siRNA, time-dependent zeta potential analyses were carried out. β-CD+/1/siRNA NPs have a zeta potential value of +4.5 mV, and no noticeable change in the zeta potential value was observed with respect to time, and the value remains the same over a time period of 6 h (time required for complete cellular internalization). This clearly confirms the structural stability of the electrostatic interaction between negatively charged siRNA and positively charged β-CD+/1 with respect to time and also rules out any premature release of siRNA from the nanoformulation (Fig. S4†).
 | ||
| Fig. 3 (a) CLSM images demonstrating cellular internalization of siRNA-FAM (top panel) and β-CD+/1/siRNA-FAM (bottom panel) NPs on HeLa cells and (b) FACS analyses for the cellular internalization of siRNA-FAM (left) and β-CD+/1/siRNA-FAM (right) NPs. | ||
We next studied the pathway of cellular internalization of β-CD+/1/siRNA-FAM NPs (β-CD+, 1: 10 μM each and siRNA-FAM: 20 nM). For this purpose, HeLa cells were pre-treated with an endocytosis inhibitor, methyl-β-cyclodextrin (MβCD, 50 μM), which prevents cholesterol-dependent endocytic processes by reversibly extracting the steroid out of the plasma membrane.35 Interestingly, MβCD-pre-treated cells exhibited decreased cellular uptake of β-CD+/1/siRNA-FAM NPs (Fig. S8†). In addition to this, cellular internalization of β-CD+/1/siRNA-FAM NPs was found to be drastically reduced for the cells maintained at 4 °C (Fig. S8†). This is in accordance with the observation that endocytosis is a temperature-dependent process, and a very poor cellular internalization is expected at low temperatures. These results collectively conclude that β-CD+/1/siRNA-FAM NPs enter the HeLa cells via an endocytotic pathway.
Typically, any NPs enter a cell via an endocytotic pathway, reach the acidic lysosomes and get degraded at the lysosome.36–41 From a therapeutic point of view, degradation of the NPs at the lysosome can cause a drastic reduction in the therapeutic efficacy of the system. In order to monitor the lysosomal localization of β-CD+/1/siRNA-FAM NPs, colocalization experiments were performed. For this purpose, lysosomes of HeLa cells were initially stained with Lysotracker deep red (LysoTracker, red fluorescence) and its colocalization with the green fluorescence of β-CD+/1/siRNA-FAM NPs (β-CD+, 1: 10 μM each and siRNA-FAM: 20 nM) was monitored. Surprisingly, no colocalization of green fluorescence of β-CD+/1/siRNA-FAM NPs with the red fluorescence of LysoTracker deep red was observed, suggesting that though β-CD+/1/siRNA-FAM NPs enter the cell via the endocytotic pathway, they undergo lysosomal escape (Fig. 4a).39 In support of this observation, the colocalization plot (Fig. 4b) and line analysis (Fig. 4c) provided a relatively low Pearson coefficient value of 0.035. Though the exact reason for the lysosomal escape of the NPs is not clear to us at this stage, we believe that this could be due to the slightly positively charged surface (+4.89 as obtained from the zeta potential studies) of β-CD+/1/siRNA-FAM NPs. Nanoparticles with positively charged surfaces are known to favor endosomal membrane destabilization followed by lysosomal escape.42
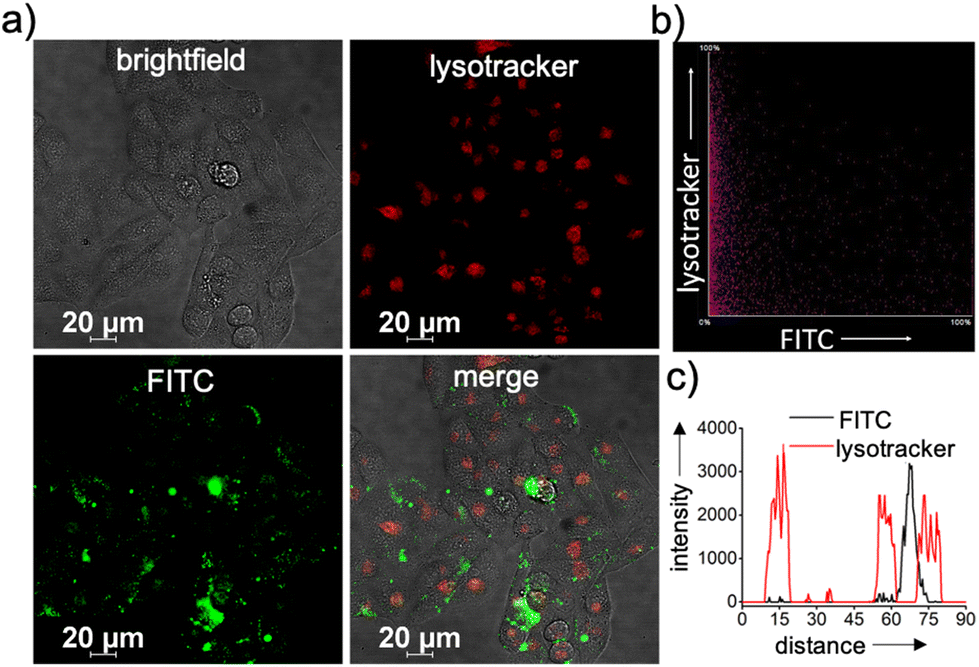 | ||
| Fig. 4 (a) CLSM images showing the lysosomal escape of β-CD+/1/siRNA-FAM NPs. The lysosomes of the cells were stained with Lysotracker deep red (red fluorescence). (b) The colocalization scatter plot and (c) the corresponding line analyses diagram. | ||
Subsequent to the lysosomal escape, β-CD+/1/siRNA NPs are mainly located in the cytosol of the cell, where they undergo disassembly upon reaction with H2O2 present inside the cells. This reaction also led to the in situ generation of quinone methide, which in turn reacts with GSH present in the cellular microenvironment and eventually depletes the GSH concentration. The GSH consumption ability of β-CD+/1/siRNA NPs was calculated using the standard GSH assay kit (from ORIGIN), which showed a 20% decrease in the intracellular GSH level compared to that of the untreated cells (Fig. S9†). The Fenton reaction of the NPs with endogenous H2O2 produces ˙OH and also leads to the release of MTH1 siRNA (siRNA). The generation of ˙OH was monitored by using 2,7-dichlorofluorescein-diacetate (DCFH-DA) as a fluorescent indicator (10 μM), which produces green fluorescent 2,7-dichlorofluorescein (DCF) upon radical detection. As shown in Fig. 5a, the β-CD+/1/siRNA NP-treated (β-CD+, 1: 10 μM each and siRNA: 20 nM) HeLa cells showed high-intensity DCF green fluorescence compared to the untreated control cells. This was further quantified by FACS analyses, which revealed a high MFI shift value for the β-CD+/1/siRNA NP-treated cells (225) when compared to the corresponding control cells (90) (Fig. 5b). These results unequivocally confirm the generation of ˙OH by the reaction of β-CD+/1/siRNA NPs with intracellular H2O2. In order to validate that the radical generation is indeed via the reaction of the NPs with H2O2, cells were pre-incubated with the H2O2 scavenger N-acetyl-L-cysteine (NAC) and then treated with β-CD+/1/siRNA NPs. As expected, a significant decrease in the intensity of fluorescence of DCF was observed in the NAC-treated cells when compared to the corresponding NAC-untreated control cells (Fig. 5c). In accordance with the CLSM analyses, FACS analyses also revealed a significant reduction in the MFI shift value of the NAC-treated cells (97), whereas a relatively high MFI shift value (297) was observed in the NAC-untreated cells (Fig. 5d). These results confirm that the intracellular radical production indeed occurs via the reaction of β-CD+/1/siRNA NPs with the endogenous H2O2 present in the cancer cells.
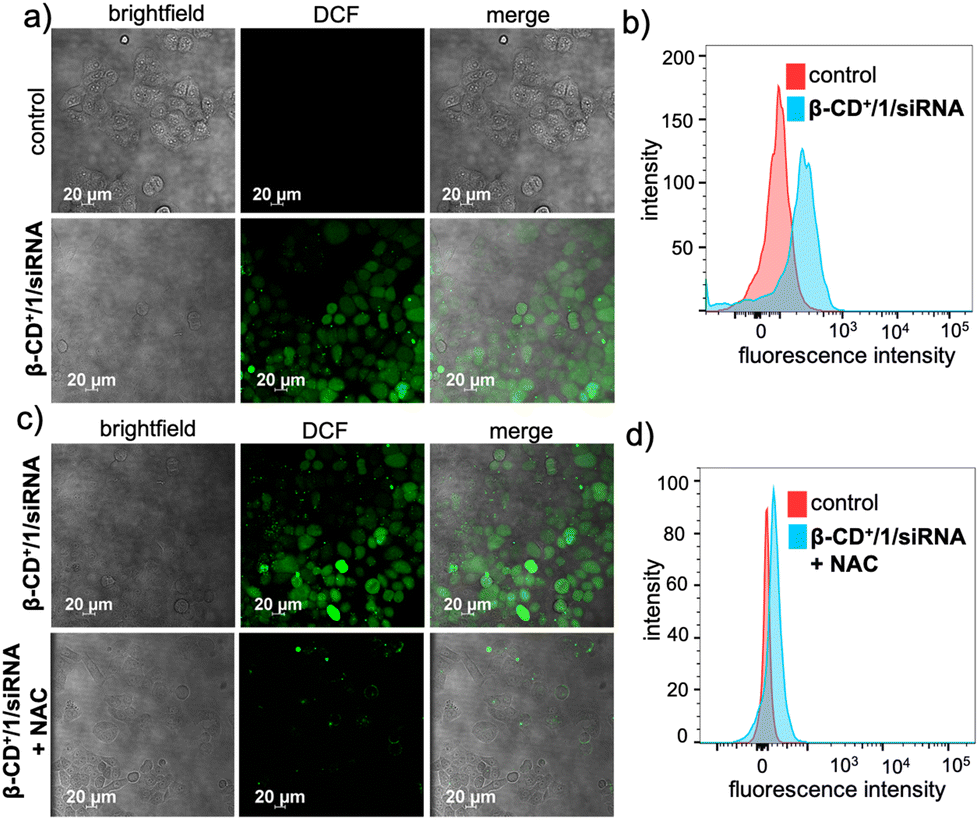 | ||
| Fig. 5 (a) CLSM images for ˙OH generation by β-CD+/1/siRNA NPs inside the HeLa cells monitored using the DCFH-DA assay and (b) the corresponding FACS analyses. (c) Comparison of CLSM images for ˙OH generation by β-CD+/1/siRNA NPs in the HeLa cells pre-treated with the H2O2 scavenger N-acetyl-L-cysteine (NAC) and the corresponding NAC-untreated cells. (d) FACS analyses for β-CD+/1/siRNA NPs in HeLa cells pre-treated with NAC. | ||
We next evaluated whether the radicals generated by β-CD+/1/siRNA NPs cause any damage to the cellular organelles including mitochondria and lysosomes. This is because the lysosomal and mitochondrial membranes are highly susceptible to radical attacks and hence can potentially be damaged by the reaction with radical species. In order to evaluate the lysosomal damage, an acridine orange (AO) assay was carried out. Acridine orange is typically used to stain lysosomes and produce red fluorescence only if the lysosomes are healthy. If the lysosomes are damaged or ruptured, a significant decrease in the red fluorescence of AO is expected. At the same time, AO can also stain the nucleus of the cells to give green fluorescence. The CLSM analyses of the β-CD+/1/siRNA NP-treated (β-CD+ and 1: 500 μM and siRNA: 1 μM) HeLa cells showed only negligible decrease in the red fluorescence of AO when compared to the untreated control cells, indicating that the lysosomes of the β-CD+/1/siRNA NP-treated cells are healthy (Fig. 6a). The FACS analyses were also in good agreement with the CLSM analyses (Fig. 6b), which revealed comparable MFI shift values of the β-CD+/1/siRNA NP-treated (340) and the corresponding untreated cells (344). This is in line with the previous observation that the NPs undergo lysosomal escape, and hence, the radical generation at the lysosomal compartments is minimal. Similarly, the mitochondrial damage caused by ˙OH was evaluated using the tetramethylrhodamine methyl ester perchlorate (TMRM) assay. For this, the β-CD+/1/siRNA NP-treated HeLa cells were stained with TMRM and the fluorescence of TMRM was monitored using CLSM analyses. TMRM exhibits orange fluorescence when accumulated in negatively charged polarized mitochondria. When the mitochondrial membrane potential collapses, the TMRM reagent is dispersed throughout the cell cytosol and the fluorescence intensity drops down dramatically. The CLSM analyses of β-CD+/1/siRNA (β-CD+ and 1: 500 μM and siRNA: 1 μM) NP-treated HeLa cells show no noticeable decrease in the fluorescence intensity in the β-CD+/1/siRNA NP-treated cells when compared to the untreated control cells, indicating the healthy nature of the mitochondria (Fig. 6c). These observations were further quantified using FACS analyses, which revealed comparable MFI shift values are associated with the β-CD+/1/siRNA1 NP-treated (18722) and untreated (18933) cells (Fig. 6d). In this case as well, the mitochondria stability toward the NPs can be attributed to the inability of the NPs to accumulate at the mitochondria. These data collectively conclude that ˙OH generated by the reaction of β-CD+/1/siRNA NPs with H2O2 does not cause any major damage to the lysosomes and the mitochondria.
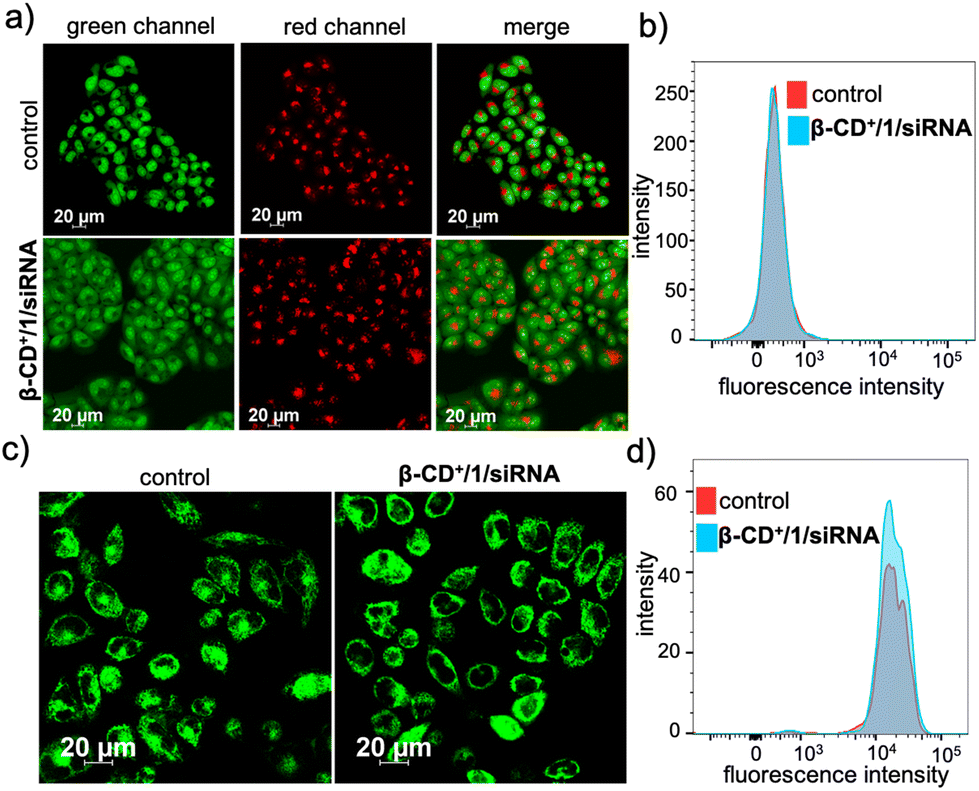 | ||
| Fig. 6 (a) CLSM images to evaluate the lysosomal degradation of β-CD+/1/siRNA NPs via the generation of ˙OH monitored by using the AO assay and (b) the corresponding FACS analyses. (c) CLSM images of TMRM assay for monitoring mitochondrial damage induced by β-CD+/1/siRNA NPs and (d) the corresponding FACS analyses. | ||
Having established the cellular internalization of β-CD+/1/siRNA NPs, cytotoxicity of the NPs towards HeLa cells was evaluated using the MTT assay. Initially, HeLa cells were incubated with siRNA (5–250 nM) and β-CD+ (50–500 μM) of varying concentrations for 24 h. Both siRNA and β-CD+ were found to be nontoxic to the cells in all the concentrations. Even at higher concentrations of siRNA (250 nM) and β-CD+ (500 μM), no significant cytotoxicity was observed and the cell viability remained more than 95% (Fig. 7a and b). The nontoxic nature of siRNA can be attributed to its cell impermeable nature, whereas β-CD+ is known to be highly biocompatible.41 Because prodrug 1 is water insoluble, the cytotoxicity of 1 alone in the cell media could not be studied. Subsequently, we have carried out cytotoxicity studies for β-CD+/1 and β-CD+/1/siRNA NPs to understand the effect of siRNA assisted enhanced cytotoxicity of the NPs. For this purpose, HeLa cells were incubated with β-CD+/1 and β-CD+/1/siRNA NPs having varying concentrations from 50 to 500 μM for 24 h. Interestingly, we could observe enhanced cytotoxicity for β-CD+/1/siRNA NPs compared to β-CD+/1 NPs for all the concentrations (Fig. 7c). An excellent cytotoxicity of 85% was observed for β-CD+/1/siRNA NPs at a concentration of 500 μM. This enhanced cytotoxicity can be attributed to the suppression of MTH1 protein synthesis with the delivery of siRNA, which in turn enhances the CDT performance of the β-CD+/1/siRNA NPs.11 Inhibition of MTH1 protein synthesis with the delivery of siRNA is not possible in the case of β-CD+/1 NPs.
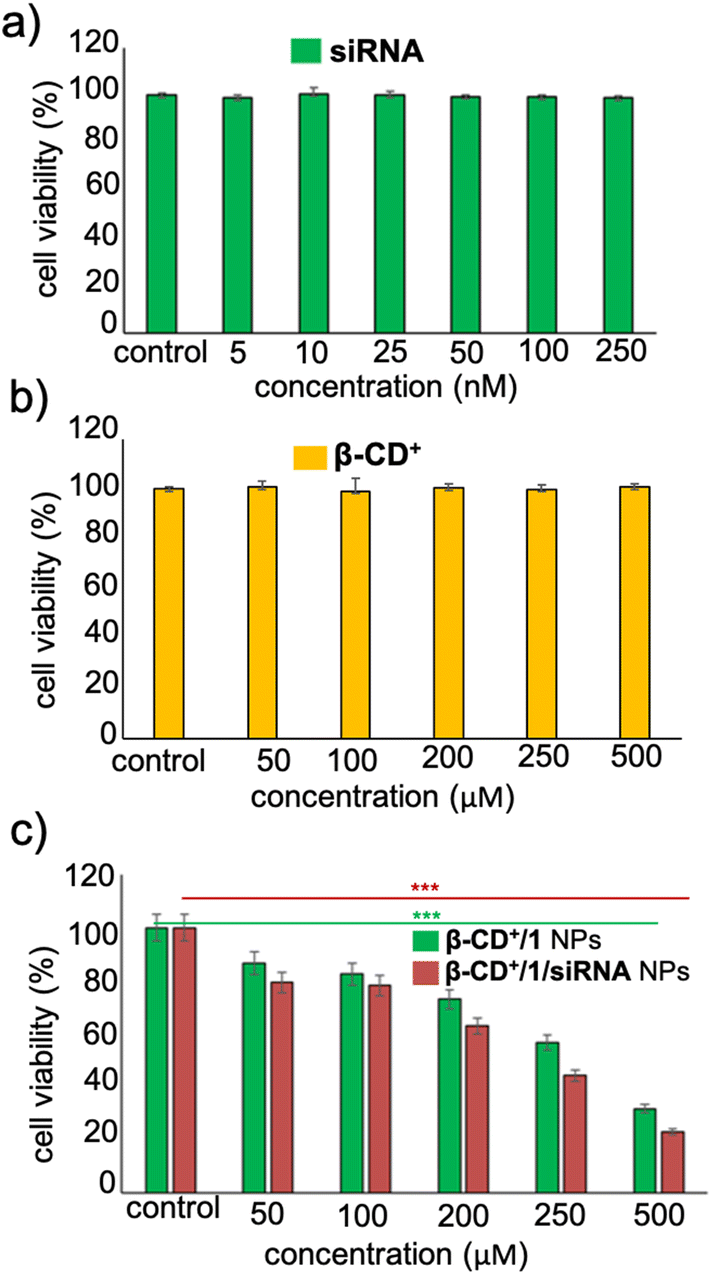 | ||
| Fig. 7 Cell viability assay (MTT) of the HeLa cells (a) with varying concentrations of siRNA and (b) β-CD+. (c) Comparison of cell viability of the HeLa cells treated with β-CD+/1 and β-CD+/1/siRNA NPs. | ||
Subsequently, the cytotoxicity of β-CD+/1/siRNA NPs was qualitatively analysed using CLSM analyses by using calcein-AM/ethidium homodimer-1 co-staining assay. Calcein-AM interacts with the intracellular esterase present in the live cells and cleaves the acetoxymethyl ester of calcein-AM to produce a green fluorescent calcein dye. However, ester cleavage cannot occur in dead cells due to the absence of cellular esterase. Similarly, propidium iodide (PI) cannot pass through the cell wall membrane of live cells, whereas it can stain the dead cells to give red fluorescence.43 As shown in Fig. 8a, untreated HeLa cells (control) showed calcein-AM staining (green fluorescence) but no red fluorescence of PI, indicating that the cells are alive, as expected. On the other hand, cells treated with β-CD+/1/siRNA (β-CD+ and 1: 500 μM and siRNA: 1 μM) NPs showed strong red fluorescence of PI, but negligible green fluorescence from calcein dye, revealing that most of the cells are dead. Finally, in order to understand the mechanism of cell death, an annexin V-FITC (AV)/PI assay was performed. As shown in Fig. 8b, the β-CD+/1/siRNA NP-treated cells (β-CD+ and 1: 500 μM and siRNA: 1 μM) exhibited strong green fluorescence of annexin V-FITC at the cell membrane and intense red fluorescence of PI from the nuclei, indicating that cell death occurs mainly via the late-stage apoptotic pathway.44,45 We confirmed this cell death mechanism using the annexin V-FITC (AV)/PI assay through FACS analyses. For this purpose, HeLa cells were treated with β-CD+/1/siRNA NPs for 24 h and analysed by flow cytometry after staining with annexin V-FITC (AV)/PI. After the treatment of the cells with β-CD+/1/siRNA NPs, the cell population mainly shifted to the quadrant that corresponds to the apoptotic pathway (Fig. 8c). These results collectively conclude that the β-CD+/1/siRNA NP-induced cell death is due to the apoptotic pathway and not due to the necrotic mechanism.
 | ||
| Fig. 8 (a) CLSM images for the live/dead cell assay of the β-CD+/1/siRNA NP-treated HeLa cells using calcein-AM/PI. (b) CLSM images for the Annexin V-FITC/propidium iodide assay of the β-CD+/1/siRNA NP-treated HeLa cells. FACS analyses of the Annexin V-FITC (AV)/propidium iodide (PI) assay of (c) the control and (d) β-CD+/1/siRNA NP-treated HeLa cells. | ||
After demonstrating the efficient CDT reaction in vitro, we then evaluated the efficiency of β-CD+/1/siRNA NPs in a multicellular tumor spheroid model using 3D cell culture. The 3D spheroids have most of the characteristics of proliferating tumor cells, and this model typically represents the in vivo conditions. Details of the synthesis of 3D spheroids are provided in the ESI.† In our study, triple-negative human breast cancer cells, MDA-MB-231, were used for the preparation of tumor 3D spheroids using the hanging drop method. The invasion potential of the spheroids was studied in the presence of β-CD+/1/siRNA NPs ([β-CD+] and [1] = 500 μM and [siRNA] = 1 μM) to understand the therapeutic efficacy of the NPs in the proliferation of the spheroids. For this, 3D spheroids were incubated with β-CD+/1/siRNA NPs at 37 °C for 24 h. Untreated spheroids were taken as control that showed a high invasion potential as they showed migration in all directions (invasion index = 1.54 ± 0.3). On the other hand, a significant reduction in the invasion index was observed for the β-CD+/1/siRNA NP-treated spheroids (invasion index = 0) due to the excellent therapeutic action of the NPs (Fig. 9). The results conclude the excellent therapeutic efficiency of the nanoformulation both in vitro and in vivo.
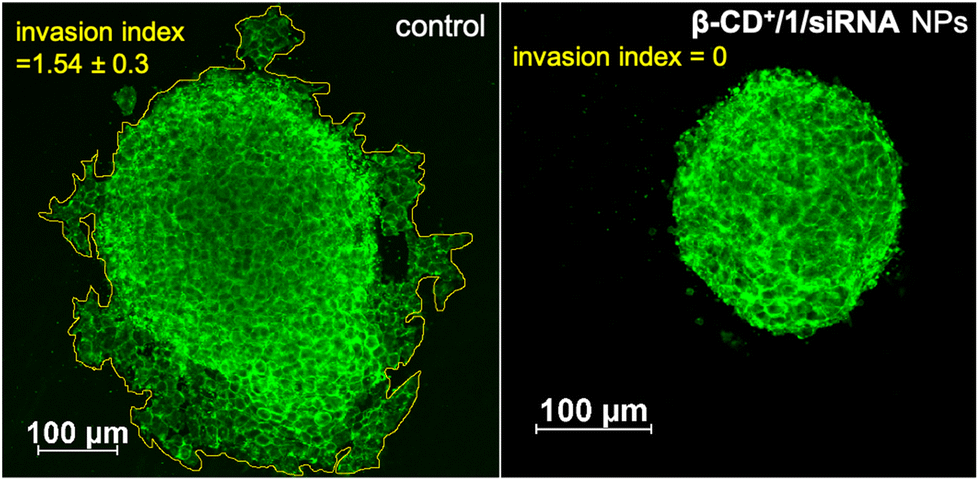 | ||
| Fig. 9 CLSM images of the β-CD+/1/siRNA NP-treated MDA-MB-231 3D spheroids demonstrating the anti-invasive effect of the CDT agent (right) and the untreated control spheroids are also shown (left). | ||
Conclusions
In summary, we have demonstrated a simple, one-step supramolecular approach for the design of a multifunctional nanoformulation for the co-delivery of MTH1 siRNA and a Fenton reagent (Fe2+) into a cancer cell. Our strategy permits the simultaneous delivery of MTH1 siRNA and a potential Fenton agent in a highly efficient manner, which is otherwise extremely difficult to achieve. A dramatic increase in the overall efficacy of CDT for cancer therapy was achieved due to the combined effect of the delivery of MTH1 siRNA (downregulating the expression of the MTH1 protein), generation of ˙OH through the Fenton reaction (CDT) and depletion of GSH by the production of an antioxidant scavenger, p-quinone methide (enhancing CDT). To the best of our knowledge, this is the first report demonstrating a supramolecular strategy for the codelivery of MTH1 siRNA and a Fenton agent in a one-step approach for enhanced CDT for cancer therapy. We strongly believe that the ease of fabrication of the nanoformulation via one-pot self-assembly, scalability of the nanoformulation without laborious synthesis and excellent CDT offer a wide range of scope for CDT-based cancer therapy.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The financial support from SERB (CRG/2022/002612) is gratefully acknowledged. The help of Sarika Mohan S. for the FACS analyses is acknowledged.References
- Z. Tang, Y. Liu, M. He and W. Bu, Angew. Chem., Int. Ed., 2019, 58, 946–956 CrossRef CAS PubMed.
- C. Jia, Y. Guo and F.-G. Wu, Small, 2022, 18, 2103868 CrossRef CAS PubMed.
- Y. Li, W. Xiu, K. Yang, Q. Wen, L. Yuwen, Z. Luo, X. Liu, D. Yang, X. Xie and L. Wang, Mater. Horiz., 2021, 8, 1264–1271 RSC.
- D. Mao, F. Hu, Z. Yi, S. Xu, S. Yan, Z. Luo, W. Wu, Z. Wang, D. Kong, X. Liu and B. Liu, Sci. Adv., 2020, 6, eabb2712 CrossRef CAS PubMed.
- H. Lin, Y. Chen and J. Shi, Chem. Soc. Rev., 2018, 47, 1938–1958 RSC.
- C. Cao, X. Wang, N. Yang, X. Song and X. Dong, Chem. Sci., 2022, 13, 863–889 RSC.
- G. Raj, V. K. K. Dommeti, V. Pradeep, A. K. Veetil and R. Varghese, Mater. Chem. Front., 2022, 6, 1533–1542 RSC.
- P. T. Sujai, S. Shamjith, M. M. Joseph and K. K. Maiti, ACS Appl. Bio Mater., 2021, 4, 4962–4972 CrossRef CAS PubMed.
- L. A. McPherson, C. I. Troccoli, D. Ji, A. E. Bowles, M. L. Gardiner, M. G. Mohsen, N. S. Nagathihalli, D. M. Nguyen, D. J. Robbins, N. B. Merchant, E. T. Kool, P. Rai and J. M. Ford, DNA Repair, 2019, 83, 102644 CrossRef CAS PubMed.
- K. V. M. Huber, E. Salah, B. Radic, M. Gridling, J. M. Elkins, A. Stukalov, A.-S. Jemth, C. Göktürk, K. Sanjiv, K. Strömberg, T. Pham, U. W. Berglund, J. Colinge, K. L. Bennett, J. I. Loizou, T. Helleday, S. Knapp and G. Superti-Furga, Nature, 2014, 508, 222–227 CrossRef CAS PubMed.
- H. Fan, L. Zhang, X. Hu, Z. Zhao, H. Bai, X. Fu, G. Yan, L.-H. Liang, X.-B. Zhang and W. Tan, Chem. Commun., 2018, 54, 4310–4313 RSC.
- W. Gao, W. Cao, Y. Sun, X. Wei, K. Xu, H. Zhang and B. Tang, Biomaterials, 2015, 69, 212–221 CrossRef CAS PubMed.
- D. Ji, A. A. Beharry, J. M. Ford and E. T. Kool, J. Am. Chem. Soc., 2016, 138, 9005–9008 CrossRef CAS PubMed.
- J. Liu, L. Song, S. Liu, S. Zhao, Q. Jiang and B. Ding, Angew. Chem., Int. Ed., 2018, 57, 15486–15490 CrossRef CAS PubMed.
- L. A. L. Fliervoet, H. Zhang, E. van Groesen, K. Fortuin, N. J. C. B. Duin, K. Remaut, R. M. Schiffelers, W. E. Hennink and T. Vermonden, Nanoscale, 2020, 12, 10347–10360 RSC.
- C. Ornelas-Megiatto, P. R. Wich and J. M. J. Fréchet, J. Am. Chem. Soc., 2012, 134, 1902–1905 CrossRef CAS PubMed.
- Y.-M. Kim, M.-R. Park and S.-C. Song, ACS Nano, 2012, 6, 5757–5766 CrossRef CAS PubMed.
- X. Liao, N. D. Falcon, A. A. Mohammed, Y. Z. Paterson, A. G. Mayes, D. J. Guest and A. Saeed, ACS Omega, 2020, 5, 1496–1505 CrossRef CAS PubMed.
- A. A. Foster, C. T. Greco, M. D. Green, T. H. Epps III and M. O. Sullivan, Adv. Healthcare Mater., 2015, 4, 760–770 CrossRef CAS PubMed.
- C. Tong, J. A. J. Wondergem, M. van den Brink, M. C. Kwakernaak, Y. Chen, M. M. R. M. Hendrix, I. K. Voets, E. H. J. Danen, S. Le Dévédec, D. Heinrich and R. E. Kieltyka, ACS Appl. Mater. Interfaces, 2022, 14, 17042–17054 CrossRef CAS PubMed.
- A. Kasprzak, M. Koszytkowska-Stawińska, A. M. Nowicka, W. Buchowicz and M. Poplawska, J. Org. Chem., 2019, 84, 15900–15914 CrossRef CAS PubMed.
- T. Auletta, M. R. de Jong, A. Mulder, F. C. J. M. van Veggel, J. Huskens, D. N. Reinhoudt, S. Zou, S. Zapotoczny, H. Schönherr, G. J. Vancso and L. Kuipers, J. Am. Chem. Soc., 2004, 126, 1577–1584 CrossRef CAS PubMed.
- M. Fathalla, A. Neuberger, S.-C. Li, R. Schmehl, U. Diebold and J. Jayawickramarajah, J. Am. Chem. Soc., 2010, 132, 9966–9967 CrossRef CAS PubMed.
- F. Wang, H. Pu and X. Che, Chem. Commun., 2016, 52, 3516–3519 RSC.
- S. Xie, J. Zhang, Y. Yuan, Y. Chai and R. Yuan, Chem. Commun., 2015, 51, 3387–3390 RSC.
- A. Harada, R. Kobayashi, Y. Takashima, A. Hashidzume and H. Yamaguchi, Nat. Chem., 2011, 3, 34–37 CrossRef CAS PubMed.
- H. Hagen, P. Marzenell, E. Jentzsch, F. Wenz, M. R. Veldwijk and A. Mokhir, J. Med. Chem., 2012, 55, 924–934 CrossRef CAS PubMed.
- S. K. Albert, H. V. P. Thelu, M. Golla, N. Krishnan and R. Varghese, Nanoscale, 2017, 9, 5425–5432 RSC.
- W. Tang and S.-C. Ng, Nat. Protoc., 2008, 3, 691–697 CrossRef CAS PubMed.
- S. Xie, J. Zhang, Y. Yuan, Y. Chai and R. Yuan, Chem. Commun., 2015, 51, 3387–3390 RSC.
- T. Auletta, M. R. de Jong, A. Mulder, F. C. J. M. van Veggel, J. Huskens, D. N. Reinhoudt, S. Zou, S. Zapotoczny, H. Schönherr, G. J. Vancso and L. Kuipers, J. Am. Chem. Soc., 2004, 126, 1577–1584 CrossRef CAS PubMed.
- C. Liu, S. Jia, L. Tu, P. Yang, Y. Wang, S. Ke, W. Shi and S. Ye, ACS Biomater. Sci. Eng., 2022, 8, 1942–1955 CrossRef CAS PubMed.
- Q. Jiang, M. Pan, J. Hu, J. Sun, L. Fan, Z. Zou, J. Wei, X. Yang and X. Liu, Chem. Sci., 2021, 12, 148–157 RSC.
- X. Fan, B. Chen, H. Xu, A. Pan, S. Liang, S. Tan and Y. He, Chem. Mater., 2023, 35, 3124–3137 CrossRef CAS.
- J. Zhou, L. Sun, L. Wang, Y. Liu, J. Li, J. Li, J. Li and H. Yang, Angew. Chem., Int. Ed., 2019, 58, 5236–5240 CrossRef CAS PubMed.
- K. Leung, K. Chakraborty, A. Saminathan and Y. Krishnan, Nat. Nanotechnol., 2019, 14, 176–183 CrossRef CAS PubMed.
- S. Walia, V. Morya, A. Gangrade, S. Naskar, A. Guduru Teja, S. Dalvi, P. K. Maiti, C. Ghoroi and D. Bhatia, ACS Biomater. Sci. Eng., 2021, 7, 5933–5942 CrossRef CAS PubMed.
- U. Singh, A. G. Teja, S. Walia, P. Vaswani, S. Dalvi and D. Bhatia, Nanoscale Adv., 2022, 4, 1375–1386 RSC.
- P. Kanjilal, K. Dutta and S. Thayumanavan, Angew. Chem., Int. Ed., 2022, 61, e202209227 CrossRef CAS PubMed.
- F. Anson, B. Liu, P. Kanjilal, P. Wu, J. A. Hardy and S. Thayumanavan, Biomacromolecules, 2021, 22, 1261–1272 CrossRef CAS PubMed.
- O. P. Oommen, C. Duehrkop, B. Nilsson, J. Hilborn and O. P. Varghese, ACS Appl. Mater. Interfaces, 2016, 8, 20614–20624 CrossRef CAS PubMed.
- T. Bus, A. Traeger and U. S. Schubert, J. Mater. Chem. B, 2018, 6, 6904–6918 RSC.
- A. Tamura, K. Nishida, S. Zhang, T. W. Kang, A. Tonegawa and N. Yui, ACS Biomater. Sci. Eng., 2022, 8, 2463–2476 CrossRef CAS PubMed.
- K. Liu, Z. Jiang, R. A. Lalancette, X. Tang and F. Jäkle, J. Am. Chem. Soc., 2022, 144, 18908–18917 CrossRef CAS PubMed.
- S. Hameed, P. Bhattarai, Z. Gong, X. Liang, X. Yue and Z. Dai, Nanoscale Adv., 2023, 5, 277–289 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2259673. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3nr06071c |
| This journal is © The Royal Society of Chemistry 2024 |