
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Active sites and mechanisms in the direct conversion of methane to methanol using Cu in zeolitic hosts: a critical examination
Mark A.
Newton
 a,
Amy J.
Knorpp
a,
Vitaly L.
Sushkevich
b,
Dennis
Palagin
b and
Jeroen A.
van Bokhoven
*ab
a,
Amy J.
Knorpp
a,
Vitaly L.
Sushkevich
b,
Dennis
Palagin
b and
Jeroen A.
van Bokhoven
*ab
aInstitute for Chemical and Bioengineering, ETH Zurich, 8093 Zürich, Switzerland. E-mail: jeroen.vanbokhoven@chem.ethz.ch
bLaboratory for Catalysis and Sustainable Chemistry, Paul Scherrer Institute, Forschungsstrasse 111, 5232 Villigen, Switzerland
First published on 28th February 2020
Abstract
In this critical review we examine the current state of our knowledge in respect of the nature of the active sites in copper containing zeolites for the selective conversion of methane to methanol. We consider the varied experimental evidence arising from the application of X-ray diffraction, and vibrational, electronic, and X-ray spectroscopies that exist, along with the results of theory. We aim to establish both what is known regarding these elusive materials and how they function, and also where gaps in our knowledge still exist, and offer suggestions and strategies as to how these might be closed such that the rational design of more effective and efficient materials of this type for the selective conversion of methane might proceed further.
 Mark A. Newton | Mark Newton is a graduate of the University of Liverpool, UK where he also obtained his PhD in surface science and catalysis, under the supervision of Prof. M. Bowker. He has worked at the University of Washington (Prof. C. T. Campbell), Seattle, USA, the University of Southampton, UK, (Profs J. Evans and B. E. Hayden), and the European Synchrotron Radiation Facility (ESRF, beamline scientist ID24/BM23), Grenoble, France. Since 2017 he has worked at the Department of Chemistry and Applied Biosciences, ETH, Zurich under the auspices of Prof. J. A. van Bokhoven and Prof. C. Coperet. |
 Amy J. Knorpp | Amy Knorpp, originally from the panhandle of Texas, received a BA in Chemistry (2008) from Colorado College, a BS in Earth and Environmental Engineering (2010) from Columbia University, and a MS in Civil and Environmental Engineering (2012) from Stanford University. She is currently a PhD candidate in the Department of Chemical and Biological Applied Engineering at ETH Zurich. Her current research interests are focused on zeolite synthesis and their performance in chemical conversion processes. |
 Vitaly L. Sushkevich | Vitaly Sushkevich received his PhD from Lomonosov Moscow State University in 2013, where he was involved in development of novel processes for the synthesis of monomers for synthetic rubbers over heterogeneous catalysts. Since 2019 he is Scientist in Paul Scherrer Institute, working on the selective methane activation problem. His research philosophy implies the combination of kinetic and spectroscopic tools under operando conditions to study the reaction mechanisms and active sites of solid catalysts. |
 Dennis Palagin | Dennis Palagin received his PhD from Technical University of Munich in 2013, where he was working on cluster-assembled materials. He then moved to the University of Oxford to learn the arts of global optimisation and college tutoring, before joining the Paul Scherrer Institute in 2016 to dedicate himself to computational catalysis. Dennis is currently leading the Computational Catalysis Project (https://www.psi.ch/lsk/theocat-project), focused on applying state-of-the-art ab initio methods to study the interplay between the structure and the chemical properties of novel catalytic materials. |
 Jeroen A. van Bokhoven | Jeroen A. van Bokhoven completed a degree in chemistry at Utrecht University (Netherlands) in 1995 and went on to obtain a PhD in inorganic chemistry and catalysis from the same university in 2000 (with honours). From 1999 until 2002 he was head of the XAS (X-ray absorption spectroscopy) users – support group at Utrecht University. In 2002, he moved to the ETH, where he worked as a Postdoc in the group of Professor Prins. In 2006 he obtained an SNF assistant professorship in the Department of Chemistry and Applied Biology. He was the 2008 recipient of the Swiss Chemical Society Werner Prize. Since 2010, Jeroen A. van Bokhoven has a Chair in Heterogeneous Catalysis at the Institute for Chemical and Bioengineering at ETH Zurich and is Head of Laboratory for Catalysis and Sustainable Chemistry at Paul Scherrer Institute. |
1. Introduction
How one might make better use of methane, the principal component of natural gas and a globally abundant but often wasted energy resource, has been, and continues to be, an intensive area of research across numerous disciplines. As a result, much has been written beforehand regarding this subject.1–10 Consequently, the details of the economic, environmental, and industrial drivers for the pursuit of a means to effectively, and selectively, convert methane to bulk chemicals of greater value and utility will not be reiterated here; for these the interested reader is directed to the many excellent reviews that have appeared previously.4–10Suffice to say that methanol is one of the most important foundation products of the chemical industry. Commercially, methanol has for a long time been founded upon the use of copper/zinc oxide/alumina catalyst formulations that catalyse methanol synthesis from syngas, a mixture of carbon monoxide and hydrogen. This process requires both elevated temperatures and pressures and the syngas is required to be synthesised beforehand from a variety of sources (e.g. natural gas, coal, biomass, and hydrocarbons).11,12 More recently there has been a great deal of interest in deriving more direct methods to produce methanol either from hydrogenation of carbon dioxide13 or directly from methane.
The selective conversion of methane to methanol presents a considerable challenge. Thermodynamically, the products of over-oxidation – e.g. carbon monoxide and carbon dioxide – are highly favoured compared to those that arise from partial oxidation, such as methanol and dimethylether (vide infra); and kinetically, methane oxidation is slower than the reactions that may subsequently lead to over-oxidation of the methanol. Obtaining high selectivity and high conversion is, therefore, a decidedly uphill battle and subject to implacable limitations.5,14–17 Nature has, however, to a degree to which chemists may currently only aspire to, resolved this challenge. Enzymes founded upon iron and copper centres (methane monooxygenases) are able to selectively convert methane to methanol under physiological conditions, i.e. ambient temperature, pressure, and in the presence of significant hydration.18,19
Amongst the various materials and chemical systems – of which there are many15,20–22 – that have been demonstrated to be able to convert methane to higher value partial oxidation products, such as methanol and dimethylether, are those based upon the exchange of copper and iron ions into microporous zeolitic hosts. Zeolites are microporous crystalline alumino-silicates that can take up a myriad of three-dimensional topologies. These topologies are founded upon the wide array of ring structures that can result from condensing aluminium and silicon as oxides around an equally wide range of structure-directing materials.23,24 As such, they can provide local coordination environments for host cations, such as copper and iron, in a manner that is, conceptually if not chemically, similar to the highly specific tertiary environments that metallo-enzymes create around well-defined metal-containing sites; a similitude that has often been noted.18,19,25
Since the first demonstration that copper hosted in zeolites could convert methane to methanol,26 considerable strides have been taken in improving the yield of this conversion. Fig. 1 illustrates this development in terms of some of the variables that these materials present. Fig. 1(a) charts the evolution of methanol yields achieved per gram of material used. Fig. 1(b) presents the methanol yield per total copper, and Fig. 1(c) shows how, from the available literature, the yield of methanol per gram of material varies as a function of Si/Al ratio for four zeolitic systems: ZSM-5 (MFI), chabasite (CHA), mordenite (MOR) and mazzite (MAZ).
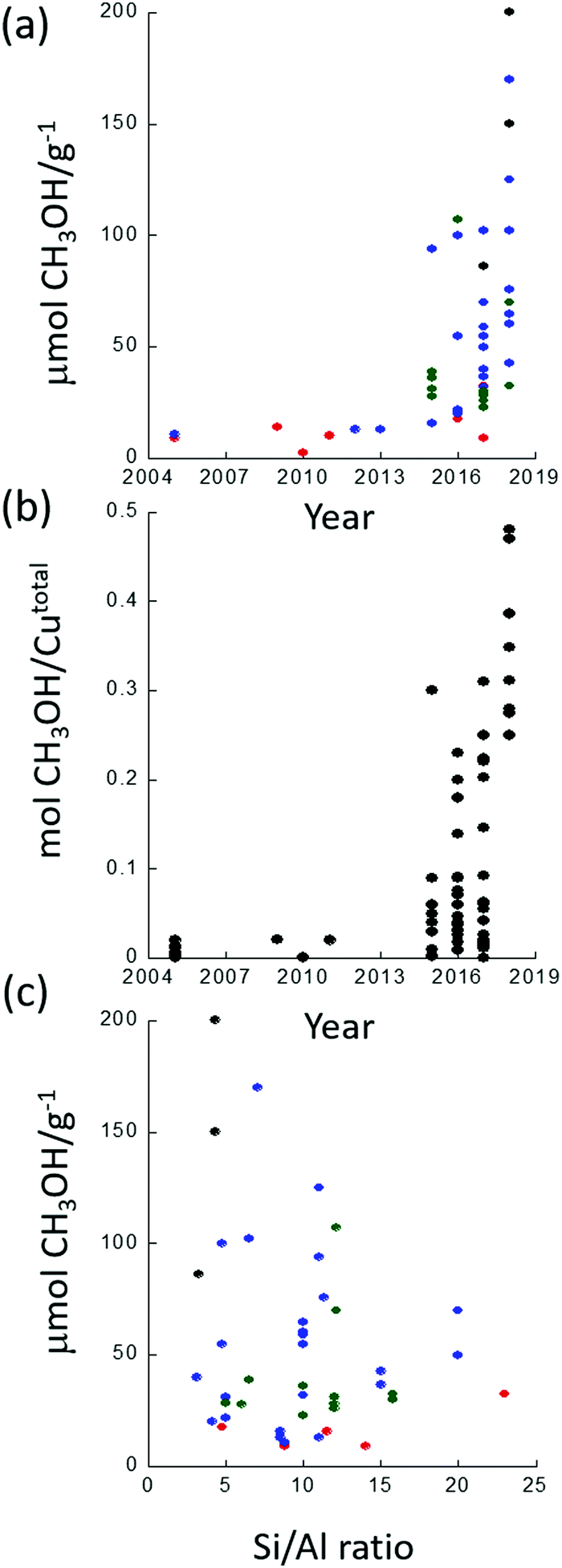 | ||
| Fig. 1 (a) Evolution of reported methanol yields demonstrated since 2005 for single reactive cycles; (b) evolution of the reported methanol yield as a fraction of the total copper content for single reactive cycles; (c) literature variation in methanol yield as a function of the Si/Al ratio reported. In (a) and (c): red = Cu/ZSM5, blue = Cu/MOR, green = Cu/CHA, and black = Cu/MAZ. | ||
Fig. 1(a) shows that methanol yields as high as 200 μmol g−1 in a single reactive cycle27 have now been demonstrated whereas yields were initially much (over an order of magnitude) lower (6–10 μmol g−1).26,28 Even more importantly, selectivity toward methanol of >80% is now regularly reported for a range of different Cu/zeolite systems.27,29–31 Moreover, in terms of the efficiency (Fig. 1(b)) of the use of the copper (i.e. how much copper is active toward the conversion of methane to methanol) significant strides have also been made and a value as high as 0.5 methanol per copper atom has been reported.31,32
Whilst none of these representations are perfect, these figures serve to make clear that there exists a considerable variation in performance and, (e.g.Fig. 1(c)), a scatter to potentially important structure function relationships that is considerably greater than ideally desired.
Therefore, though the trend is very much toward more active and efficient materials, clear indications as to how to optimise these materials in terms of their structure and composition are still lacking in many ways. Moreover, there remains considerable debate surrounding the basic mechanisms and active sites at work present in these materials. Concomitantly, a logical framework that might serve as a foundation for the rational optimisation and design of these materials might still be considered in its infancy; though, as we hope to delineate, it is growing up rapidly.
This review is, therefore, dedicated to examining what we know about the structure of copper hosted in zeolites and how the selective conversion of methane to methanol is mediated, how we come to this understanding, and what areas we do not yet have an adequate understanding of. We shall critically compare competing ideas regarding the active sites, and discuss the fundamental nature of the basic chemical considerations – copper redox, oxyl radical formation, the nuclearity of the active sites, the role of both zeolite topology and aluminium content, and sensitivity to overall reaction conditions – that must come together to affect this challenging conversion. In doing so, and concentrating on systems based upon mordenite (MOR) and chabasite (CHA), for which the most extensive studies exist, we examine the experimental evidence put forward for the various propositions that have come to the fore in recent years. We shall also critically investigate the concurrences and differences that exist between experiment and theory and to attempt to distil what we actually know regarding fundamental aspects of this conversion. From this foundation we shall put into perspective where our knowledge is still lacking, and suggest means by which these deficits might be resolved, such that functional systems superior to those that exist now are discovered, to further improve routes to selective oxidation of methane beyond what has been achieved in the last fifteen years.
2. Processes, active sites, mechanisms, zeolite composition and topology: experiment and theory
The ability to convert methane to methanol in a selective manner has its origins in a capacity to stabilise intermediate species, such as methoxy, and thus protect them from over-oxidation. For copper zeolites, the copper must be activated by an oxidant, in order to create one or more active sites. These sites subsequently serve both to activate the C–H bond (at relatively low temperatures, typically between 423 K and 473 K) and then to stabilise the methoxy species thus derived against further oxidation. From this property of copper-containing zeolites, two processes have emerged for the conversion of methane to methanol on copper zeolites: a stepwise – and stoichiometric – approach that utilises successive activation–reaction cycles (“chemical looping”),26,28,33–35 and a catalytic system based upon continuous (gas) flow with either oxygen36,37 or di-nitrous oxide38 employed as the oxidant. Catalytic processes have also been demonstrated for iron-containing zeolites39–42 and then extended to copper zeolites using an aqueous medium and employing hydrogen peroxide as the oxidant.39 This approach, however, cannot be considered practical, as the peroxide is more expensive than methanol. Later, Narsimhan et al.36 developed a continuous flow catalytic system that allows direct methane partial oxidation with a mixture of water, oxygen and methane after an initial high temperature activation. This process is truly catalytic but can only be achieved selectively at methane conversion levels of well below one percent; it therefore comes at a significant cost in terms of achievable methanol yields.Fig. 2 illustrates two step-wise routes by which methane to methanol conversion has thus far been achieved along with “notional” ways that various of the commonly observed products of this reaction might arise, along with the total number of electrons required to be handled for their production. Herein it should be noted that these are formulated as simplified, indicative, descriptors of the possible ways in which some of these products may be formed. For instance, only the redox chemistry of copper is considered, whereas, in the zeolitic case, Lewis and Brønsted acid functions are also present, and products, such as water and methanol, can also lead to a myriad number of further possible reaction schemes. Of note however, is that the production of products other than methanol within such schemes, along with the number of electrons required to be transferred for their production (up to eight) necessarily implies a certain mobility of reactive species within the zeolite framework for their synthesis to be achieved.
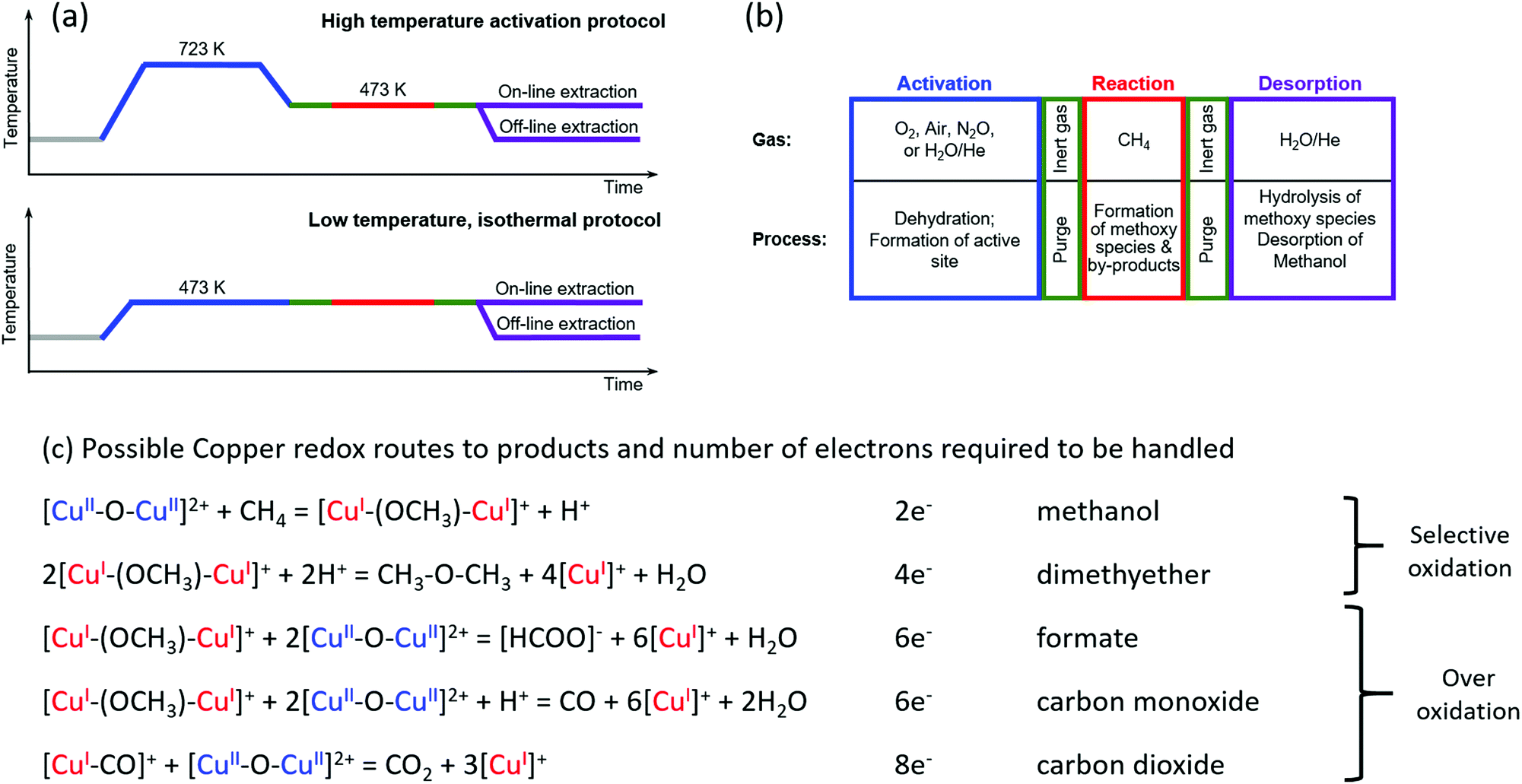 | ||
| Fig. 2 (a) Schematic of two procedures for conversion of methane to methanol. The high temperature activation procedure typically uses low pressure during activation and reaction (1 bar or less).26 The isothermal approach uses a reduced temperature of activation (473 K) but requires high pressure (up to 30 bar) of methane during the reaction to achieve high methanol yields.43,44 (b) Description of possible oxidant gases and types of process occurring within the step-wise approaches to methane oxidation over copper containing zeolites. (c) Notional descriptions of how methane may be converted to both selective and on selective oxidation products along with the total number of electrons required to be handled for each product. | ||
These step-wise routes employ successive activation, reaction and product extraction steps that are repeated to yield a cyclic process. The high temperature activation procedure has been developed with the oxidation step typically performed at >673 K in oxygen followed by the methane reaction step at 423–473 K, and remains the most frequently employed approach to the study of this chemistry using copper–zeolites.26,28,29,43–46 Additional permutations of this procedure have also included the use of different oxidants, such as nitric oxide43 and water.30 A further stepwise procedure has recently been demonstrated, that of low-temperature isothermal cycling.47–49
In this procedure, all steps are performed at 473 K, and the reaction step is conducted under methane at elevated pressures (up to 30 bar). Such a process may improve the applicability of the stepwise procedure. The isothermal procedure has not been studied in as much depth as the high activation temperature route, and relatively little is known about the active sites and reaction selectivity and kinetics involved in this approach.
Recently, Sushkevich et al.30 reported a new “anaerobic” approach, which uses water as the source of oxygen for oxidation of methane to methanol over copper-exchanged mordenite. Isotope experiments with H218O confirmed the insertion of labelled oxygen into methanol; in situ XAS showed the change of copper oxidation state upon reacting with methane and water; and mass spectrometry showed the formation of hydrogen. Combining the experimental data with DFT calculations, the authors suggested a reaction mechanism in which methane reacts with active copper–oxygen species giving methanol, surface methoxy species and reduced Cu(I). The latter then interacts with water to re-oxidise the copper into Cu(II) and releasing molecular hydrogen, which itself is a valuable chemical. Later on, it was shown that not all the copper-exchanged materials are active in anaerobic oxidation of methane and water plays an essential role in stabilising the newly formed Cu(II) species.30,50,51 A specific configuration of active sites, comprising copper–oxo oligomers, such as dimers and trimers, is required to enable the Cu(I) re-oxidation with water. Equally, however, copper monomers, which are stabilised in mordenite with high Si/Al ratio of 46, are more selective in the conventional “aerobic” oxidation with molecular oxygen, with respect to copper-exchanged mordenite with low Si/Al ratios. This difference is most probably associated with the stabilisation effect of the water molecules interacting with active copper sites and forming hydroxides and additional hydrogen bonds.
To date the vast majority of scientific attention, particularly in regards to reaction mechanisms and active site, has been given to the stepwise, high temperature, activation procedure and it is from within this paradigm that the majority of information regarding the mechanisms and active sites at work in these materials has been drawn.
Throughout the development of these procedures, a wide array of zeolites frameworks26,29,31,45,46,52,53 and amorphous supports54,55 have been tested and evaluated for the conversion of methane to methanol. Fig. 3 collates the results obtained for a range of zeolite types and in terms of the methanol yield per unit copper present.
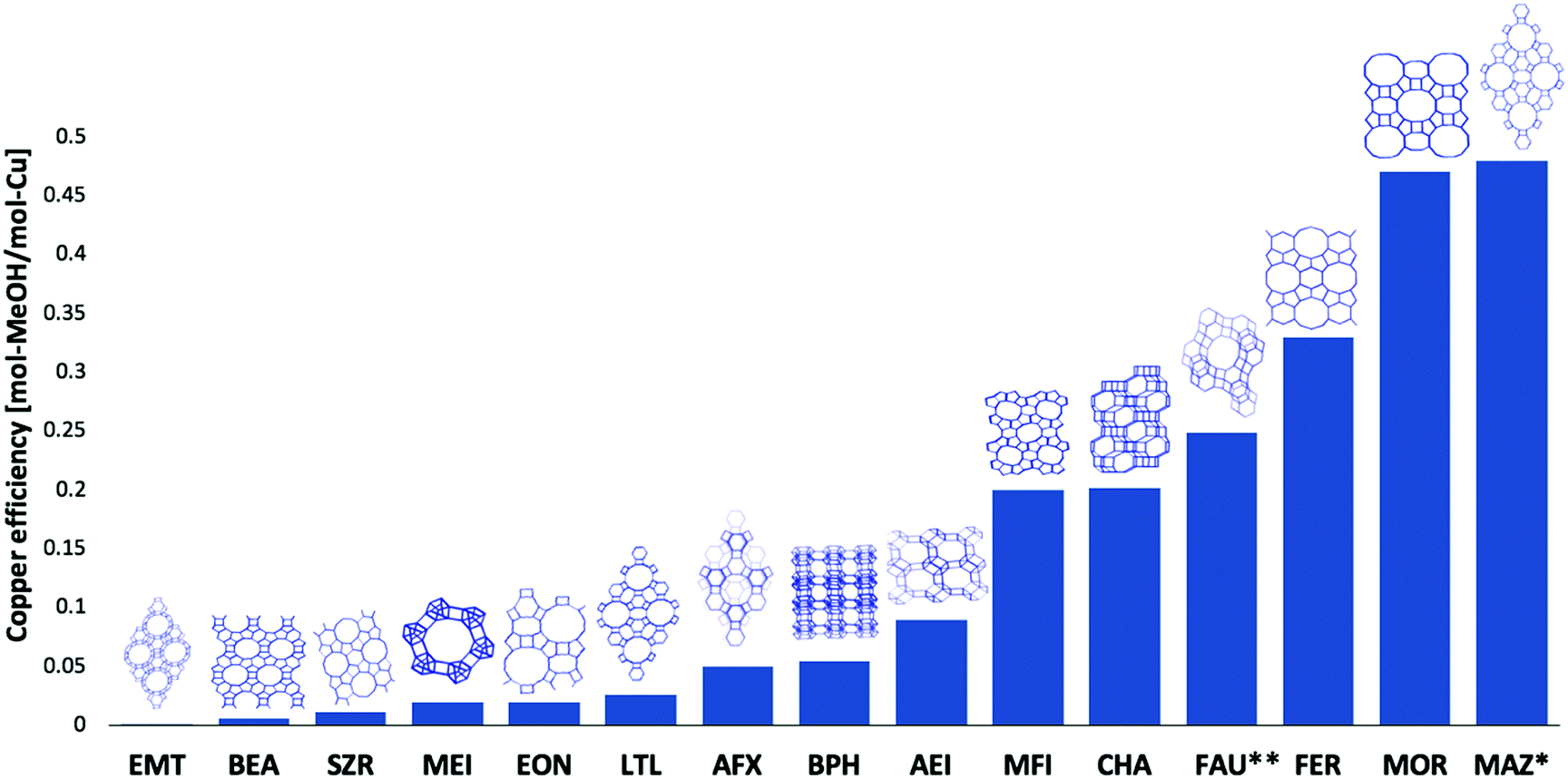 | ||
| Fig. 3 Performance of the range of copper zeolite systems tested for methane to methanol conversion along with schematic illustrations of the unit cell topology for each zeolite.26–29,31,47,48,52,53,56 and for the high temperature, step-wise, approach to activation with subsequent reaction with methane at 473 K and at 1 bar pressure. This except for: FAU** where an optimised isothermal route was used with activation and reaction was conducted at 633 K and 15 bar total methane pressure. * For MAZ, only the active copper was accounted for. Framework drawings reproduced from ref. 23. | ||
The best performing zeolites, in terms of methanol per copper atoms present, are MOR, CHA, ZSM-5, and MAZ. In terms of methanol produced per unit mass of the material tested, Cu/MAZ has recently been shown to outperform Cu/MOR or Cu/CHA, though unlike these latter two cases, Cu/MAZ has yet to be demonstrated to be a stable system under conditions of reactive cycling.27,53 On the other hand, zeolites, such as faujasite (FAU) and beta (BEA), yield little or no methanol for the high activation temperature procedure with a low (473 K) reaction temperature. However, it has recently been shown that with higher reaction temperatures (633 K), the methanol yield for faujasite can be greatly improved and lead can lead to a copper efficiency (molCH3OH/molCu, Fig. 3) similar to chabazite and even higher methanol yields per gram zeolite than for MAZ and MOR.56
One inference that could be drawn, therefore, is that the larger pore zeolites (those based on ten membered rings or greater) are, in some way, much less able to present and stabilise the copper sites required for effective and selective formation of methanol at low reaction temperatures. Moreover, we might intuit that, in general, zeolite rings must be larger than six-members to achieve activity.46,57–59
As ever, the recent demonstration that FAU56 can be induced to yield the highest level of methanol per reaction cycle thus far achieved, when activation and reaction with methane are made isothermally at much higher temperature (633 K) and at increased methane pressure (15 bar), shows that such a general conclusion cannot be arrived at: FAU being composed of twelve membered ring channels and nominally inactive (six and four membered rings) sodalite cages.
Equally, however, this observation highlights the potential importance of the framework in stabilising the active site, with some frameworks being more active/selective for the conversion of methane to methanol than others. It also indicates that, within the parameterisation of the conventional, high temperature, activation procedure, there appears to be an optimal range for the size of the pore or channels required to achieve an effective conversion of methane to methanol. Equally, however, such a comparison does not take into account any possible differences that may exist in the reducibility of the copper species present within each framework system; a parameter which might also be thought of as critical in determining whether a system is active/selective or not within a given reaction protocol. Furthermore, recent works have found that methane can be converted to methanol on non-zeolitic material like amorphous silica,54 mesoporous silica (SBA-15),60 and alumina.55 The zeolite structure is therefore not a requirement for direct methanol synthesis from methane, though methanol yields thus far demonstrated are much lower than now regularly reported for copper-exchanged zeolites.
But what of the active copper sites themselves? One of the on-going debates in the study of copper-exchanged zeolites regards the precise configuration of the available active sites: what is their nuclearity (number of copper atoms) and overall geometry; which are active; which are inactive; and under what conditions is selectivity to methanol, or over-oxidation to unwanted products, achieved?
Originally, di-copper sites (bis μ-oxo di-copper), comprising two copper atoms connected with oxo bridges, were proposed as active structures.26,61 This derivation was made largely on the basis of a specific UV-vis signal observed at 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700 cm−1,26,28,43,62,63 and owed much to the biological study of dicopper metallo-enzymes wherein UV-vis is a widely used method to determine types of copper speciation.64–66 More recently, and as we shall discuss in more depth in subsequent sections of this review, debate regarding the nature of the active sites has opened up. This has resulted from a great deal of subsequent experimentation and a considerable – and growing – influence of theoretical studies. These have resulted in a preference for mono(μ-oxo) di-copper species, rather than the bis(μ-oxo) form.43
700 cm−1,26,28,43,62,63 and owed much to the biological study of dicopper metallo-enzymes wherein UV-vis is a widely used method to determine types of copper speciation.64–66 More recently, and as we shall discuss in more depth in subsequent sections of this review, debate regarding the nature of the active sites has opened up. This has resulted from a great deal of subsequent experimentation and a considerable – and growing – influence of theoretical studies. These have resulted in a preference for mono(μ-oxo) di-copper species, rather than the bis(μ-oxo) form.43
Trimeric ([Cu3(μ-O)3]2+) sites, however, have also been invoked in both MOR29 and ZSM-567 and have more recently been considered for other large pore systems such as those based upon MAZ.68 In the case of MOR, this assertion has been made on a threefold basis: firstly, that, up to a point, as copper is added to the MOR, the yield of methanol per unit copper is a linear function of gradient 1/3;69 secondly, the analysis of EXAFS obtained in the activated and reacted states within the high temperature activation protocol;29 and, thirdly, through DFT calculations67,70 that have indicated that such sites are more stable than di-copper–oxo species during the required activation process.
It is the case that, as with the dimers, trimeric copper centres are suggested to be present in some enzymatic systems.71,72 Equally, however, whether they are present in methane monooxygenases is a matter of ongoing debate in the biological community, in a situation that parallels the current debate that exists for the copper–zeolite systems under consideration in this review.71–74
Moreover, the demonstration of the isothermal stepwise process (Fig. 2) has shown that methanol can be produced by these systems under conditions in which the “typical” di-copper and tri-copper sites should not be formed.48,49 In the isothermal procedure, the activation step occurs at 473 K, well below the required temperature (>553 K) for the formation of μ-oxo-di-copper28 and the trimer sites.69 In spite of this, methanol conversion has been demonstrated for various zeolite framework types using this lower temperature approach to activation and reaction.48,49,75–77 This suggests that the nature of the copper active species might not be limited to dehydrated di- or tri-copper oxo sites, and that a wider spectrum of active site motifs might exist.
As has been mentioned above, DFT calculations indicated that tri-copper sites are more stable than di-copper species under reactive pre-treatment conditions.29 Theory has further suggested that formation energies per CuO formula unit are comparable for CunOn clusters with n up to 9 in the case of Cu/CHA (Cu–SSZ-13).78 Our own recent theoretical investigation suggested that tetramers and pentamers are potentially more stable than any smaller species.70 Using DFT based on global geometry optimisation, we were able to identify a general trend of greater stability as the nuclearity of the copper oxide cluster increased. For instance, the identified ground-state structures of tetra- and pentamer copper clusters of CunOn2+ and CunOn−12+ stoichiometries embedded in an 8 MR channel of MOR exhibit higher relative stability compared to smaller clusters. Therefore, from a theoretical point of view, dimer and trimer are not necessarily the most thermodynamically stable species. However, within a given range of copper and aluminium loading, zeolite pore size and experimental conditions, such structures are predicted to be viable.
On the other side of the complexity spectrum, Kulkarni et al. have recently presented a comprehensive analysis of various mono-copper species as potential active sites for partial methane oxidation in Cu-exchanged SSZ-13 (CHA) zeolite.79 The variety of suggested copper oxide active structures that has emerged from these studies (Fig. 4) is thus striking in its complexity.
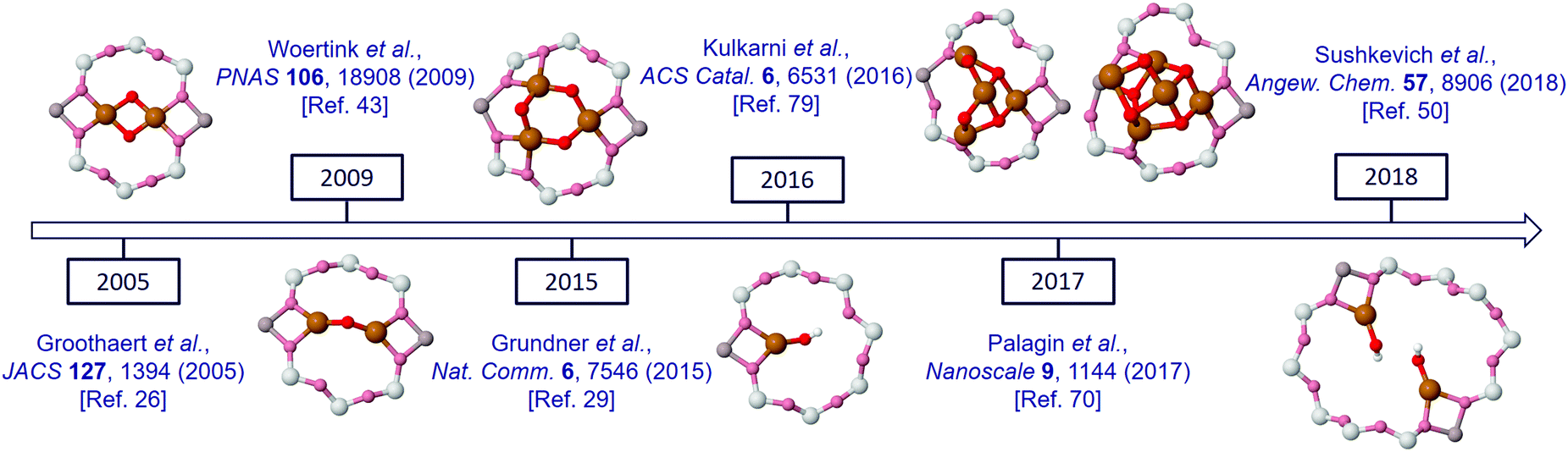 | ||
| Fig. 4 Timeline of various suggested configurations of the copper oxide active species in Cu/MOR. | ||
Taking into account that different zeolite frameworks are capable of stabilising different active species, unravelling the configurational manifold of the copper oxide centers becomes extremely challenging. Indeed, several groups have suggested transforming the discussion of a well-defined selected active site to a discussion of the mixtures of sites, or dynamic co-existence of various states.80,81 For instance, it has been shown that dynamic, multinuclear, sites can by formed by mobilised copper ions during NOx selective catalytic reduction.82,83 There, copper ions can travel through zeolite windows and form transient ion pairs that participate in an oxygen-mediated Cu(I) to Cu(II) redox step integral to this selective catalytic reduction (SCR) process.
Moreover, the structure and dynamics of various types of copper ions can be strongly influenced by the environment they experience, such as the presence of water,30 or ammonia.84 For instance, it has been shown that the strong interaction of the CuII oxidic species with water in zeolites is a unique feature that helps to thermodynamically stabilise active centres, e.g. through the formation of highly stable Cu–(OH)2–Cu dihydroxo species.30 It has further been suggested that, alongside with the stabilising effect, water might also play an important role in the mechanism of the reaction itself, such as in the case of anaerobic methane oxidation.85 In the case of ammonia, it has been shown that it liberates copper atoms from the framework and greatly enhances their mobility, which leads to the formation of the mobile copper active sites.84 At the same time, a simple mixture of active sites might also be responsible for the observed experimental features. For instance, it has been shown that both monomers and dimers have comparable binding energies towards the lattice.86 While for the monomer species similar adsorption energies are obtained with the different zeolite frameworks, an optimum Al–Al distance is, however, also required for the dimeric species to exist. Furthermore, DFT studies support the self-organisation of mononuclear oxygenated/hydroxylated cations in zeolite frameworks toward oxygen-bridged binuclear complexes.46
In regards to the reaction mechanism of the direct conversion of methane to methanol, a classical assumption is that that the reaction might proceed via homolytic C–H bond dissociation followed by direct radical rebound, resulting in CH3OH adsorbed on the reduced dual Cu(I) sites.43 From the analysis of the reaction kinetics Woertink et al.43 deduced that C–H bond breaking is involved in the rate-limiting step of the oxidation of methane. DFT calculations revealed that hydrogen atom abstraction is endothermic by only 57.7 kJ mol−1, reflecting the difference in bond dissociation energy of H–CH3 compared with that of the [Cu–OH–Cu]2+ intermediate that would be generated. Within this point of view, the strong O–H bond of the [Cu–OH–Cu]2+ species helps drive the reaction. In the subsequent step, rebound of the hydroxyl radical (leaving two Cu(I) atoms), leads to coupling with the methyl, thus completing the reaction. Thus, oxygen-activated Cu/ZSM-5 can abstract a hydrogen atom from methane through a low activation barrier.
Zhao et al. have theoretically studied reaction profiles on both mono(μ-oxo) and bis(μ-oxo) di-copper sites in MOR using nitrous oxide and oxygen as oxidants.87 The mono(μ-oxo) di-copper site is more likely to be present after the activation process on the basis of thermodynamic considerations, in agreement with the work of Alayon et al.80 In addition, these calculations suggest that methane activation is only feasible for mono(μ-oxo) di-copper site, whereas bis(μ-oxo) di-copper site has a low rate of methane conversion. In fact, methane activation by Cu–O–Cu has a free energy barrier of 99 kJ mol−1, which corresponds to a rate of 0.8 s−1. Various positions for the location of the Cu–O–Cu cluster were considered and similar barriers were found for methane activation in all cases except for the 8 MR ring, which was suggested to be due to steric hindrance in this small pore. The formed methanol binds rather strongly to Cu/MOR, leading to a desorption free energy of 50 kJ mol−1. This energy is reduced through co-adsorption of water so that methanol can be removed through treatment with water.44
Li et al. have shown that the nature of the exposed bridging oxygen ligands in trinuclear [Cu3(μ-O)3]2+ clusters is similar to that in the binuclear complex.67 The pronounced radical character of bridging oxygens is necessary to promote the homolytic activation of C–H bond in methane. The authors revealed that, unlike the case of the dimer, successful radical recombination of the products of methane oxidation by the [Cu3(μ-O)3]2+ to form methanol does not require a spin-crossing transition to yield methanol. The spins of the CH3˙ and bridging OH species are antiparallel in the high- and low-spin states. Accordingly, direct methanol formation is thermodynamically strongly favoured for these electronic configurations. Besides the direct rebound route, the authors also considered alternative paths for radical recombination over the trinuclear oxygenated copper cluster. Both reactions of CH3˙ with basic oxygens of the zeolite lattice (“adsorption” route), and the extra-framework Cu cations (“heterolytic” route), are thermodynamically much less favourable than direct formation of molecular methanol over [Cu3(μ-O)3]2+. These paths are, respectively, 94 and 90 kJ mol−1 less exothermic than the direct rebound mechanism.
More recently, Kulkarni et al. presented an analysis of various mono-copper species as active sites in CHA (Cu–SSZ-13) zeolite.79 Using periodic density functional theory calculations combined with a thermodynamic analysis of the oxygen activation process, they concluded that [CuOH]+ in the eight-membered rings is responsible for the experimental activity of Cu-exchanged SSZ-13. The process starts by C–H bond cleavage of CH4, via a feasible 110 kJ mol−1 free energy barrier, and the proposed reaction mechanism is consistent with the spectroscopic data and experimental observations. Furthermore, the proposed mechanism successfully explains: (i) the necessity of hydration during the methanol extraction step and; (ii) the lower activity of the high-Al SSZ-13 sample. The effect of aluminium content and distribution is especially interesting and leads to a general design principle: that 6-membered rings are detrimental for the reaction, while 8-membered rings are desirable and favour the formation of the active [CuOH]+ species. Specifically, the statistical analysis of Bates et al. suggests that increasing the Al content in CHA increases the probability of forming two “paired” Al atoms in the 6 MR.88 This strongly favours the formation of bare Cu(II) cations that are unreactive for C–H activation and inhibit [CuOH]+ formation in the 8 MR. It was predicted that <5% of the copper cations belong to the 8 MR–[CuOH]+ species therefore that the lower performance for CHA with lower Si/Al ratios is due to the smaller number of available [CuOH]+ active sites and is not due to an inherent decrease in reactivity.
The invocation of such a wide range of potential active sites has considerable implications for how the conversion of methane is achieved, i.e. by what mechanism? From the diverse considerations listed above, two general mechanistic possibilities may be synthesised (Fig. 5).
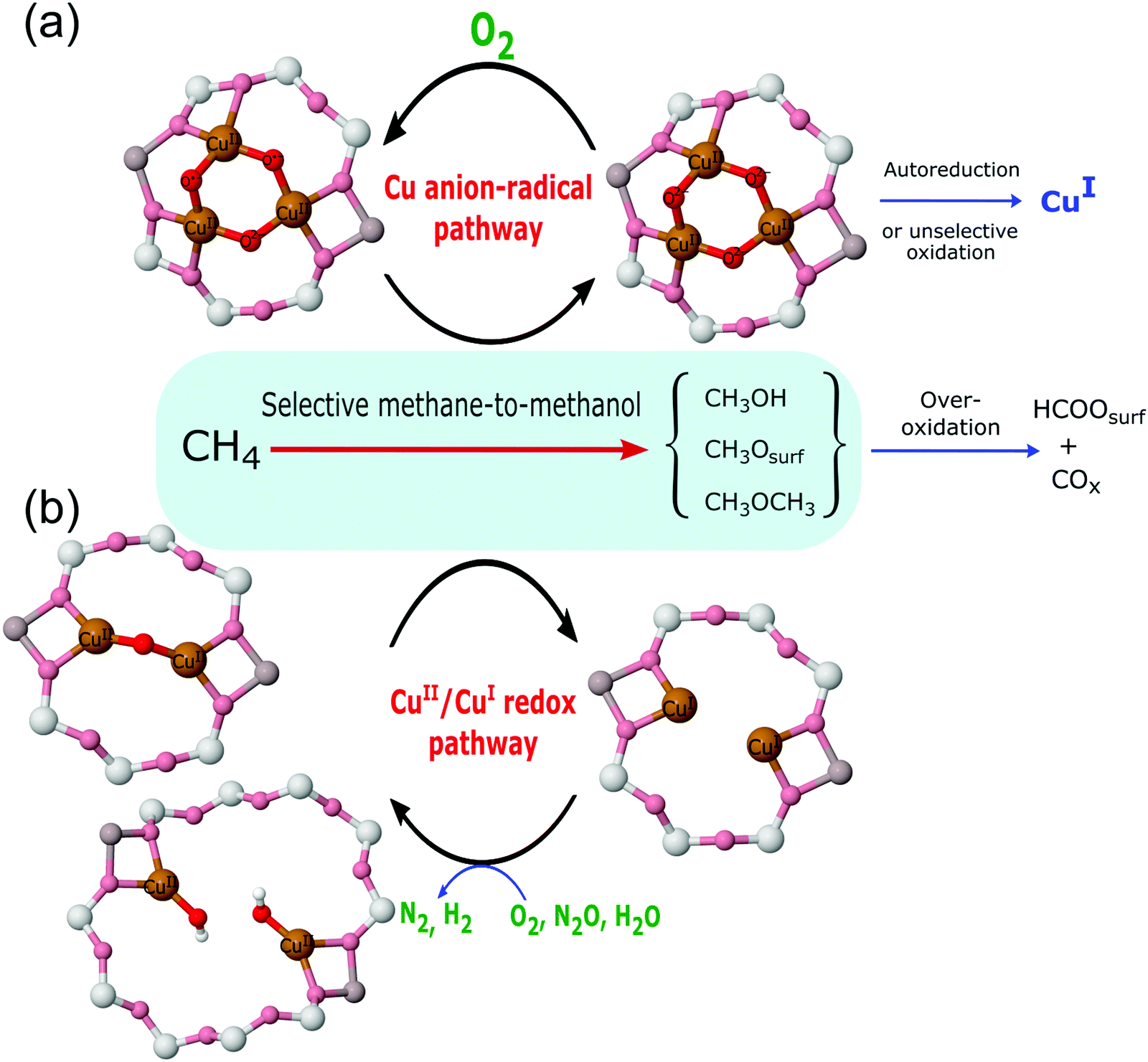 | ||
| Fig. 5 A schematic illustration of the two reaction pathways derived for the selective conversion of methane to methanol over copper containing zeolites: (a) the mechanism wherein the required electrons are handled via the formation of an oxyl radical species; (b) the mechanism localised at the copper centres with concurrent reduction of two Cu(II) to two Cu(I). | ||
The redox mechanism shown at the bottom of Fig. 5 might be considered the traditional view of how this and other copper facilitated selective oxidation processes are mediated. In this mechanism the two electrons required to be handled to effect the conversion of methane to methanol are localised on the copper atoms and the copper is reduced from Cu(II) to Cu(I); this mechanism therefore requires the participation of two copper atoms to produce one methanol molecule.
The second mechanism is a more recent derivation that has its origins in theory,67 and has been derived for both di-copper and tri-copper sites. In this mechanism, the electrons required for the oxidation of methane end up localised at oxygen atoms within the active site as oxyl radical species.
These reaction pathways are, therefore, very different in terms of where the electrons required to be transferred, to yield the varied products observed (see Fig. 2), during reaction with methane end up; they are also radically different in how they view the role of Cu(I) in determining the selectivity of the methane to methanol process. In the oxyl radical based proposal, there is no role for Cu(I) in the selective oxidation of methane. By inference, therefore, the formation of Cu(I) during the reaction can be associated with the formation of unwanted over-oxidation products and reduced selectivity toward the desired conversion, or the often observed phenomenon of auto-reduction. As will be shown (vide infra Section 4.5) the available evidence (for Cu/MOR) suggests that, within the aerobic (oxygen activation) high temperature activation protocol, auto-reduction can be neglected as the source of Cu(I).
On the other hand, in the more traditional copper redox mechanism, Cu(I) is central to both selective and (again by inference) non-selective reactions. Within this paradigm the only difference between these two possibilities is in the number of electrons required to be handled for each product to be formed i.e. two for methanol, six for formate and carbon monoxide, and eight for carbon dioxide. Therefore, these two reactive possibilities lead to radically different expectations for the relationship (or otherwise) between methanol yield and selectivity and any associated production of Cu(I). As discussed in later sections these different expectations, given that we can quantitatively measure both Cu(I) formation and reactivity, lead to experiments that are able to decide which of these possibilities is reactively dominant in these systems.
From this brief discussion, it is evident that, despite intensive research and a radically improved performance of copper containing zeolites for the selective oxidation of methane to methanol, many fundamental questions regarding this conversion remain.
In subsequent sections, we shall consider in more detail the experimental and theoretical bases for all these propositions in an effort to examine the evidence that underpins each of them in a critical manner. From these bases we shall then ask as to whether we are able to make some more concrete assertions and to therefore understand the behaviour of these complex systems in a less equivocal way. Before we do this however, it is worth spending some time to consider some of the specific challenges that these systems present both from experimental and theoretical points of view.
3. Specific challenges that these systems present
3.1 Experimental considerations for in situ study and the derivation of reliable structure function relationships
The study of methane to methanol conversion by metals hosted in zeolites poses a number of unusual (if not unique) challenges in respect of in situ study and the restoration of reliable and in depth structure–function relationships; and these should be explicitly pointed out if only as, by and large, in the extant literature, they are not.Most important is the fact that the very nature of the zeolites means that the products of methane oxidation are retained within the material. As such, they can only be extracted and quantified post factum using either steaming at elevated temperature (via à visFig. 2), or aqueous extraction at ambient temperature or slightly above. Whilst a variety of methods, such as UV-vis,26,28 Raman,43 and X-ray spectroscopy29,80 (vide infra) can be used to follow aspects of the reactive chemistry occurring within copper containing zeolites, none of these are capable of quantifying the products formed, or selectivity achieved, at the same time. Ordinarily, the go-to technique for such assessments would be mid-range infrared spectroscopy as it may identify, if not easily quantify, the speciation formed within the zeolites. However, even this method has only been used post factum due to the overwhelmingly large contributions derived from gas phase methane and the relatively small and overlapping signals that may be derived from species of relevance, such as adsorbed methoxy, methanol, carbonate, carbon monoxide, and carbon dioxide.30,50
As such, unlike the vast majority of catalytic gas–solid conversions, where specific performance indicators can be relatively easily derived in parallel with other measurements of the state of the material, for the selective conversion of methane to methanol by chemical looping, this has not been achieved. As a result, aspects of structure and function are, for the most part, derived from entirely separate experiments and (often) using very different equipment. This decidedly sub-optimal situation begs a simple but fundamentally important question: how can one be sure that a valid measurement, one that truly represents the nature of the materials one is studying, has been made?
This is a general problem, and one that may be thought of as at its most critical where synchrotron-based studies are undertaken. These sorts of experiments are conducted away from a home lab, within limited time allocations, and possibly using equipment that is not so well known to the experimenters. It is also the case, more often than not (though exceptions to this have recently appeared),31 that post factum analysis of the result of an X-ray based experiment in terms of the product yields and selectivity achieved, is not entered into.
This situation is made worse by the intrinsic nature of the species under study. Cu(I) is extremely sensitive to the presence of oxidants, such as oxygen and water, and can be facilely re-oxidised to Cu(II). Therefore, as well as being in a reactively (in terms of the molecular products formed) “blind” situation, experimental protocols must be extremely robust and well understood to ensure that trace levels of these oxidants do not interfere with the chemistry at hand. Critically, isothermality within a reactor bed must be achieved to an acceptable degree in order to avoid thermal gradients yielding incorrect or misleading results from what are either, point measurements made within the body of a much larger sample, or measurements that report an average view over a large (a few millimetres) portion of a sample bed.89
In the case of the use of synchrotron radiation, there is also the possibility that the X-rays themselves may induce unwanted changes in the systems under study, either through local heating effects, or from the production of electrons. Both of these possibilities have the potential to promote reduction of Cu(II) to Cu(I) wherein none should ordinarily occur. Such effects are known and have been demonstrated for copper in other (predominantly) homogeneous systems.90,91 To date, however, we are unaware of any quantitative assessment of whether these affects are present or of reactive significance in copper/zeolite systems. We might also note that X-rays are not the only probe that has the potential to alter these systems; the power densities often applied in spectroscopies such as Raman also have the capacity to induce unwanted localised heating effects that have (at least notionally) the capacity to perturb the system under study in potentially misleading ways.92
Lastly, and as mentioned in the previous section, the phenomenon of auto-reduction of Cu(II) to Cu(I) in these systems has often be observed.32,93–95 This behaviour (vide infra) has been attributed to numerous factors and its contribution to subsequent methane activation and methanol production remains a vexed question, further complicating the process of understanding this process and how these materials mediate it.
3.2 Challenges these systems present to theory
From the point of view of theory, modelling zeolites is also a challenging problem. Fundamental understanding of these complex materials requires, on the one hand, a carefully refined structural model, that captures the essential chemistry of the system,96 and, on the other hand, an appropriately accurate description of the experimentally observed spectroscopic features of the system. Furthermore, a suitable model describing the reaction mechanism, that includes identification of the configuration of every intermediate, as well as the transition states, has to be developed to yield a predictive quality modelling of the process, as well as deep understanding of the underlying chemistry. The following features of Cu–zeolites specifically render the above tasks challenging.In order to decide, in an unambiguous fashion, whether any theoretically predicted structures exist in reality, an interplay and exchange between experimental and theoretical chemistry approaches is required. Theoretical methods do not only yield geometrical and electronic density information, but are also able to model many experimentally observed features of the system, such as vibrational, optical absorption, X-ray photoelectron spectra, Cu K-edge XANES, NMR shifts, etc., that can be directly compared to experiment. An ideal approach towards solving catalysis-related questions is therefore to build a dynamical feedback system, where experimental evidence is initially used to build an appropriate theoretical model, which can be used to screen the configurational and chemical possibilities available to the system in question. The latter may then guide further experimental efforts, starting the second cycle of theoretical approximation.
4. Experimental studies of copper containing zeolites in respect of the selective conversion of methane to methanol
In this section, we consider the experimental methods that have been most commonly applied to the understanding of the distribution and specific locations of copper species within zeolites systems in relation to the conversion of methane to methanol. It is from these studies that we shall attempt to distil which aspects of the copper speciation might be considered as known and understood. Equally, we shall determine what remains to be clarified in respect of the mediation of effective methanol production from a methane feed.4.1 High resolution X-ray diffraction (XRD)
The crystalline nature of zeolites, with their myriad of extended three dimensional structures, has resulted in XRD playing a central role in the investigation of their structure and properties at many levels.24,102 XRD permits the specification of symmetry, and identification of the zeolite, as well as detailed structural analysis of the zeolite pores and channels and how they interconnect. Lastly, the resolution of the detailed geometric structure of the sites occupied both by templating molecules and, subsequently, cations exchanged into the zeolite structure, may be derived from suitable diffraction data and analysis.57,103 Of these capacities it is this last, and most demanding, possibility that is of most relevance to the current discussion.XRD is a high-energy X-ray method and therefore, due to the ability of X-rays of this energy to penetrate matter, applicable under a wide range of in situ conditions. Moreover, modern day synchrotron and lab sources, together with state-of-the-art detectors, means that it can be operated with varying degrees of resolving power (Å−1) and time resolution. However, by its very nature, XRD is reliant on a high degree of translational symmetry being present in any sample for that power to be realised. As such, it is ultimately the structural order intrinsic to the materials themselves that dictates how much information may be derived from its application.
To obtain quantitative information regarding the site occupancy of an element such as copper in a zeolite material then data of a quality suitable for Rietveld analysis must be arrived at. From such analysis local electron density maps can then be constructed. As the intensity of X-ray scattering is fundamentally related to electron density then, if sufficiently high-resolution data can be obtained, the crystallographic location of copper being a relatively electron rich element within a matrix comprised of less electron rich elements – such as aluminum, silicon, and oxygen – may therefore be specified using this approach.
In respect of the current discussion, the most extensive XRD-based studies have, thus far, been made on Cu/CHA. Cu/CHA appears to be particularly amenable to the application of high level XRD analysis57,82,104–106 and has been much studied in this respect for the selective reduction of nitric oxide by ammonia (SCR process). We might note that, in the case of ZSM-5, similar XRD based data is also available.107 Equally, however, for the two other most active Cu/zeolite systems for methane conversion to methanol (Cu/MOR and Cu/MAZ), high-level information regarding the location of copper under relevant conditions remains absent.
As an example of the application of high resolution XRD to locating the copper sites present in CHA, and also in respect of NO-SCR catalysis, Deka et al.104 established that, under conditions of reaction, the 6 MR rings of CHA host isolated Cu(II) cations. From a combination of in situ XRD and EXAFS, they were then able to propose a reactive mechanism, schematically illustrated in Fig. 6. This assignment built upon previous XRD due to Fickel et al.57 who, two years previously, had considered thermally induced dehydration of two Cu/CHA samples and whose Rietveld refinements of XRD placed the observable copper ions within the 6 MR rings. Fig. 6 illustrates the copper ion in the 6 MR and how this copper ion bonds ammonia and is displaced at enhanced temperature, derived from the application of combined XRD and EXAFS measurements.
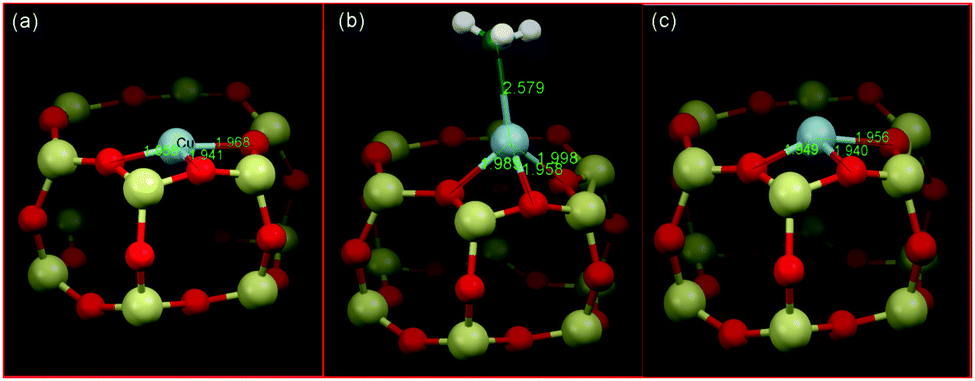 | ||
| Fig. 6 Schematic illustrations of the local copper environment in 6 MR subunit of CHA. (a) Local structure after calcination with copper on the plane and slightly distorted from the centre of the d6r subunit of CHA; (b) interaction with ammonia at ∼125 °C under SCR conditions resulting in a coordination geometry change; (c) under SCR conditions above 250 °C. The local environment is obtained from EXAFS fits using an initial model proposed from the refinement of the corresponding diffraction data collected at the different temperatures. Reproduced from ref. 104 with permission from the American chemical society, copyright 2012. | ||
However, the site occupancy established from this study accounts for only 25% of the total copper present. Therefore, be it as a consequence of its non-crystalline distribution, the resolution of the in situ experiment, or the quality of the sample, the location of the remaining copper, and its possible contribution to the chemistry under study, could not be resolved.
A subsequent study105 using a maximum entropy method to enhance the electron density maps derived from Rietveld analysis established that the copper missing from the study of Deka et al.104 could be found. From this study it was established that, alongside the copper present in the 6 MR rings, the majority of the copper occupies (as monomeric species) the 8 MR rings (Fig. 7).
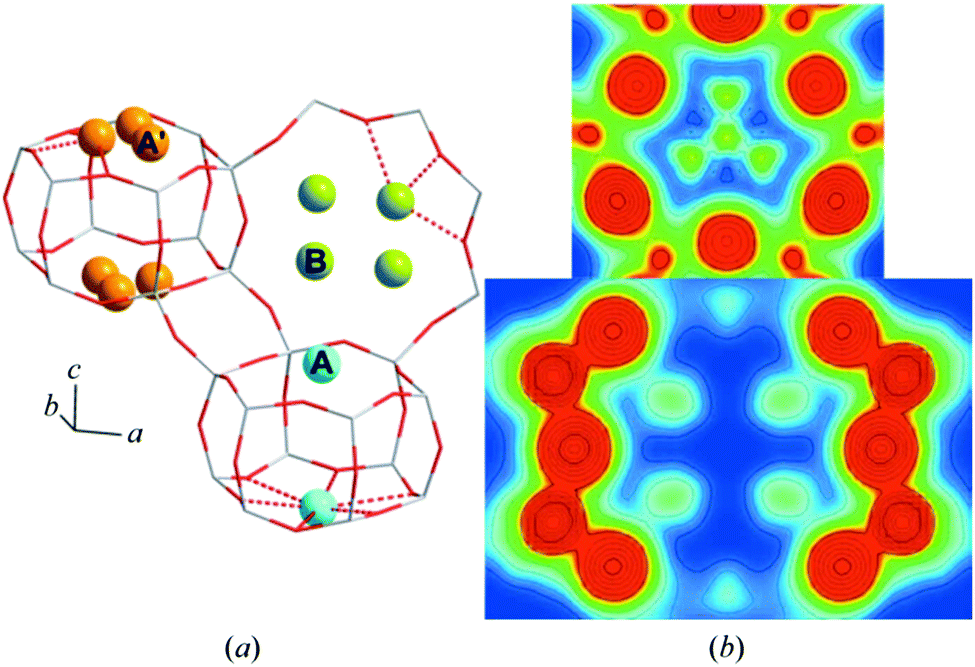 | ||
| Fig. 7 (a) High resolution XRD data together with details of elements of the fitting procedure and the residuals remaining after fitting of the data using Rietveld analysis. (b) Schematic illustration of the resulting models for the occupancy of copper atoms within the CHA framework for the hydrated sample at close to ambient temperature (far left), and then the three sites after the CHA has been fully dehydrates at 723 K in oxygen before being cooled to 323 K for measurement. This analysis restores a single occupancy of the 6 MR ring whilst the majority of the copper is shown to occupy two different sites within the 8 MR rings. Reproduced from ref. 105 with permission from the International Union of Crystallography, copyright 2014. | ||
Regarding the relative contributions made by the three types of copper resolved by the work of Anderson et al.,105 and as an exemplar of how labile site occupation can be in copper zeolites under reaction conditions, operando XRD was subsequently used to show a temperature dependent migration of copper between 6 MR and 8 MR ring sites.82 Moreover, this variable site occupation could be directly correlated with the ability of Cu/CHA to facilitate SCR catalysis.
In respect of selective conversion of methane to methanol, and as part of a larger work that included UV-vis, Raman, and reactivity testing, Ipek et al.106 again used XRD and Rietveld analysis to delineate the site occupation of copper within Cu/CHA samples within both hydrated and high temperature activated (723 K, oxygen) Cu/CHA, post factum at 323 K. An example of their data, and the fitting of it, as well as a schematic illustrating the 6 MR and 8 MR occupancy they obtained from it is given in Fig. 8.
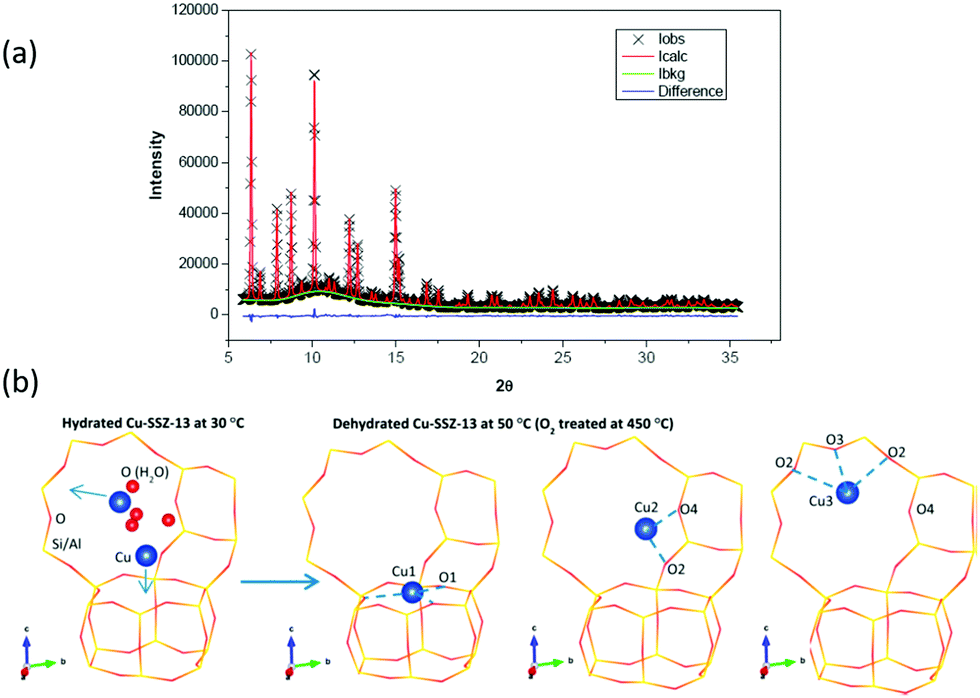 | ||
| Fig. 8 (a) High resolution XRD data together with details of elements of the fitting procedure and the residuals remaining after fitting of the data using Rietveld analysis. (b) Schematic illustration of the resulting models for the occupancy of copper atoms within the CHA framework for the hydrated sample at close to ambient temperature (far left), and then the three sites after the CHA has been fully dehydrates at 723 K in oxygen before being cooled to 323 K for measurement. This analysis restores a single occupancy of the 6 MR ring whilst the majority of the copper is shown to occupy two different sites within the 8 MR rings. Reproduced from ref. 106 with permission from the American Chemical Society, copyright 2017. | ||
Herein, therefore, we also find a fine example of the combined use of methods to establish as much information as possible from what are undeniably complex materials. The high resolution XRD is able to locate all the copper atoms in this material. However, it is only with the subsequent application of symmetry-based arguments arising from the use of Raman spectroscopy (vide infra) that it can be deduced that some of the 8 MR copper atoms co-exist in the same 8 MR ring and can form the di-copper oxo species inferred to be the active sites for methanol formation from methane.
Lastly, if we then cross reference these data to the methanol yields obtained from these samples, following high temperature activation (723 K, oxygen) and reaction with methane at 473 K, we understand that most of the copper in these materials is inactive (22.7 < CH3OH (μmol g−1) < 28.5; 0.03 < CH3OH/Cu < 0.05). We might further deduce, that alongside the inactive copper present in the 6 MR rings, the probability of finding two copper atoms in an 8 MR ring that are able to form the dimeric copper–oxo sites indicated to be the active sites is rather small, and that they are very much a minority species and therefore not detected by diffraction.
As we shall see throughout this review this sort of multiple site occupancy, within which only some sites are active, is very typical and makes the quantitative establishment of structure activity relationships by any single method fraught with pitfalls and challenges.
Where these XRD based studies have, thus far, been applied to the selective oxidation of methane by copper containing zeolites, they have been restricted to materials of relatively low methanol yields and where most of the copper present is inactive and, therefore, where accessing the location and structure of the sites that are active is correspondingly difficult.
Advanced XRD studies of this nature have yet to be reported for much more active CHA,46 MOR,29,31 and MAZ27,108 systems, wherein the likelihood that this powerful method may be able to resolve the nature of the active copper component appears much higher. Such studies, were they to be achieved, would appear to be one of the best routes available for the resolution of this fundamental issue.
4.2 UV-visible spectroscopy
Since the discovery of the methane conversion into methanol by Groothaert et al.,26 UV-vis spectroscopy is extensively used for the investigation of copper sites hosted in zeolites. Cu(II) di-cations that are normally present in the activated copper-exchanged zeolites prior to the reaction with methane have an open d-electron shell, which results in various absorption bands in the electron spectrum. However, the vibrational–rotational structure leads to a diffuse broadening of UV-vis bands at ambient temperature, decreasing the resolution and making the determination of the number and exact positions of the bands difficult. The use of UV-vis spectroscopy to access the active sites of zeolites in the process of methane conversion into methanol was first reported for Cu–ZSM-5, and has been followed by multiple studies (Table 1).26,28,33,43,44,58,63,109 However, to date, this extensive use has yet to result in the establishment of a clear and unambiguous database of UV-vis bands which might serve as diagnostic when studying zeolite-based materials.| Material | Conditions | Ref. | ||
|---|---|---|---|---|
| Cu/ZSM-5 | Bands | Assignment | Develops upon heating in oxygen, reacts with methane | 26 |
| Cu/MOR, Cu/ZSM-5, Cu/BEA, Cu/FAU | 22700 cm−1 |
CT band in Cu–(O)–Cu | Appear after treatment in oxygen at high temperature. Reactive towards methane, but at different temperatures | 28 |
| 14000 cm−1 |
Isolated Cu(II) | |||
| 34000 cm−1 |
CT Ozeolite → Cu(II) | |||
| 6200 cm−1 | N/A | |||
| Cu/ZSM-5 | 21800 cm−1 |
CT band in Cu–(O)–Cu, copper mono-μ-oxo | Reactive towards methane at >175 °C | 33 and 43 |
| Cu/ZSM-5 | 14000 cm−1 |
d–d transition Cu(II) in pseudo-octahedral coordination | Established linear correlation between intensity of 22700 cm−1, copper loading and amount of extracted methanol |
63 |
| 22700 cm−1 |
Cu–(O)–Cu, copper mono-μ-oxo | |||
| Cu/ZSM-5 | 29000 cm−1 |
CT π*σ to Cu(II) in Cu–(O2)–Cu, di-copper peroxo | Forms after treatment of reduced material in oxygen, converts into 22700 cm−1 upon heating |
33 and 111 |
| Cu/MOR | 22700 cm−1 |
Copper mono-μ-oxo | Disappears during the reaction with methane at 200 °C | 44 |
| Cu/MOR | 13600 cm−1 |
Cu(II) coordinated in 6 MR with 2Al | Assignment of the location of the copper atoms, together with EPR spectroscopy. More than 60% of binuclear sites | 109 |
| 16750 cm−1 |
Square planar configuration in 5 OR 6 MR with 1Al | |||
| 22200 cm−1 |
Binuclear, unknown location | |||
| Cu/ZSM-5 | 22000 nm |
N/A | Reversibly reactive towards nitrous oxide and methane | 47 |
| Cu/MOR | 13300 cm−1 |
d–d in square pyramidal configuration of Cu(II) | 22200 cm−1 was found exclusively on Cu/MOR without Na+ cations, while 12500 cm−1 was present on CuNa-MOR |
110 |
| 16700 cm−1 |
d–d in square planar Cu(II) | 9600 cm−1 decays upon introduction of methane after activation | ||
| 22200 cm−1 |
Copper mono-μ-oxo | |||
| 12500 cm−1 |
N/A | |||
| 9600 cm−1 | N/A | |||
| Cu/SZ-13, Cu/SAPO-34, Cu/ZSM-5 | 11000, 13600, 16500 and 19700 cm−1 |
Located in 6 MR of the zeolite | 23000 and 23350 cm−1 found over Cu/SAPO-34 and Cu/ZSM-5 react with methane |
45 |
| 23550 and 16500 cm−1 |
||||
| 23000 cm−1 |
||||
| Cu/MOR | 31000 cm−1 |
New CuxOy species, trimers | No 22700 cm−1 |
29 |
| Cu/MOR | 21900 cm−1 |
Copper mono-μ-oxo, type 1 | Broad band at 22200 cm−1 splits into two upon treatments. 21900 cm−1 is unstable towards heating, while 23100 cm−1 persists to 450 °C. Both sites react with methane, however, showing different activation energies |
81 |
| 23100 cm−1 |
Copper mono-μ-oxo, type 2 | |||
| Cu/MOR | 13500 cm−1 |
Cu(II) species | No bands ∼22000 cm−1 after activation at 200 °C |
48 |
| Sh. 16750 cm−1 |
||||
| Cu/MOR | 11600 cm−1 |
Hydrated Cu(II) (H2O)6 | Extensive UV-vis study of copper species over Cu/MOR | 112 |
| 16700 cm−1 |
Square planar Cu(II) | |||
| 30000–40000 cm−1 |
CuO large clusters | |||
| 34000 cm−1 |
CuO species | |||
| Cu–MOR | 22500 cm−1 |
Copper mono-μ-oxo | Intense bands were observed for the samples prepared by solid-state ion exchange, for solution IE almost no bands were detected | 113 |
| 31000 cm−1 |
Copper oxo trimers | |||
| Cu/SSZ-13, Cu/SSZ-39 | 13600 cm−1 |
CuxOy clusters | 13600, 16600 and 20000 cm−1 do not change upon contacting methane. Broad band between 30000–35000 cm−1 decreases, being active towards methane |
38 and 106 |
| 16600 cm−1 |
||||
| 20000 cm−1 |
||||
| 10900 cm−1 |
||||
| 30000–35000 cm−1 |
||||
| Cu/SSZ-13 | 29000 cm−1 |
29000 cm−1 reacts with methane while 16400 cm−1 is not active |
114 | |
| 16400 cm−1 |
||||
Originally, the 22700 cm−1 absorption feature, that could be associated with both the activation process and the subsequent reaction, was used to characterise active copper sites.26,28,33,43,44,58,63 Initially, this band was attributed to charge transfer arising from bis-μ-oxo di-copper species; an assignment made by analogy to enzymes, and, specifically, methane monooxygenase.66 However, in later studies the attribution of this band was re-considered. Using resonant Raman (rR) spectroscopy the assignment to di-copper bis-μ-oxo sites was excluded (vide infra), and a new attribution to the di-copper mono-μ-oxo site was proposed.43 Further investigations revealed the formation of the intermediate Cu–(O2)–Cu peroxo species characterised by a 29000 cm−1 band due to charge transfer from π*σ to Cu(II).58 These species were suggested to form at the first step of activation in oxygen and to later disproportionate to yield the mono-μ-oxo di-copper sites and lattice oxygen (Fig. 9). Importantly, the transition at ∼22700 cm−1 has been observed for a number of different zeolites, including Cu/MOR, Cu/BEA and Cu/FAU.28 However, these materials show very different activity towards methane and require different temperatures to achieve reaction. The loading of copper loading has also been found to have a significant effect on the structure of UV-vis spectrum. At low copper loadings (<Cu/Al = 0.2) in ZSM-5, there are no observable absorption features except for a weak absorption at 13300 cm−1, and an intense band around 40000 cm−1, due to charge transfer transitions from the oxygens of the zeolite lattice to Cu(II). As soon as the Cu/Al ratio exceeds 0.2, a new absorption band appears at 22700 cm−1 that increases in intensity with Cu/Al ratio. In addition these features were only present for Cu/ZSM-5 samples with a Si/Al ratio between 12 and 30.26
 | ||
| Fig. 9 Oxygen activation pathway suggested for copper-exchanged ZSM-5. Reproduced from ref. 58 with permission from the American Chemical Society, copyright 2010. | ||
Importantly, among the purely spectroscopic observations, the systematic work by Beznis et al.63 established a linear correlation between the intensity of the peak at 22700 cm−1, copper loading in Cu/ZSM-5 and the amount of methanol resulting from reaction with methane. Their work confirmed the assignment of this UV-vis band to an active species and also showed the presence of inactive Cu(II) sites in pseudo-octahedral coordination characterized by the band at 14000 cm−1. However, the absolute amounts of active sites present were not clearly understood.
Shortly after, an extensive study of Cu/MOR, which has a structure comprising 12-MR channels and 8 MR side pockets, made use of UV-vis in an attempt to describe the location of Cu(II) cations in the zeolite framework. Vanelderen et al.109 suggested that bands at 13600 and 16750 cm−1 corresponded to isolated copper ions in MOR pores that contain two and one aluminum atoms, respectively.
However, the location of binuclear copper sites characterised by the band at 22200 cm−1, that according to electronic paramagnetic resonance (EPR) spectral analysis accounts for 60% of all copper loaded into zeolite, was not determined.109 A similar exercise, conducted using Cu/CHA, a zeolite that contains both 6 MR and 8 MR rings, revealed a number of further bands (at 11000, 13600, 16500 and 19700 cm−1). These were identified as corresponding to isolated copper ions present in the 6 MR of this zeolite.45 Notably, the influence of zeolite topology on the structure of UV-vis spectrum was also demonstrated, and revealed that a unique set of bands exists for Cu/SAPO-34, Cu/ZSM-5, Cu/MOR, Cu/SSZ-13, Cu/SSZ-16 and Cu/SSZ-39 materials (Table 1).
Narsimhan et al.110 also showed that introduction of a co-cation, such as sodium, can affect the UV-vis spectrum and lead to the appearance of different features. Thus, for Cu/MOR without sodium the band at 22200 cm−1 was observed, while for Cu/Na-MOR a band at 12500 cm−1 was present. Both bands diminished in intensity in the presence of methane. A new band, at 9600 cm−1, that also responded to activation and reaction with methane was reported, though not assigned to any specific copper site.
Later on, careful investigation of the behaviour of the band at 22200 cm−1 in Cu/MOR under reactive conditions showed that upon reaction with methane it splits into two bands centered at 21900 and 23100 cm−1, respectively.81 Both sites corresponding to these bands were reactive towards methane and showed measurably different kinetic character. Moreover, the sites that give rise to the band at 23100 cm−1 appeared relatively stable and persisted in Cu/MOR up to 723 K, while those responsible for the 21900 cm−1 feature easily decomposed upon heating.
This study, therefore, clearly showed the presence of at least two distinct copper–oxo sites in Cu/MOR, both of which are active towards activation of methane.
Subsequently, copper–oxo sites consisting of three copper atoms were suggested to be present in Cu/MOR, and associated with a further band at 30000–31000 cm−1.29,115 Importantly, no bands around 22000 cm−1, as had been observed previously in Cu/MOR, were found in this study. Support for this proposition subsequently came in the publication by Le et al.,113 where solid-state ion exchange (SSIE) was used for the synthesis of Cu/MOR. In this case, the authors observed two bands (at 22500 and 31000 cm−1) in differently prepared Cu/MOR samples. The latter absorption, however, was only observed for the samples prepared by SSIE, which concurrently showed twice the yield of methanol (ca. 55 μmol g−1) compared to the samples prepared by conventional solution ion exchange, (ca. 28 μmol g−1) per cycle.
This is a revealing observation. Whilst each of these samples is verifiably Cu/MOR of very similar composition and copper loading, the copper speciation and reactivity obtained are markedly different. Moreover, taken together, they could imply the possibility that multiple active sites, that possess different local structure and nuclearity, might be able to exist within notionally very similar host materials.
Finally, Ipek et al.38,106 investigated the small pore (CHA) zeolites, such as Cu/SSZ-13 and Cu/SSZ-39, and found multiple absorption bands at 13600, 16600, 20000 cm−1, none of which were reactive towards methane. Simultaneously, however, further broad features in the 30000 to 35000 cm−1 region, found in samples with high copper loading and low Si/Al ratio, responded to the presence of methane. These high wavenumber bands were subsequently ascribed to the presence of extra-pore (extruded) copper oxide clusters,106 pointing to the importance of the Si/Al ratio and copper distribution in respect of the formation of CuxOy species. Increasing the Cu/Al ratio for SSZ-13 (Si/Al = 12) resulted in enhanced absorption between 30000 and 34700 cm−1, indicating a higher concentration of copper oxide clusters. The bands at about 13600, 16600, and 20000 cm−1 also showed increased intensity with increasing Cu/Al ratio.
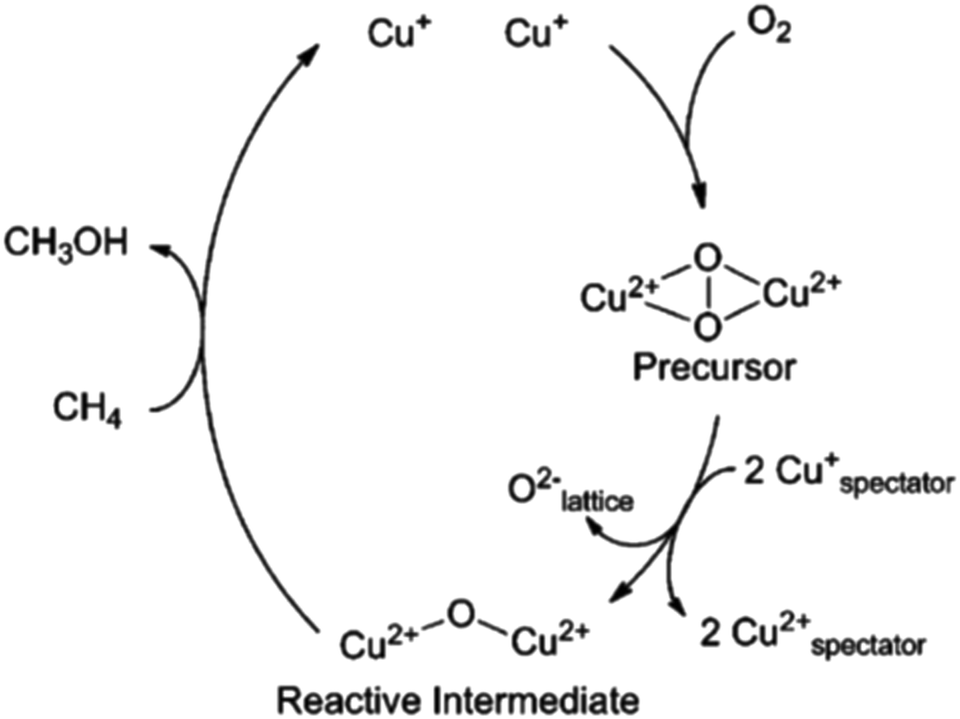
Recently, Kim et al.112 have attempted to systematically analyse what has become a veritable menagerie of UV-vis bands observed in copper-exchanged zeolites. Using two types of oxidants, oxygen and nitrous oxide, they proposed a plausible scheme for processes taking place in Cu/MOR during the activation that is shown in Fig. 10. In this scheme, dehydration of the samples in absence of oxidant leads to the formation of inactive CuxOy clusters, reduced Cu(I) species, and fully framework-balanced Cu(II) cations. On the other hand, activation in oxygen results in copper–oxo trimers that originate from Cu–(O2)–Cu intermediate species of the type originally described by Smeets et al.58 However, the precise nature and mechanisms of interconversion between copper–oxo sites, and to what extent these studies may be carried over to other zeolites, are issues that remain unclear.
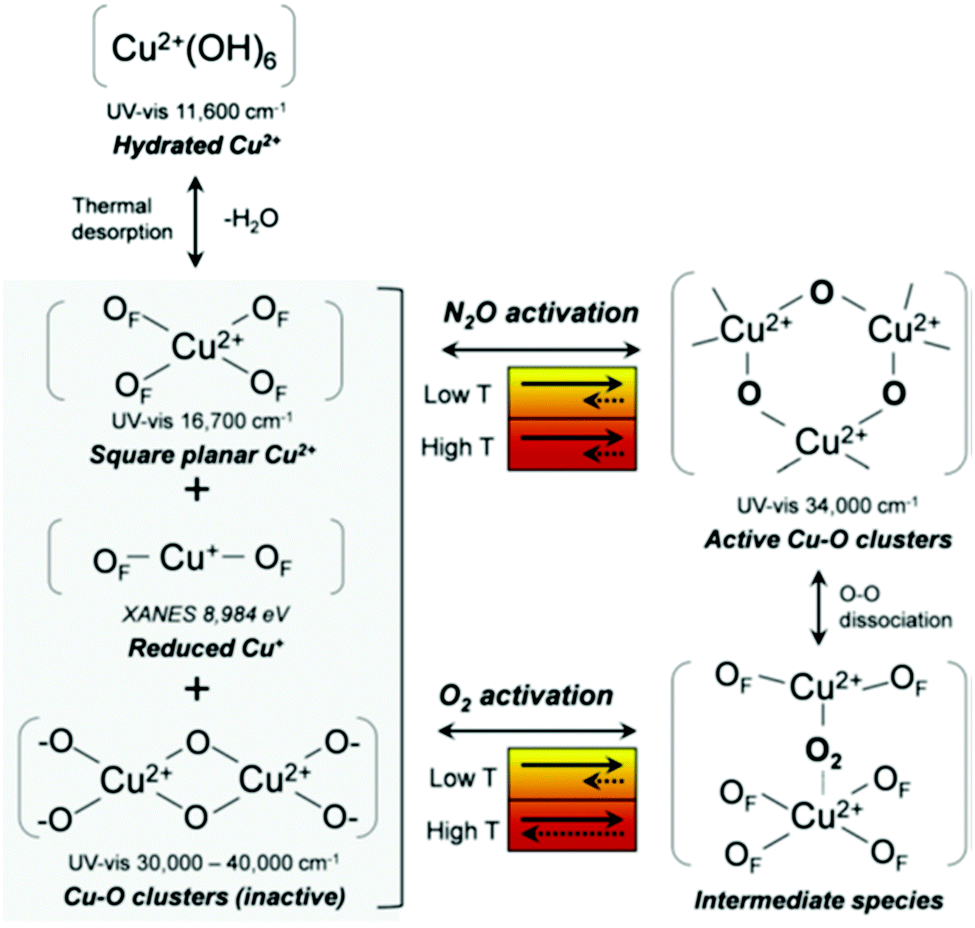 | ||
| Fig. 10 A possible mechanism for the formation of copper species during nitrous oxide and oxygen activation. OF is an oxygen atom in the zeolite framework as suggested by Kim et al.112 Reproduced from ref. 112 with permission from the Royal Society of Chemistry, copyright 2017. | ||
In summary, UV-vis spectroscopy is the method that has been most extensively used to study copper-exchanged zeolites and, specifically, the selective conversion of methane to methanol over them. A plethora of data now exists that reveals a cornucopia of distinct UV-vis transitions in copper containing zeolites of different topologies. However, no consensus or unambiguous specification as to the nature of the copper species that are selectively active in the methane to methanol process has resulted solely from the application of UV-vis.
At present, the only UV-vis band that has a definitive, experimentally derived attribution is that observed at ∼22700 cm−1 in Cu/MOR and Cu/MFI. That this is due to a di-copper mono-μ-oxo species, has been demonstrated by subsequent application of resonant Raman spectroscopy (vide infra) and DFT calculations.81 Moreover, the validity of this assignment has been subsequently confirmed in numerous independent works.26,28,29,33,43,45,61,111,113 In contrast, the attribution of other commonly observed bands at 12000–14000 cm−1 to isolated Cu(II) cations, and at ∼31000 cm−1 to copper trimers remains speculative.
Equally, however, UV-vis studies have shown that the synthesis procedure of the samples, as well as the activation and the reaction conditions, can all have a significant effect on what is observed by this method. By implication, therefore, all these experimental variables can also influence the multiplicity and structure of the copper sites present. For Cu/MOR, the use of the sodium or the proton form for aqueous ion exchange leads to the appearance of completely different bands upon activation: 22700 cm−1 is found for the sample prepared from sodium form and assigned to copper mono-μ-oxo species, while the use of proton form leads to development of the band at 31000 cm−1. Currently, proposed assignments would translate this observation directly to active copper species of different nuclearity (i.e. di-copper and tri-copper respectively).
To conclude, the application of UV-vis to the problem at hand has resulted in a mass of information regarding both the likely speciation of copper species present, within the paradigm of the high temperature route to the activation of copper–zeolites, and in respect to their reactivity with methane. Most of the deductions made, however, rest substantially upon inference and precedent. Only in a few cases (for the 6 MR monomeric sites existing in CHA, and the mono-μ-oxo di-copper species present in MOR) might these assignments be considered as verified by other, structurally direct or symmetry based, techniques (XRD105 and resonant Raman106). What these numerous UV-vis studies have, however, established, is the shear multiplicity of species that can be formed in these systems. Even within a single zeolite topology, the presence of multiple copper sites is almost ubiquitous and seen to vary with the synthetic method applied.
4.3 Raman spectroscopy
Raman spectroscopy is used in catalysis to provide a structural fingerprint by which certain species can be identified. It relies on inelastic scattering, or Raman scattering, of monochromatic light, derived from a laser source in the visible, near infrared, or near ultraviolet range. The laser light interacts with molecular vibrations, phonons, and other excitations in the system, resulting in scattered photons being shifted up or down in energy. As mentioned previously, a potential caveat exists in regard to the use of this method that arises from its non-linear nature: the necessary use of lasers can result in high local power densities being applied to a sample that may, or may not, have the potential to alter the behavior of the system being investigated.That being said, resonant Raman (rR) spectroscopy is an elegant and potentially very powerful method to establish local site symmetry and bonding motifs. The resonant approach makes use of the tunability of the laser source to excite the system at the frequency of the UV-vis transition of interest. This selective excitation results in a vibrational spectrum that is specific to the species giving rise to the UV-vis visible band. As a result, this method greatly enhances the specificity of the UV-vis observation; the vibrational spectrum may be analysed on the basis of symmetry, rather than analogical assignment or inference that is used to interpret UV-vis spectra. This approach has now been used in several cases to elucidate the nature of the Cu oxo species present in various copper–zeolite systems in situations of relevance to the current discussion (Table 2).
| Material | Bands | Assignment | Conditions | Ref. |
|---|---|---|---|---|
| Cu/ZSM-5 | 456 cm−1 | ν symm(Cu–O–Cu) | No in situ rRaman, results for the samples heated in oxygen | 43 |
| 870 cm−1 | ν asymm(Cu–O–Cu) | Assignment confirmed by 18O2 isotope shift | ||
| 1725 cm−1 | 2νsymm(Cu–O–Cu) | |||
| 237 cm−1 | δ symm(Cu–O–Cu) | |||
| Cu/ZSM-5 | 736 cm−1 | ν(O–O) | Found in di-copper peroxo precursor, decomposes into mono-μ-oxo species | 58 |
| 269 cm−1 | ν(Cu–Cu) | |||
| Cu/MOR | 465, 450 cm−1 | ν symm(Cu–O–Cu) | Observation of two types of copper oxo species over Cu/MOR at different conversion levels. Both are reactive towards methane | 81 |
| 850, 870 cm−1 | ν asymm(Cu–O–Cu) | |||
| Cu/SSZ-13, Cu/SSZ-39 | 616 cm−1 | Mono-μ-oxo | First species are stable upon heating, the last three decomposes, being precursors of active CuxOy | 106 |
| 511, 574 cm−1 | ν(Cu–O) of trans-Cu–(O2)–Cu peroxo | |||
| 213, 240, 360 cm−1 | δ(Cu–O) of trans-Cu–(O2)–Cu peroxo | |||
| 836 cm−1 | ν(O–O) of trans-Cu–(O2)–Cu peroxo | |||
| Cu/CHA | 1100–1155 cm−1 | Cu–O–O˙ radical | Formation of two intermediates of copper–oxo species from molecular oxygen | 45 |
| 830 cm−1 | ν(O–O) in Cu–O–O˙ radical | |||
| 507–580 cm−1 | ν(Cu–O) Cu–O–O–Cu peroxo species | |||
| 618 cm−1 | ν(Cu–O) in Cu–O–Cu |
In 2009, work from the Solomon group43 collated the characteristic resonance Raman vibrations of all known structures in Cu–O chemistry, and then compared them to data obtained from oxygen-activated Cu/ZSM-5. In the rR spectrum of activated Cu/ZSM-5, the most intense isotope sensitive vibration is observed at 456 cm−1 (Δ16/18O = 8 cm−1). There is also a weak vibration at 870 cm−1, which exhibits a large 16/18O isotope shift (40 cm−1). Based on frequency, isotope shift and analysis of the angle in Cu2O core, these bands were assigned to symmetric and asymmetric vibrations of mono-μ-oxo di-copper sites (Fig. 11).43
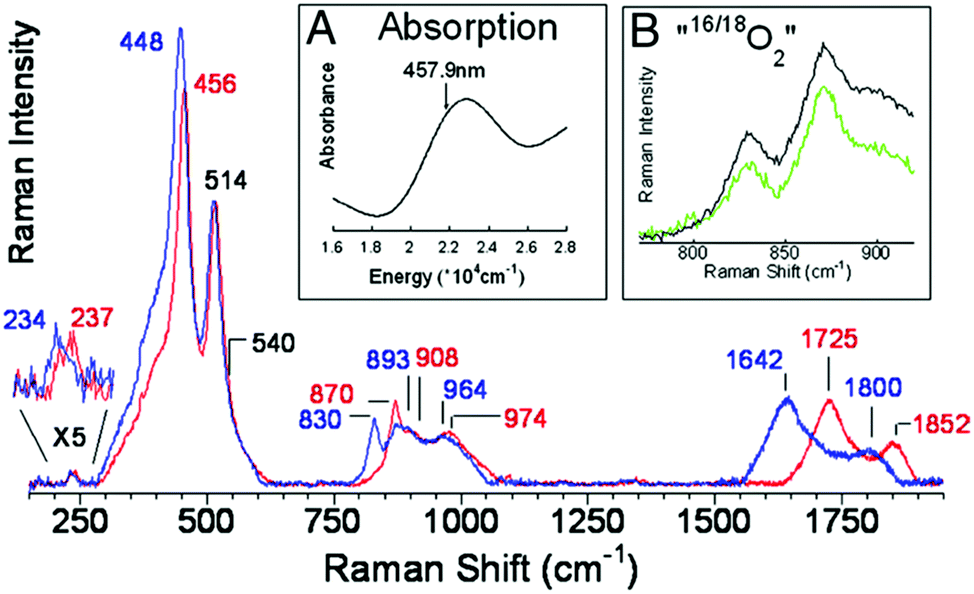 | ||
| Fig. 11 rR spectra (ex. 457.9 nm) of Cu/ZSM-5 16O2 (red), 18O2 (blue). Inset A: Absorption spectrum of oxygen activated Cu/ZSM-5. Inset B: “16,18O2” (green), and 1:1 normalized sum of 16O2 and 18O2 (black). Reproduced from ref. 43 with permission from the United States National Academy of Sciences, copyright 2009. | ||
That the active sites are bis-μ-oxo species, as had been originally suggested, was excluded by rR. No intense 16/18O-sensitive vibration at 600 cm−1, which would be characteristic of a bis-μ-oxo di-copper site, was observed. This deficit, along with an absence of other bands expected from the bis-μ-oxo motif, rule this species out.
Vanelderen et al.81 then showed that rR can be useful in distinguishing different types of copper mono-μ-oxo species found in copper-exchanged mordenite. Both of these species are reactive towards methane upon heating (from UV-vis, vide supra), and were shown to have different apparent activation energies (46 and 61 kJ mol−1 respectively). However, the spectral resolution obtained was not sufficient to observe the expected doublet lines in Raman (465 vs. 450 cm−1 and 850 vs. 870 cm−1); and this multiplicity was only revealed by partial conversion of the more reactive species with methane. Notably, the difference in activity of two copper mono-μ-oxo species was ascribed to the influence of the zeolite lattice that, either directed the approach of methane to the active sites, or altered the stability of the products and transition state.
In more recent work,116 the same group gone further and addressed the localisation of these two copper–oxo sites. This was achieved through monitoring the reaction of both sites with organic substrates of different size: methane, tetrahydrofuran and 2,3-dimethylbutane. This elegant experiment is conceived on the basis that these probe molecules would show different abilities to access copper sites located in 8 MR channels and side pockets of the MOR structure on the basis of their different sizes and steric requirements. Methane, being much smaller, on the other hand, would be able to interact with all copper sites with an essential equanimity. rR spectroscopy showed that no reaction of copper mono-μ-oxo sites resulted from exposure to either tetrahydrofuran or 2,3-dimethylbutane, whereas this species was again shown to be highly reactive toward methane. The copper mono-μ-oxo active sites must therefore exist within a sterically constrained environment within the MOR, and most likely within the 8 MR rings and/or 8 MR side pockets that this structure provides. Furthermore, these measurements provided evidence that van der Waals forces, arising from the approach of methane to the most reactive, and sterically hindered, copper core, have a significant role to play in methane activation and reaction.
Resonant Raman spectroscopy has also been used to study the activation (using oxygen) of copper-exchanged zeolites that must be achieved prior to reaction with methane. Considering that the mono-μ-oxo di-copper sites cannot be formed via simple insertion of dioxygen molecule into the Cu(II) motif, several groups have tried to find the intermediate species responsible for oxygen activation. Smeets et al.58 discovered that the interaction of pre-reduced (to Cu(I)) Cu/ZSM-5 with oxygen at ambient temperature leads to the appearance of rR bands at 736 and 269 cm−1 attributed to di-copper peroxo sites. Similar bands were previously found in μ-2:2 side-on peroxide-bridged copper dimers stabilised by tris(pyrazolyl)borate ligand.117 Later, Ipek et al.106 and Pappas et al.46 found multiple bands (Fig. 12) that they assigned to different types of copper peroxo species and radicals formed within Cu/CHA. Interestingly, the positions and shape of the bands observed over similar materials were not identical (Table 2 and Fig. 11), which is ascribed by the authors to the partial hydration of the sample106 and/or adventitious heating of the sample as a result of the use of the Raman laser.46
 | ||
| Fig. 12 rR spectra for Cu/CHA materials before and after activation in a flow of oxygen at 450 °C.46,106 Reproduced from ref. 46 and 106 with permission from the American Chemical Society, copyright 2017. | ||
In summary, rR spectroscopy has been particularly useful in giving assignments of speciation observed using UV-vis a more thorough and fundamental foundation; this has been especially the case for the UV-vis band at 22700 cm−1 for which kinetic data regarding reactivity toward methane have also been obtained. Taken together, UV-vis and rR have, in this instance, permitted unambiguous assignment of the above mentioned band to the mono-μ-oxo di-copper species, shown that it is manifestly reactive toward methane over a broad temperature range, and provided significant indications as where it may be located within the MOR structure.
Being a resonant technique, however, rR suffers from a fundamental deficit in regards to the quantification of the number or fraction of total copper sites to which a certain band corresponds. As such, even the most intense band might correspond to only a small fraction of the species present in the zeolite, and the potential for misinterpretation of the true import of the spectroscopic results exists. In this respect, the combined use of UV-vis and rR43,81,106 has much to recommend it: quantitative information regarding the relative concentrations of species can be derived from the former spectroscopy, whilst the latter provides the symmetry based specificity that the former lacks.
4.4 Mid-range infrared spectroscopy
FTIR spectroscopy is a versatile and robust characterisation tool for studying zeolite-based materials. It can be used either as a direct probe, through observation of the bands typical for surface species, such as reaction intermediates, spectators, and OH groups, or indirectly via the use of suitable probe molecules which can penetrate the porous system of zeolite and interact with the copper sites present.118–120 FTIR spectroscopy of chemisorbed probe molecules, such as carbon monoxide and nitrogen monoxide, can be used to infer the oxidation state and coordination of transition metal sites, which is particularly useful for studying the speciation of copper in zeolites.121,122 An important issue, which should be kept in mind during the selection of probe molecules for IR studies, is the kinetic diameter of molecules, which must be smaller than the size of the pores of zeolite.Copper-exchanged zeolites have been extensively studied using infrared spectroscopy of adsorbed probe molecules, mainly in respect of their use as catalysts for the selective catalytic reduction (SCR) of NOx by ammonia (NH3-SCR).123–128 Different probe molecules are able to reveal different aspects of the investigated species.93,129–136 The use of weakly interacting probes, such as molecular nitrogen and hydrogen, minimises the potential for perturbation of the system by the probe molecule itself and, it may be argued, enables better/more reliable discrimination between adsorption sites of similar structure, as compared to strongly interacting probes. In respect of the latter, carbon monoxide permits a titration of Cu(I) sites, whereas nitrogen monoxide probes both Cu(I) and Cu(II) sites and therefore aspects of site multiplicity.
Copper-exchanged zeolites are usually prepared via aqueous ion exchange from cupric salts, resulting in the introduction of Cu(II) cations into the framework. Upon thermal activation, cupric ions can undergo auto-reduction to cuprous species, resulting in Cu(I) loaded in zeolites.94 Carbon monoxide has been used to confirm the high coordinative unsaturation of Cu(I) cations hosted in zeolites. Different carbonyl complexes can be formed at Cu(I) sites at different pressure and temperature. For instance, relatively stable adducts such as mono-carbonyl Cu(CO)+ and di-carbonyl Cu(CO)2+ complexes are observed in the 2150–2190 cm−1 region at room temperature. These then transform into tri-carbonyl Cu(CO)3+ species when the system is cooled to liquid-nitrogen temperature (Table 3).
| Material | Probe molecule | Bands | Assignment | Conditions | Ref. |
|---|---|---|---|---|---|
| Cu/ZSM-5 | Pivalonitrile | 2280 cm−1 | C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N coordinated to the surface Cu atoms N coordinated to the surface Cu atoms |
After activation in O2, no correlation with methanol yield | 94 |
| Cu/ZSM-5 | Nitrogen monoxide | 1813 cm−1 | Cu(I)(NO) nitrosyl | Adsorption at RT, linear correlation of the sum of intensities with methanol yield | 63 |
| 1907 cm−1 | Cu(II)(NO) nitrosyl | ||||
| Cu/MOR | Nitrogen monoxide | 1804 cm−1 | Cu(I)(NO) nitrosyls | Inter-conversion of Cu(I) associated bands into Cu+ during the reaction with methane and water | 30 |
| 1731 and 1827 cm−1 | Cu(I)(NO)2 di-nitrosyl | ||||
| 1909, 1948, 1995 cm−1 | Cu(I)(NO) nitrosyl | ||||
| Cu/MOR | Nitrogen monoxide | 1908 cm−1 | Cu(I))(NO) nitrosyls in copper oxo monomer | Different nuclearity of copper–oxo species which depends on Si/Al ratio in the zeolite | 50 |
| 1950, 1995 cm−1 | Cu(II)(NO) nitrosyls in copper oxo oligomers | ||||
| Cu/MOR | Nitrogen monoxide | 1906, 1950, 1995 cm−1 | Cu(II)(NO) nitrosyls in copper oxo species | The copper speciation does not depend on the amount and nature of co-cations (Na+ or H+) in mordenite | 100 |
| Cu/MOR | Carbon monoxide | 2159 cm−1 | Cu(I)(CO) carbonyl | Bands are present on activated sample due to autoreduction, increase the intensity upon reaction with methane and decrease the intensity being reactivated in water | 50 |
| 2179 and 2151 cm−1 | Cu(I)(CO)2 dicarbonyl | ||||
| Cu/FER | Carbon monoxide | 2157 cm−1 | Cu(I)(CO) carbonyl | Found in the spectra after activation at 450 °C. Intensity increases with the increase of copper loading | 52 |
| 2178 and 2151 cm−1 | Cu(I)(CO)2 dicarbonyl | ||||
| Cu/MOR | Hydrogen | 4031, 4063 cm−1 | H2 coordinated to copper–oxo-oligomers | Only oligomers can adsorb molecular hydrogen at low temp, no bands over copper oxo monomers | 50 |
| Cu/CHA | — | 3650 cm−1 | ν(OH) in Cu(II)–OH+ species | Activity goes up together with the intensity of this band | 46 |
| Cu/SSZ-13 | 3654 cm−1 | ν(OH) in Cu(II)–OH+ species | Are responsible for activity, correlate with methanol yield | 114 |
Dinitrogen interacts with cuprous (Cu(I)) ions inside zeolites and leads to a ν(NN) band in the 2300–2290 cm−1 region, which is shifted with respect to gas phase (Raman active/IR inactive) band at 2321 cm−1. Donor–acceptor interactions with Cu(I) ions weaken the NN bond and result in the induction of a dipole that makes the resulting vibrational structure observable in IR and causes the observed red shift in the Cu(I)⋯N2 adducts relative to the gas phase vibration. The interaction of N2 with Cu(II) is considerably weaker.
Nitrogen monoxide, being isoelectronic to carbon monoxide, can also interact with copper species in zeolites. The main difference between CO and NO is that the additional electron in nitrogen monoxide occupies the π*-orbital. This causes greater sensitivity for NO to the electronic state of the copper cation during the formation of Cu⋯NO bond, and leads to structure-sensitive bands in the 1700–2000 cm−1 region of the IR spectrum. NO is currently used to infer the oxidation state of copper cations in zeolites, due to its ability to form stable nitrosyl adducts with both Cu(II) and Cu(I) cations. To suppress NO decomposition, however, NO must be dosed at liquid-nitrogen temperature to be used as a probe molecule on copper sites.
In work by Beznis et al.,63 two probe molecules having different kinetic diameter (pivalonitrile and nitrogen monoxide) were used to differentiate the copper species located in the pores and on the surface of Cu/ZSM-5 activated at 623 K.
While for pivalonitrile an absorption band at 2280 cm−1, attributed to the interaction with copper surface sites, showed no correlation with methanol yield, the sum of intensities of the bands due to Cu(II) and Cu(I) nitrosyls showed a linear correlation with methanol yield extracted after reaction. However, as the experiment was made at room temperature, therefore NO disproportionation and reaction with copper sites may have occurred, leading to potential complications to the interpretation of the result.137,138
Sushkevich et al.30 have recently gone further to study a Cu/MOR sample after activation, reaction with methane, and re-activation with water, during anaerobic methane conversion into methanol. Through the use of carbon monoxide and nitrogen monoxide as probe molecules (Fig. 13), they showed that the activated sample is composed of a mixture of Cu(II) and Cu(I) sites. Cu(II) sites adsorbed NO yielding bands at 1909, 1948 and 1995 cm−1, and Cu(I) sites were characterised by a set of the bands at 1731, 1804 and 1827 cm−1 for adsorbed NO, and 2151, 2159 and 2179 cm−1 for CO). In this case the source of Cu(I) was thermally/vacuum induced auto-reduction (Table 3). Analysis of the spectra obtained after the different steps of the subsequent reaction and reactivation process showed that the Cu(I) sites, formed by reduction of initial Cu(II) by methane, could be rejuvenated using water as an oxidant. Moreover, this was achieved with no change in the IR frequency of carbonyl and nitrosyl vibrations showing that the copper speciation during the stoichiometric cycle also remained unchanged; the overall activation/reaction cycle was therefore shown to be ostensibly reversible. The reoxidation of Cu(I) to Cu(II) with water was, however not complete due to the weak oxidation properties of the latter. However, no certain assignment of the IR bands to the particular copper sites was made.
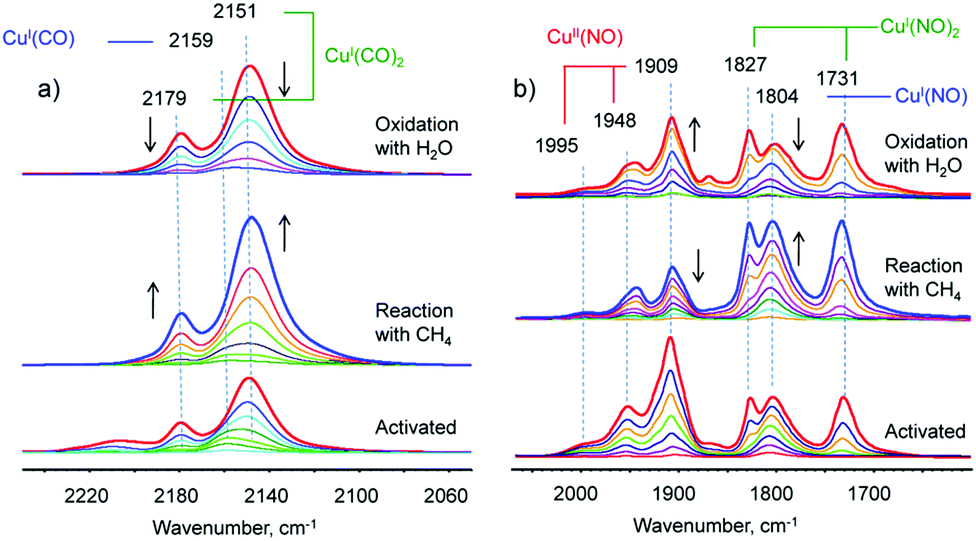 | ||
| Fig. 13 (a) FTIR spectra of CO adsorbed at 100 K onto Cu/MOR that was vacuum-activated (bottom), reacted with methane (middle), and reoxidised with water vapor (top). (b) FTIR spectra of NO adsorbed at 100 K onto Cu/MOR that was vacuum-activated (bottom), reacted with methane (middle), and reoxidised with water vapor (top).30 Reproduced from ref. 30 with permission from the American Association for the Advancement of Science, copyright 2017. | ||
Pappas et al.52 have also used the infrared spectrum of adsorbed carbon monoxide for studying the copper site in Cu/FER (Fig. 14). After the saturation of the samples with CO at ambient temperature, three bands at 2157, 2151 and 2178 cm−1 due to the copper mono and di-carbonyls were found. Importantly, the copper loading shows a significant influence on the intensities of the bands: the higher copper content is, the more intense bands develop in the IR spectrum.
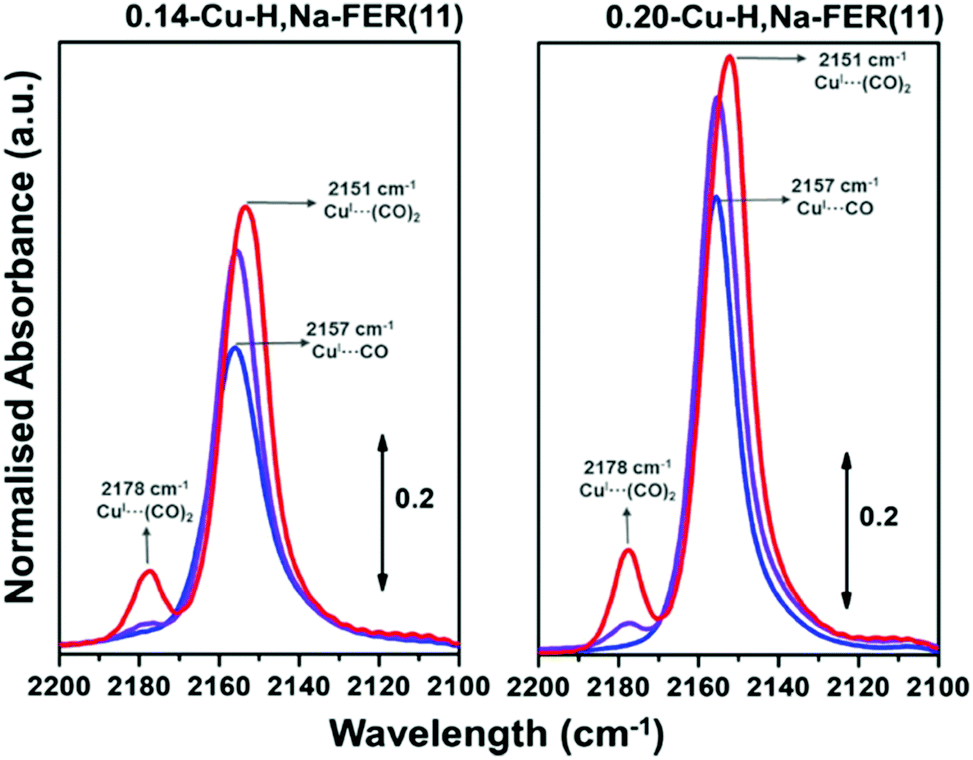 | ||
| Fig. 14 IR spectra of CO dosed at ambient temperature on 0.14Cu-H, Na-FER(11) and 0.20Cu-H, Na-FER(11). Blue, purple and red lines correspond to low, medium and high PCO. The samples where activated in vacuum at 150 °C for 1 h, 300 °C for 1 h and 450 °C for 1 h.52 Reproduced from ref. 52 with permission from Wiley, copyright 2018. | ||
In addition, Sushkevich et al.30 demonstrated that FTIR spectroscopy can be used for the in situ monitoring of the activation of methane to yield methoxy species (characterised by set of bands at 2980, 2963, 2869 and 2856 cm−1), which are stabilised by the zeolite. They found a linear correlation between the number of Brønsted sites generated through reaction with methane with the relative amount of methoxy species that permitted a further clarification of the reaction mechanism of methane activation. This measurement verified that hydrogen atoms abstracted from the methane molecule during activation by copper centres could then be transferred to an Al–O–Si to create Brønsted acid sites.
In contrast, and for the case of the small pore Cu/CHA system, Pappas et al.46 suggested copper monomers, in a form of Cu–OH+, to be related to the efficient activation of methane. Using FTIR spectroscopy they showed the band at 3560 cm−1, assigned to the O–H stretching vibrations in Cu–OH+ motif, to decrease in intensity as the activation (in oxygen) temperature was increased. Over the range of temperatures investigated (623–773 K) the methanol yield was observed to augment by a factor of ca. 2.5; an inverse relationship between the IR band at 3560 cm−1 and methanol yield was, therefore, demonstrated.
Whilst the authors concluded that Cu–OH+ species were unlikely to be the species directly responsible for methane activation, this relationship prompted them to suggest that they might act as a precursor to the active species through some form of temperature dependent evolution in oxygen. However, no quantitative link between the diminution of this IR feature and, for instance, methanol yield or the formation of Cu(I) (from XAFS, vide infra), could be established and therefore any idea of how much of the copper was originally present as Cu–OH+.
This unfortunate deficit, however, does allow for another interpretation; that the removal of this band during activation might result from the annealing away of any extra-framework aluminium present in the samples during the activation process. The Al–OH vibration due to such aluminium appears in a very similar part of the IR spectrum139 and, a priori, it is not an easy matter to decide between the Cu–OH+ and Al–OH possibilities.
The relationship observed between this OH band and methanol yield could, therefore, also be the result of the extra-framework Al being reincorporated into the zeolite framework. Once re-established within the framework this Al may contribute to the creation of new active sites and, therefore, contribute to methane activation and methanol production. The precise and unequivocal determination of how this band, and the speciation that accompanies it, relates to successful and selective oxidation of methane in the Cu/CHA system therefore awaits definitive confirmation.
In a further study, the use of adsorbed nitrogen monoxide was extended to investigate the nature of copper speciation in Cu/MOR samples across a range of different Si/Al ratios (Fig. 15a).50 From Fig. 1(c) we have already seen that the role of this compositional variable is one of the least well defined aspects of these systems. Hypothetically, increasing the Si/Al ratio diminishes the probability of Al pairs existing within any given zeolite pore. As a result, and on the assumption that ligation of copper to the pore walls requires the presence of aluminum, the probability of forming multinuclear copper–oxo species should also diminish.
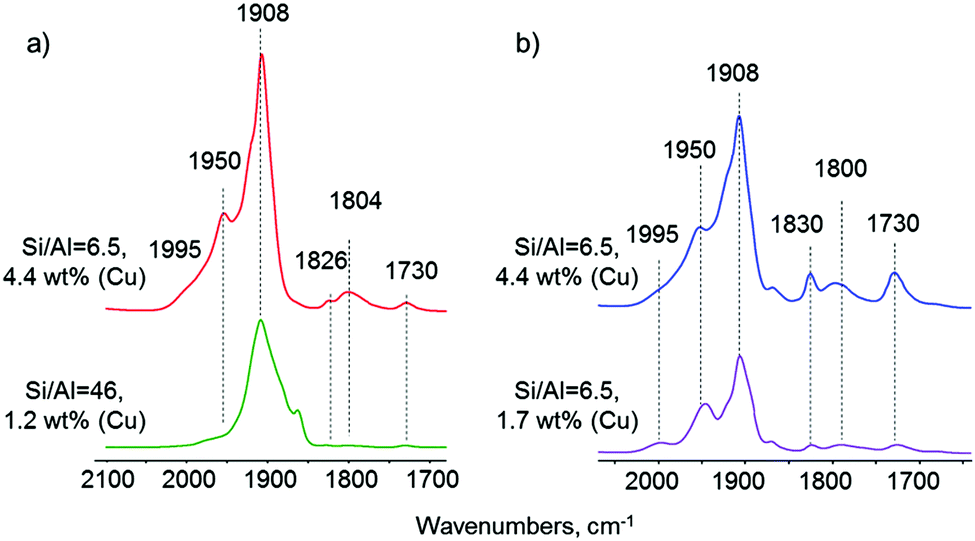 | ||
| Fig. 15 FTIR spectra of NO adsorbed over Cu/MOR samples with (a) different Si/Al ratio and (b) different copper loading at 77 K.50 Reproduced from ref. 50 with permission from Wiley, copyright 2018. | ||
It was found that the Si/Al ratio has a strong effect on the IR spectra of adsorbed nitrogen monoxide. In a sample having a Si/Al of 46 the IR of adsorbed nitrogen monoxide is dominated by a band at 1908 cm−1 ascribed to the presence of copper monomers. For samples with progressively lower Si/Al ratio this band is joined by other, higher frequency absorptions at 1950 and 1995 cm−1. In other words at lower Si/Al ratios a mixture of copper–oxo sites is observed, with species of higher copper nuclearity joining the population of monomers.50
Considering the high activity (per copper) and selectivity to methanol of the Cu/MOR sample, a model involving “monomer pairs”, rather than pre-existing di-copper species, was suggested.50 According to this model, copper monomers can be present in the Cu/MOR pores at distances that are short enough to enable reaction with methane, and the required electron and proton transfer, as confirmed by DFT.
Importantly, decreasing the copper loading in MOR does not lead to any significant change in IR visible speciation. Furthermore, adjustment of the copper salt concentration, to achieve the copper loading of 1.7 wt%, i.e. similar to that in Cu–MOR with Si/Al = 46, does not result in a significant variation of the IR spectrum either, and only the intensities of the bands change (Fig. 15b). This observation suggests the considerable importance of the Si/Al ratio for the structure of copper sites hosted in zeolites.
The IR of adsorbed molecular hydrogen was also shown to be useful in studying the aggregation of copper–oxo sites in zeolites: monomeric copper sites are unable to stabilise molecular hydrogen. On the other hand, multi-nuclear copper–oxo species, such as dimers and trimers, can adsorb molecular hydrogen to yield IR bands around 4031 and 4063 cm−1.50 The latter were visible for the Cu/MOR samples with a low Si/Al ratio, and indicate the presence of copper–oxo oligomers; the sample with high Si/Al of 46, on the other hand, predominantly yielded bands at ∼4100 cm−1 that correspond to the interaction of hydrogen with isolated and bridged OH groups of zeolite.
Using nitrogen monoxide adsorption, it has also recently been shown that, in contrast to variation of Si/Al ratio, the variation of amount and nature of co-cation in Cu/MOR does not affect the nature of copper–oxo sites.100 Presence of sodium cations left in zeolite pores after the cation exchange does not lead to any significant change of the IR spectrum of adsorbed nitrogen monoxide, as compared to that resulting from Cu/MOR prepared from a pure proton form. We note here that previous observations reported a strong effect of the co-cation on the UV-vis spectrum of Cu/MOR (vide supra).110 The source of this disparity cannot be precisely defined, though most likely has its origin in the different sensitivities of the respective techniques in respect of the local electronic and coordination environments of the copper.
IR spectroscopy, as applied to study aspects of the methane-to-methanol process, provides an opportunity to study the copper sites in the zeolite and to monitor the surface species formed during the reaction. At present, only a relatively small number of works devoted to this field exist, almost exclusively for the Cu/MOR case. A more extensive utilisation of infrared spectroscopy, both in situ, and via the use of probe molecules, would appear both warranted and highly informative.
4.5 X-ray spectroscopic studies of speciation and reactivity in copper-containing zeolites for methane to methanol conversion
Cu K-edge X-ray spectroscopy (XAS) is extensively used to investigate the conversion of methane to methanol in zeolitic systems.29,44,46,75,140–142 Uniquely, XAS can address the Cu in an elementally specific and in situ manner. It offers the possibility of quantitative determination of copper oxidation states (XANES) and insight into the local structure/ligand coordination and geometries (EXAFS) of the copper under any given circumstance.143,144Table 4 summarises the features that may be observed in Cu K-edge XANES, their origins, and the types of speciation they may be associated with.| Sample/standard | Oxidation state | 1s → 3d (eV) | 1s → 4p (eV) | Absorption maximum (eV) |
|---|---|---|---|---|
| Copper foil | 0 | 8980.5 | 8993.3 | |
| Cu2O | 1 | 8981.1 | 8994.4 | |
| CuO | 2 | 8977.5 | 8984.8 | 8996.2 |
| Cu(OH)2 | 2 | 8977.0 | 8996.9 | 8996.9 |
| Cu/MOR | 2 | 8977.2 | 8986.3 | 8997.0 |
| 1 | 8983.6 |
The weak 1s → 3d pre-edge transition is dipole forbidden – but quadrupole allowed – and is present in spectra of CuII and CuIII but not CuI or Cu0. This feature is sensitive to the symmetry of ligand binding around the copper and can shift by up to ca. 1 eV depending on the ligand field strength and symmetry.145–147 In CuIII it typically shows a binding energy that is ca. 2 eV higher compared to CuII.146
The 1s → 4p transitions are combined with ligand related shakedown of electrons into the Cu 3d core hole149 and are thus sensitive to both the core-hole charge (oxidation state) and the effect of the ligand environment. In the case of copper hosted in zeolites these features are firstly – for CuII – sensitive to the degree of hydration of the copper and, secondly, the formation of CuI. As a result, the position (and indeed intensity) of these features can therefore change significantly compared to bulk standard compounds. For instance, there exists a ca. 1 eV shift in the 1s → 4p transition due to CuI between the standard CuI reference (Cu2O) and that observed for Cu/MOR.80,142
The traditional step-wise and high temperature, approach for methane oxidation starts with the activation of copper-exchanged zeolite in oxygen at elevated temperature that leads to the formation of copper-containing sites active for the desired conversion. Being interested in determination of the structure of these sites, specifically copper oxidation state and local geometry, numerous researchers have monitored this process using XANES, that have resulted in similar conclusions.27,29,31,32,44,115 Briefly, as-made materials contain Cu(II) in a fully hydrated state, yielding the peaks at ∼8997 eV in XANES and an average first shell Cu–O coordination close to 6. The absence of any well-defined second coordination sphere in the EXAFS indicates a high level of disorder (mobility) of the hydrated copper, and the presence of highly symmetrical Cu(II)(H2O)6−n(OH)n ions in the non-calcined materials.
Heating of this material (in either oxygen or helium) initiates a gradual dehydration and the formation of more rigidly anchored copper species. This is evidenced by the evolution of the shoulder at 8986 eV in XANES spectrum together with the appearance of more defined scattering shells beyond the first in EXAFS.
At temperatures above ca. 723 K in oxygen, complete dehydration is achieved. In this state EXAFS (vide infra) reports Cu–O coordination numbers within the range of 3 to 4 and suggests the anchoring of copper–oxo species to the zeolite framework.
However, if high temperature activation is done in inert environment, i.e. flow of helium or vacuum, the reported results differ significantly and Cu(I) formation is observed.31,32,80,95 For some samples, this occurs at temperatures as of ca. 473 K and the pre-edge feature at 8983 eV, indicative of Cu(I), progressively develops till 723–773 K. As such, under anaerobic activation conditions, reduction of the copper is readily achieved. Quantitation by linear combination analysis (LCA) of XANES spectra shows that at 773 K, the Cu(I) content can reach >80%. Simultaneously, the first shell coordination number decreases to 2.5–3 indicating the loss of oxygen by copper species, in line with appearance of XANES signatures of Cu(I). For other samples, the appearance of Cu(I) is evident only from 673–723 K, and the total amount does not exceed 50–60% even at 1000 K.95,150–152 In most cases, this behaviour has been associated with “autoreduction”. However, as was recently pointed by Occhiuzzi et al.153 and Sushkevich et al.94 the reduction of copper at low temperature can be associated with the interaction of CuII with residual carbonaceous species left in the materials after the synthesis. Using EPR, XAS and on line mass-spectrometry they show that as-made copper exchanged zeolites still contain a considerable amount of carbonaceous deposits, which during the heating in inert gas interact with copper species to yield Cu(I) and carbon dioxide. In light of that, the high temperature activation in oxygen is essential for complete removal of these carbon-containing species and must be included into all synthetic protocols for the materials tested in methane conversion to methanol. In situ oxidation therefore is preferred as it minimises the possibility that such reduced copper species will form adventitiously. By doing this one can exclude the impact of potentially confusing and deleterious side reactions that interfere with the production of active CuII species. Furthermore, the oxidation of carbon impurities instead of methane could impact the overall production of carbon dioxide observed and result in an underestimation of the selectivity achieved.
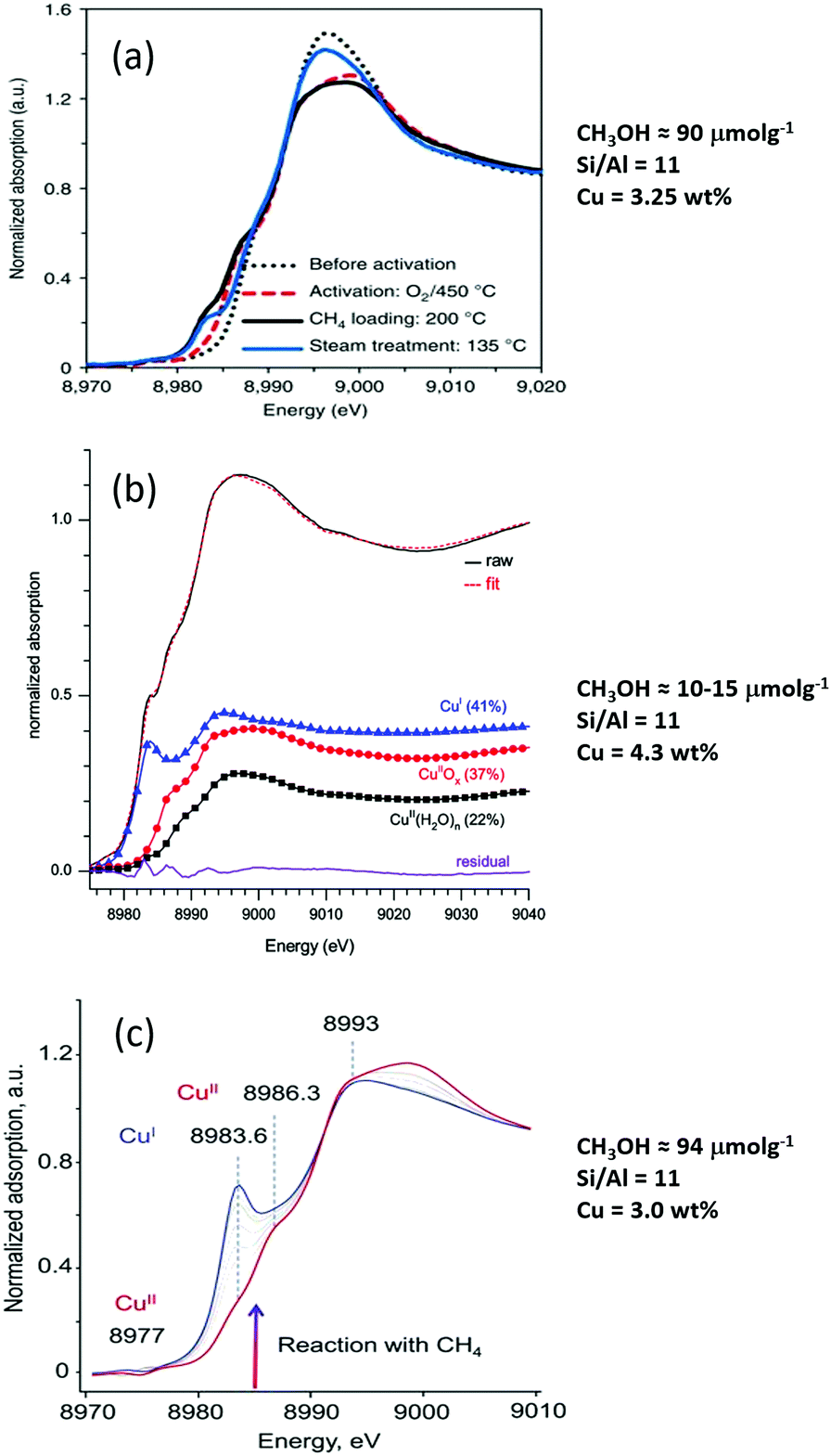 | ||
| Fig. 16 Examples of Cu K-edge XANES derived from similar Cu/MOR samples subjected to high temperature activation in oxygen at 673–723 K and then expose to methane at 473 K along with their independently derived yields of methanol from separate reactivity (batch and flow) studies. (a) From Grundner et al.;29 (b) from Alayon et al.;44 and (c) from Sushkevich et al.30 Individual graphs reproduced from ref. 29, 44, and 30 with permission from Nature publishing Group, The Royal Society of Chemistry and the American Association for the Advancement of Science, copyrights, 2012, 2015, and 2018. | ||
In Fig. 16 we see that all these features are present in the spectra from various Cu/MOR samples, and they occur with a wide range of intensities according to the example and the treatment applied.
Indeed, the outstanding feature of this side-by-side comparison lies in the remarkable variation in the results obtained from systems that are nominally very similar in terms of their composition (if not their established reactivity), and the treatments that have been applied to them. Moreover, and of most import to the current discussion, this variation contains within it potential evidence for the two most currently debated reaction mechanisms (vide supra, Section 2, Fig. 5). In two of these cases the Cu K-edge EXAFS (vide infra) has also been analysed to provide experimental evidence for the formation of different (dimer80 and trimer29) active copper sites.
Two of these cases ((a) and (b)) might be considered as providing evidence in favour of the DFT derived (for both dimer and trimer active sites) Cu–O˙ (oxyl) radical based mechanism.29,67 In (a)29 a high yield (and selectivity) of methanol is equated with a low yield of CuI; in (b),44 a low yield of methanol (and, implicitly, an enhanced selectivity for over-oxidation products) can be equated with much more significant formation of CuI. Both of these observations would be compatible with the general expectations of the Cu–O˙ radical based mechanism wherein CuI formation is associated with over-oxidation of methane and not with its selective oxidation to methanol. Case (c),30 on the other hand, produces both a high yield and selectivity for methanol yet, at the same time, a high yield of CuI; this case would be far more in keeping with the expectations of the more conventional CuII/CuI redox mechanism as the foundation for the selective conversion of methane to methanol.
Firstly, given the similarity of the samples in each case, it seems unlikely – though we cannot in any absolute sense rule this out – that the source of the observed variations lies with the different Cu/MOR materials used. Indeed, the fact that notionally similar zeolite materials can yield some degree of variability in copper speciation has been reported (vis à vis evidence from UV-vis110 – see Section 4.2, though no such differences could be noted from subsequent IR studies using NO – Section 4.4). It is the case that each experiment shown in Fig. 16 utilised a different commercial MOR source and the protocols (starting counter ion and copper salt used) for the exchange of copper were different. However, given the extreme nature of the differences observed in these three XAS experiments, we must consider other, potentially more likely, reasons for these different results.
Secondly, therefore, we might consider the possibility that the formation of Cu(I) in these systems from Cu(II) could be influenced by the application of the X-rays themselves. It is the case for homogeneous copper systems that the formation of Cu(I) from Cu(II) precursors as a result of conducting X-ray experiments at the Cu K-edge has been observed and, in some cases, quantified.91 For copper–zeolite systems the phenomenon of auto-reduction of some portion of CuII initially present in a system has also been reported93,94,152–155 though not necessarily ascribed to any influence of X-rays. Aside from these observations, that the application Cu K-edge energy.
X-rays might adversely affect measurements in these sorts of systems, has not, to our knowledge and to date, been openly discussed or investigated in the literature. In this respect, therefore all that we may currently do is to compare the nature of the beamlines involved i.e. (a) B18156 at the Diamond Light source, UK, and ((b) and (c)) SuperXAS157 at the Swiss light source. If we do this we find, that in terms of the magnitude of the power densities they apply to the sample, given the types of beam sizes used in the studies shown in Fig. 16, these two X-ray sources are rather similar at the Cu K-edge and therefore, unlikely to be the source of the observed variations.
Lastly the possibility that some of these experiments have fallen foul of the numerous sensitivities of copper hosted in zeolites outlined in Section 3, i.e. inaccurate or unreliable temperature measurement (too high or too low), insufficiently pure gases and/or the presence of leaks, or a combination of these, must be admitted. At this point, however, any possible explanation for this remarkable considerable variation can only remain just that; a possibility. Moreover, whilst recognition of these possibilities is objectively required, it does not help us understand which, if any, of these experiments have yielded a valid and useful result? As such, another way to resolve this rather fundamental issue is required.
To go further, and also to provide a foundation for a later discussion of the information that can be derived from analysis of Cu K-edge EXAFS, we must firstly remind ourselves of the general expectations of the two opposing mechanisms that are currently debated and that were outlined in Section 2 (Fig. 5). The most significant difference between these mechanisms is the implied role of CuI in the methane to methanol process. Within the mechanism founded upon the handling of electrons via a Cu–O˙ radical, this is implicitly associated with the formation of over oxidation products; in the CuII/CuI redox model it is associated with both the formation of methanol and over-oxidation products. As such, given a knowledge of the number of electrons required to be handled for the different products to be (see Section 2), a rational basis for deciding which mechanism is at work can be arrived at; but only if we can reliably and quantitatively assess both reactivity and the levels of CuI formation that might accompany that reactivity.
As already noted in Section 3, the intrinsic nature of these materials has thus far prevented a single in situ experiment achieving this desirable objective, as product formation may only be established post factum. Fig. 17, however, collates the results of a recent study that attempts to make the required correlation, using independently made measurements of methanol production and CuI formation for the same treatments, and for wide range of samples and approaches to activation.75,142 For the detailed elucidation of the steps required to achieve these data, and most specifically the accurate quantification of CuI under reliable conditions of experiment, the reader is referred to ref. 142.
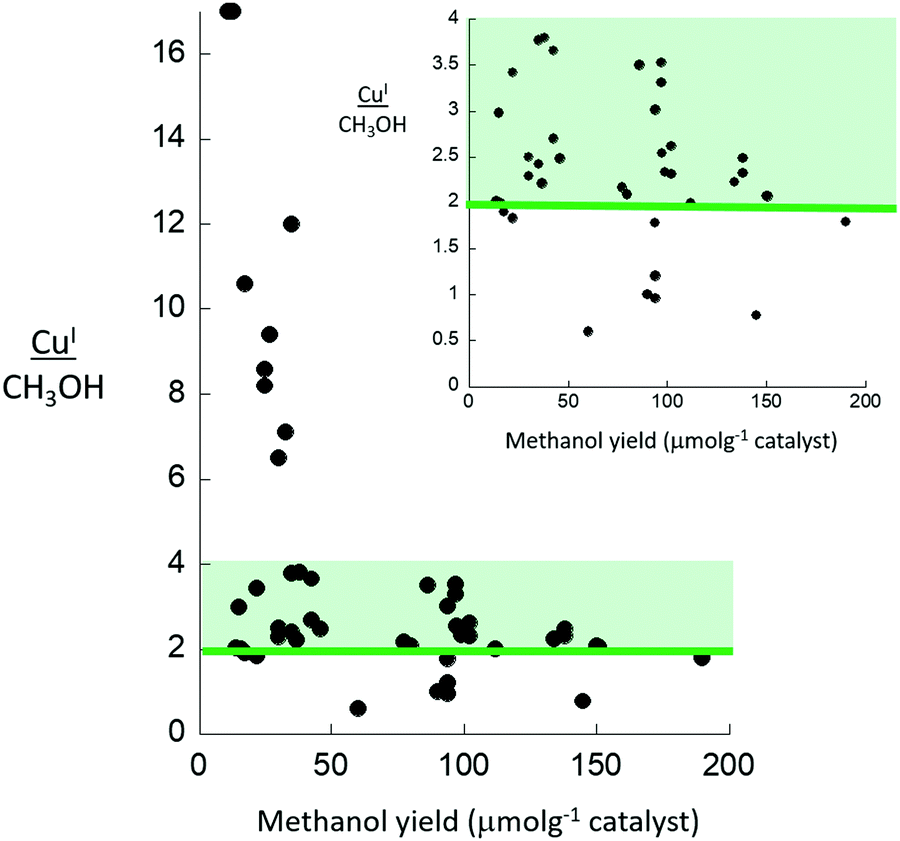 | ||
| Fig. 17 Collation of literature available data relating CuI formation as measured by Cu K-edge XAS to independently established methanol yields for both high temperature and isothermal routes to methane conversion to methanol for Cu/MOR, Cu/MAZ, Cu/ZSM-5, and Cu/CHA systems. CuI/CH3OH = 2 is the expectation value for a CuI/CuII redox mechanism that involves the transfer of 2e−, and the participation of two copper atoms, and that is 100% selective for methanol. The inset shows an expanded region of the plot highlighting the predominance of results in the 2 < CuI/CH3OH < 4 range along with a small cluster of results that have returned anomalously low CuI/CH3OH ratios. Data extracted from ref. 27, 29, 35, 46, 75 and 148. Adapted from Newton et al.142 | ||
Recalling that, for a mechanism that has CuII/CuI redox as being the central element to effective conversion of methane, a 100% selectivity for methanol (a 2e− process) should result in a ratio of CuI formed (from Cu K-edge XANES) to methanol obtained (from reactivity studies) of 2, then the implications of this study are clear.
A considerable majority (>70%) of the systems studied, be they under conditions of high temperature or isothermal approaches to activation, produce CuI/CH3OH ratios of between 2 and 4; precisely in line with the expectations of the CuI/CuII redox mechanism for a production of methanol that is highly, if not 100%, selective. Furthermore, for some other cases wherein a relatively high ratio is indicated, it can be shown142 that these high ratios result from low selectivity rather than any other factor. Once the relative electron counts for methanol and carbon dioxide production are taken into account, then these instances are also found to be consistent only with the CuI/CuII redox model.142
What further emerges from this study is that we can now put the individual results shown in Fig. 16 in a deeper perspective. Both the results shown in Fig. 16(a)29 and Fig. 16(b)44 are now seen to be anomalies within a considerably expanded population of experiments. It is only the result shown in Fig. 16(c)30 that can now be seen as commensurate with the vast majority of results shown in Fig. 17.
These results establish that the fundamental basis of the selective conversion of methane to methanol in copper exchanged zeolites is a CuII/CuI redox couple that involves two Cu atoms per methanol molecule produced. Concurrently no evidence for the opposing Cu–O˙ radical based mechanism is forthcoming from this study. However, this does not exclude the involvement of Cu–O˙ radicals within the overall reaction mechanism as short-lived intermediate. Moreover, DFT calculations67 predict the existence of the resonant Cu–O˙ anion-radicals in equilibrium with conventional Cu(II)–O2–Cu(II) sites. As such, radical-like species might still be found to play a key role in the activation of methane.
However, the approach given in Fig. 17 does not shed any light onto the structure of the species responsible for the conversion. Whilst Cu K-edge XANES can provide a quantitative handle on the different CuI oxidation states present as well as their relative proportions, and general statements can be made regarding the likely coordination and geometry of CuI complexes as opposed to CuII complexes, at this level of interrogation the XANES itself can go no further and we must consider the EXAFS.
 | ||
| Fig. 18 Top: Cu K-edge EXAFS data from Grundner et al.29 pertaining to high temperature activated Cu/MOR considered as due to trimeric (Cu3O3)2+ species. Bottom: Cu K-edge EXAFS derived in the study of Alayon et al.80 for a similar Cu/MOR sample subjected to the same activation protocol, and considered as due to di-copper species. Reproduced from ref. 29 and 80 with permission from Nature publishing Group, The Royal Society of Chemistry, copyright 2015, and 2018. | ||
What is evident is that the EXAFS envelopes, in considerable contrast to the XANES shown in Fig. 17, appear similar in each case. As such, we are bound again to ask the question: can both of these studies be correct in their assertions regarding the structural information contained within these datasets?
Fig. 19 goes a step further to compare directly the Cu K-edge XANES (Fig. 19(a) to the k3-weighted Cu K-edge EXAFS (Fig. 19(b) and (c) obtained from four different Cu/MOR samples in their oxygen activated states. Each of these samples (one measured on two separate occasions on two different beamlines), have been activated in oxygen at 723 K and then reacted with methane at 473 K. The levels of CuI produced within this sample vary from ca. 5 to 50% when placed in contact with methane, a fact that also implies, given the results shown in the previous section, that the activated samples comprise anywhere between 95 and 50% of copper that is inactive toward methane activation at this temperature.
 | ||
| Fig. 19 (a) Cu K-edge XANES derived from four different Cu/MOR samples after high temperature activation in oxygen and subsequent exposure to methane at 473 K. (b) the k3-weighted Cu K-edge EXAFS derived from each sample under oxygen at 723 K. (c) The Fourier transform of the k3 weighted data for each case. (Adapted from ref. 142.) | ||
As such, these systems must be initially composed of a mixture of CuII sites. Yet, the Cu K-edge EXAFS obtained from their activated states is, to all intents, the same in each case. That this can be so is strongly suggestive that, in these cases, Cu K-edge EXAFS has no powers of discrimination between those CuII sites that react with methane to form CuI sites and those that do not. Indeed the only insight that might arise from these data is that the active and inactive CuII species appear very similar to each other within the decidedly local field of view afforded by the EXAFS experiment.
Moreover, the similitude that exists between the Cu K-edge EXAFS derived from the topologically rather different zeolites (CHA and MOR), and across the stages of high temperature activation, methane exposure, and methanol extraction, that comprise the methane to methanol cycle, has also recently been noted and commented upon141 and a summary of these results is given in Fig. 20.
 | ||
| Fig. 20 Top panel: Copper K-edge XANES spectra derived from differently loaded (L = low, H = high) Cu/CHA and Cu/MOR samples under steady-state conditions (a) at the end of the oxygen activation step at 500 °C, (b) at 200 °C in helium, after the oxygen activation step and subsequent cooling down in oxygen; (c) at 200 °C at the end of the methane loading step; (d) at 200 °C at the end of the methanol extraction step. Middle panel: The corresponding Fourier transforms of the k2-weighted Cu K-edge EXAFS. Bottom panel: Coordination numbers and bond distances extracted from the EXAFS for Cu–O, Cu–Cu, and possible Cu–Si/Al scattering shells.141 Reproduced from ref. 141 with permission from Elsevier, copyright 2019. | ||
Once more, it is evident that even though the Cu-XANES shows decided differences, specifically in terms of the levels of Cu(I) present at each stage of the methane to methanol process, and between sample types, the EXAFS envelope remains remarkably consistent, as do the extracted coordination spheres.
The authors further admit that they cannot rule out that the sphere of coordination they associate with Cu–Al/Si bonding (i.e. to the wall of the zeolite; ca. 2.8 Å) is not convoluted with further shell due to Cu–Cu scattering.141 On this point it is very worthy of note that most structures for dimeric and trimeric Cu sites derived from DFT studies (see for instance Section 2) contain within them Cu–Cu and Cu–Al co-ordinations in and around 2.8 Å from the Cu atom and that differ from each other in terms of bond distance <0.1 Å. It is also worth noting, as circumstantial evidence for the likely nature of the species present after activation, which in both these studies (Fig. 19 and 20) the first shell Cu–O coordination sphere returns coordination numbers that are consistently closer to 3 than to 4. Such a situation, given that in these cases the copper is present solely as Cu(II), can only be achieved, on the basis of the DFT generated structures given in Fig. 4, by monomers, the mono-μ-oxo di-copper species, or mixtures thereof.
Moreover, that this shell can be effectively modelled using a single Cu–O bond distance of between 1.9 and 2 Å is, at first sight, at odds with the nature of the Cu–O bonding indicated to be present within DFT generated structures: this apparent discontinuity we shall consider further in a later section.
The sum of these examples, along with two more recent analyses of Cu K-edge EXAFS for nominally similar Cu/MOR wherein the second shell coordination is fitted to of either Cu–Al32 or Cu–Cu29,115 scattering systems, strongly suggests that the use of EXAFS, at least as it is commonly analysed, is hobbled by numerous factors. The result is that reliable extraction of the true composition of the second coordination shell of the Cu sites (both active and inactive), and therefore their structure, is not possible. Consequently, any specification of active site structure using this method must be considered as unfounded.
The factors that contribute to this impasse are many fold. Chief among them, however, would be that the vast majority of evidence (vide supra) suggests that we are faced with multiple, rather than single, site occupation by the copper. This, allied to thermally induced disorder, and a lack of any other information that may be used to narrow down the available parameter space – this is particularly the case when considering the presence of Cu–Cu and Cu–Al scattering paths that may overlap to a great degree – conspire to render the otherwise powerful spectroscopy of little utility in these circumstances.
In respect of the thorny issue of the structure and nuclearity of the active copper sites, therefore, and whilst the correlation of methanol production with the formation of CuI demonstrated above (Fig. 17) strongly indicates the we are dealing with a 2e−, two copper centre redox (CuII/CuI) conversion, EXAFS is of little definitive help.
Pappas et al.31 then applied these methods to high-energy resolution fluorescence detection (HERFD) XANES, measured for Cu/MOR materials engaged in the selective conversion of methane to methanol using the high temperature activation route. HERFD is an approach to XANES that relies upon the detection of copper K-edge fluorescence using crystal detectors placed before the fluorescence detector itself.162 In doing this one pays a considerable price in terms of detectable X-ray flux but the energy resolution of the XANES measurement is enhanced beyond that achievable in a transmission-based experiment. This, in turn allows the resolution of detail present in the XANES that is washed out as a result of core-hole lifetime broadening effects in the standard transmission experiment.
Fig. 21 gives an example of how the HERFD is combined with MCR to yield a far greater insight into the number of copper species present and how they behave during activation of a Cu/MOR sample using either helium or oxygen as the activating feed. Combining the enhanced energy resolution of the HERFD with subsequent analysis using MCR leads to the extraction of five spectroscopically discrete copper species, along with their interconversion as a function of temperature, using both aerobic (oxygen) and anaerobic (helium) approaches to high temperature activation of Cu/MOR.
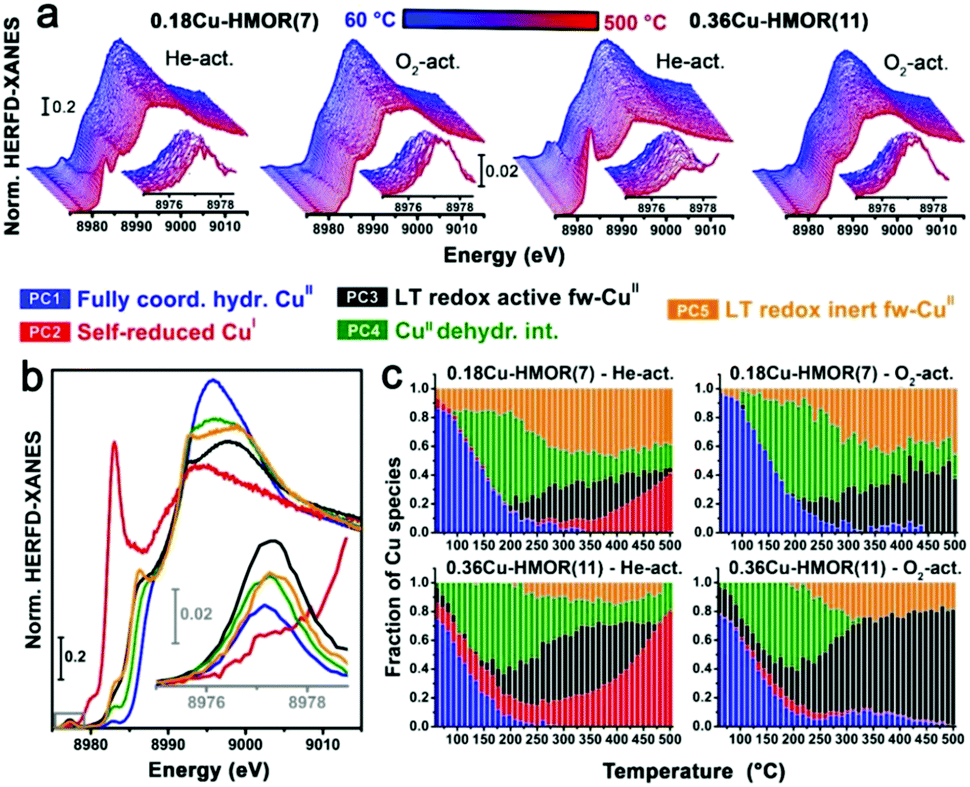 | ||
| Fig. 21 (a) Time-dependent HERFD XANES collected on two different Cu/MOR samples (0.18Cu-HMOR(7), left panels, and 0.36Cu-HMOR(11), right panels) during thermal treatment in oxygen and He gas flow from 60 °C (blue curves) to 500 °C (red curves), using an heating rate of 5 °C min−1. The insets report a magnification of the weak pre-edge peak mostly deriving from the dipole forbidden 1s → 3d transition in d9 Cu(II) centers. (b) Theoretical ‘pure’ HERFD XANES spectra of Cu-species from MCR analysis of the dataset in part (a). (c) Corresponding temperature-dependent concentration profiles of each Cu-species. Pseudo-octahedral Cu(II) aquo complexes (PC1) undergo partial dehydration to four-coordinated Cu(II) species (PC4). These CuII dehydration intermediates reach maximum concentration around 200 °C, and then progressively convert into framework-interacting Cu(II) species (fw-CuII). Among these, a low-temperature (LT) redox-active component (PC3) is found, efficiently undergoing reduction to Cu(I) (PC2) in inert atmosphere from 250 °C upwards. A LT redox-inert component is also identified (PC5): it remains stable in He up to 400 °C and is more abundantly formed in the highly active 0.18Cu-HMOR(7) material.31 Reproduced from ref. 31 with permission from The American Chemical Society, copyright 2018. | ||
The MCR based approach details the significantly different behaviour in terms of copper speciation induced by each approach to activation. Specifically, that activation in helium significantly promotes the formation of Cu(I) through auto-reduction, possibly as a result of the presence of carbonaceous deposits left within the materials post synthesis (vide supra, Section 4.5). Under aerobic activation auto-reduction to yield Cu(I) is virtually absent at higher temperatures and is only transiently observed at lower temperatures. Furthermore, different activation conditions alter the balance between two types Cu(II) species, that form from a four-coordinate Cu(II) intermediate species (PC4), and that display significantly different propensities to resist auto-reduction (denoted in Fig. 21 as PC3 and PC5). The sum of this evidence, when correlated to reactivity studies, indicates that of these species it is PC5 – the Cu(II) species that is most resistant to auto-reduction using anerobic activation – that is the species responsible for the activity of Cu/MOR for the selective oxidation of methane to methanol.
In the previous examples given (see Fig. 17) the formation of CuI was quantitatively derived from transmission copper K-edge XANES. This information was then related to the levels of methanol produced by a range of copper–zeolite systems to reveal the nature of the mechanism underlying the conversion of methane to methanol. The adoption of the HERFD methods and MCR analysis demonstrated by Pappas et al.31 achieves a very similar end but, as a result of the application of HERFD and MCR, the information restored regarding the multiplicity of copper states, and how they interconvert to yield the active material, is greatly enhanced. This work is therefore able to go further, starting from understanding which of these CuII centres are most prone to yield CuI through self-reduction in either helium or oxygen environments, and how these species are distributed in two different Cu/MOR samples that display significantly different reactive behaviour toward methane and subsequent production of methanol.
Fig. 22 summarises how this new information can relate the activity of four different copper/MOR systems for methanol production, to the fraction of active sites present and, most importantly, their nuclearity.
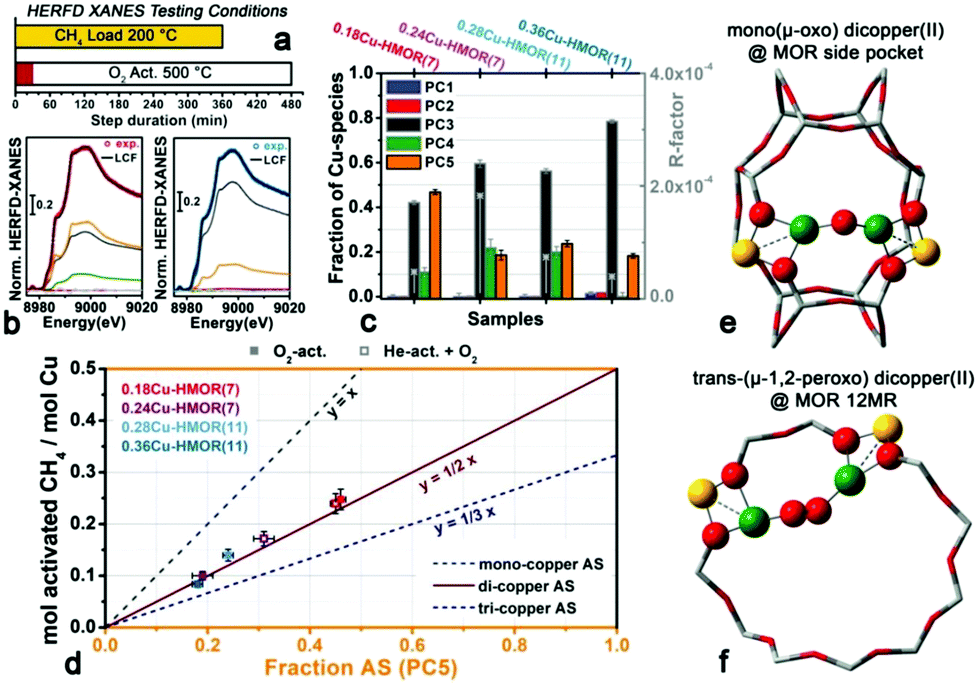 | ||
| Fig. 22 (a) Bar plot representing the duration of the oxygen activation and methane loading steps under HERFD XANES testing conditions, adopted into parallel laboratory tests to correlate spectroscopy and performance. (b) Left hand panel, XANES of representative oxygen-activated Cu–MOR samples, namely 0.18Cu-HMOR(7) and 0.36Cu-HMOR(11); right hand panel, best fit curves from LCF analysis, using the ‘pure’ spectra from MCR analysis as references. For each fitted spectrum, the LCF components scaled by their respective optimized weights. (c) Cu-Speciation in the oxygen-activated Cu–MOR series as determined from LCF analysis of HERFD XANES spectra. The Cu-species (PC1–PC5) are denoted using the same colour code as used in the previous figure (Fig. 20). The LCF R-factor is also reported (grey stars, right ordinate axis). (d) Quantitative correlation between the normalized productivity evaluated at the HERFD XANES testing conditions and the fraction of LT redox-inert fw-CuII (PC5 – our presumed active site (AS)) from LCF analysis (oxygen-activation: full coloured symbols; He-activation + oxygen: empty coloured symbols). The experimentally determined values match the ideal trend line for stoichiometric conversion over a di-copper AS, reported as a full dark red line. (e and f) Illustrations of possible Cu2Ox active sites in the MOR framework compatible with the experimental results reported here: (e) a mono(μ-oxo) di-copper(II) core in the MOR side pocket and (f) a trans-(μ-1,2peroxo) di-copper(II) core in the MOR 12 MR. Atom colour code: Cu, green; O, red; Si, grey; Al, yellow.31 Reproduced from ref. 31 with permission from The American Chemical Society, copyright 2018. | ||
Through correlation of the methanol produced in each sample to the fractions of the different components present revealed using MCR/HERFD a clear and quantitative relationship is established between one of the elements shown to be formed during activation (denoted as PC5, see Fig. 21 [ref]) and the methanol yield. This relationship can then be tested against the expectations of models for the nuclearity of the active site that involve one (y = x, monomer), two (y = 1/2x, dimers of pairs), or three (y = 1/3x, trimers) copper atoms. The result of this exercise is very clear and a relationship that emerges between methanol production and PC5 over the four different samples tested that falls along the line expected from a reactive system whose active sites comprise two copper atoms. Suggestions for the nature of these active sites are then given in Fig. 22(e) and (f)) i.e. a mono-μ-oxo CuII (e) or trans-(μ-1,2, peroxo) (f) dimers.
Here, it is also important to note that, on the basis of these measurements alone, these active sites remain very much postulates rather than verifiable fact. Whilst this is a very powerful demonstration of the application of advanced analytical methods to XAFS data, that allows a very detailed insight into the numerous copper environments present in these systems, and quantitatively links one of them to the desired reactivity, it still cannot specify the precise geometrical nature of this site. As such, the particular characteristics of this site that engender activity and selectivity for the conversion of methane to methanol, once more evade unequivocal resolution.
Alternative methods and strategies that might permit us to go further in this respect such that the essence of their activity may yet be further elucidated will be briefly discussed further toward the end of this review (vide infra).
5. Summary of evidence regarding reaction mechanisms and the nature of the active site
Having made our way through some of the copious literature regarding the subject of the selective conversion of methane to methanol by copper exchanged zeolites, but before we discuss the challenges that remain to be overcome, it is appropriate that we summarise what we have learned.Four critical aspects can be elucidated: how many sites are present, and of those how many are active; what is the underlying mechanism of reaction; what is/are the specific nature(s) of the active sites; and, lastly, what role(s) do(does) the zeolite topology and aluminium play in determining reactivity and the product distribution.
Taking the multiplicity of sites first. Whilst the synthesis of single site Cu/MOR materials has been claimed,29,69 the vast majority of evidence from all the methods and systems we have discussed in direct relation to methane to methanol conversion, suggests that a multitude of copper sites, only a fraction of which can convert methane to methanol, exist when copper is exchanged into zeolites. Moreover, the precise number of sites present can vary according to the topology of the zeolite as well as the silicon to aluminium ratio used. To illustrate this, and to help understand why this is an issue which may be considered as both intrinsic to zeolites, but also a highly variable parameter within this family of materials, Fig. 23 shows how such site multiplicity can originate in one of the most studied zeolite systems for the conversion of methane to methanol, Cu/MOR.68
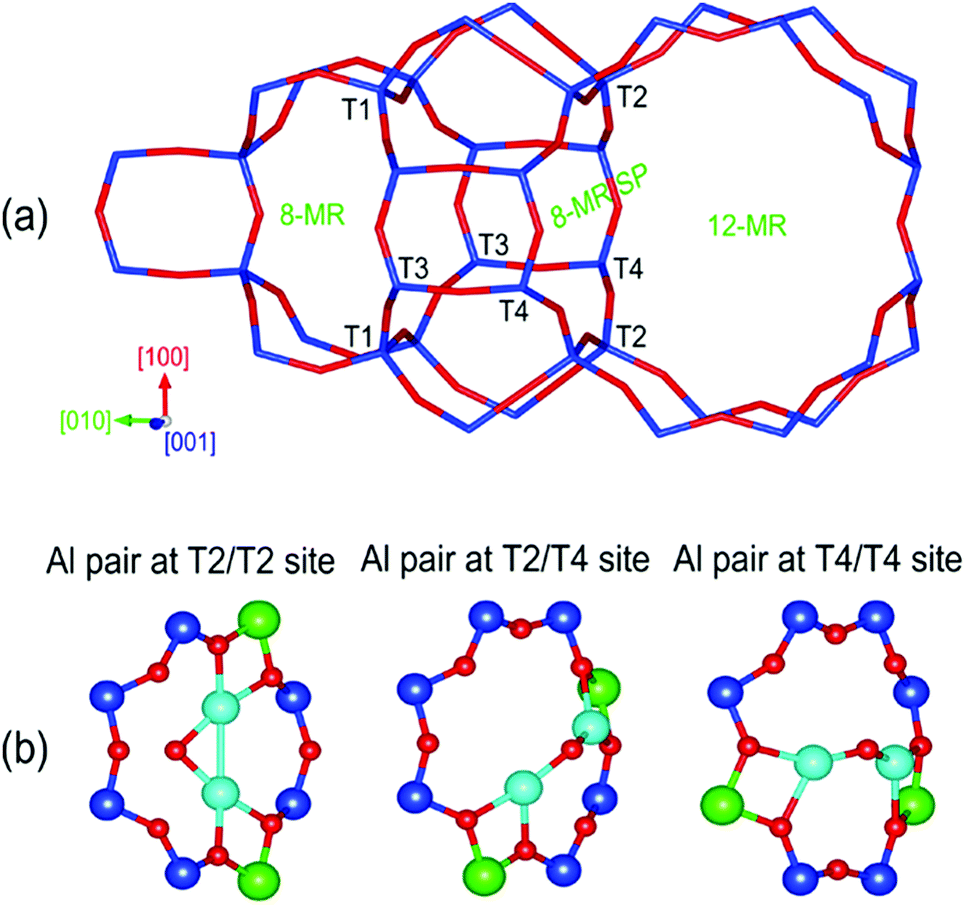 | ||
| Fig. 23 (a) A schematic illustration of the topological structure of MOR and specifies both the ring structure present (12 MR, 8 MR, and the 8 MR SD (“side pocket”) and indicates the position of the 4 possible positions where T-site aluminium atoms may exist. (b) Three examples of di-copper sites that could conceivably exist within this structure.68 Reproduced from ref. 68 with permission from The American Chemical Society, copyright 2018. | ||
From this representation of the MOR structure, and with specific reference to the number of different placements this structure permits for the siting T-site aluminium, we can see that four discrete T sites emerge from this structure that in turn yield a total of 48 different possible sites within the MOR structure.23 It is evident that a number of possible adsorption sites are notionally tenable and that the likelihood of multiple site occupation is inherent.
From a conceptual standpoint, therefore, not only is a multiplicity of states to be expected in these systems, but it is also evident that this multiplicity will vary according to the specific topology of the zeolite in question and the amount of aluminium present. As we have seen, the plethora of structures that have thus far been assigned to copper species using UV-vis,26,28,33,45,63,109 the use of probe molecules in infrared,30,50,100,122,132,137 the results of MCR analysis as applied to XAS for Cu/MOR,31 and the quantitative XRD106 for dehydrated Cu/CHA, bears witness to this multiplicity.
Indeed, we might go so far as to say this inherent property of zeolites has made getting to the fundamentals of how this conversion works in these systems such a challenging process. More importantly it is this undesirable property of these materials that would explain why only a fraction of the copper present in any system thus far studied is active for the conversion of methane to methanol; it is, therefore, one of the most fundamental issues to be eradicated by design if significantly more active/efficient systems are to be developed.
Secondly, there is the fundamental basis of the reactive chemistry in terms of the role of the copper. The traditional viewpoint, in this respect, that of a two electron, two copper atom, Cu(II)/Cu(I) redox couple – that necessarily requires the participation of two copper centres – is vindicated on the basis of recent quantitative XAS measurements.27,31,75,142 These have quantitatively related the formation of Cu(I) to the production of methanol or other products in a manner that conform to the fundamental expectations of this reactive model.
Moreover, overwhelming evidence from these studies comes down in favour of this being the fundamental driving mechanism at work across both high temperature and isothermal/high pressure approaches to methanol synthesis and across a range of zeolite types (MOR, CHA, MAZ, and ZSM-5). In this respect, therefore, and despite the great variation in zeolite architectures and compositions, a degree of universality to this aspect of the chemistry is demonstrated.
These results show that the active site – and indeed any active site derived from theoretical considerations – must involve, at least, two copper atoms that must be able to support the reversible reduction of them both from Cu(II) to Cu(I). However, it still does not allow us to rule out the participation of radical species within the overall process of methane conversion to methanol, though to date no experimental evidence has been produced that might confirm either their presence or whether they play any role at all in this chemistry. These results also do not permit us to specify the nuclearity, detailed geometry, or bonding within these sites.
Thirdly, therefore, there is the issue of the nuclearity of the active copper site. The most robust evidence for the dimeric nature of one possible active site, comes from resonant Raman experiments.43,81,106,111 The word “robust” is used as the resonant Raman experiment is the only spectroscopy thus far applied to this problem that permits deductions to be made upon bases that do not rely on assertions based on precedent or analogy. It is in this last sense that information, and therefore deductions derived from, for example UV-vis and mid-range infrared, must be seen as yielding a different type of information; one that relies on assignment and, to a significant degree, analogy to, what are often very different materials and circumstances. Symmetry, on the other hand, has mathematical foundations and is correspondingly hard to cheat. Hence, the evidence derived from the resonant Raman experiments, which clearly points to mono-μ-oxo di-copper species as being active for methanol production in Cu/MOR, must be considered as the best and most detailed indicator for the nature of an active site thus far derived.
That said the Raman experiments are not quantitative; in the sense that they cannot specify how much of this species is actually present amidst the general population of copper sites present. UV-vis, though being less specific, is both quantifiable and extremely sensitive to changes in local coordination. The real strength and elegance of the work of Vanelderen et al.81 therefore lies in the combined application of these methods to provide the most solid overall description of the active sites present in Cu/MOR.
At this point, the claims for single site Cu/MOR materials warrant closer examination. It is upon this basis that trimeric [Cu3O3]2+ were first put forward as active sites, and a radical, rather than copper redox mechanism,29,67 put forward as the foundation of the ability of copper exchanged zeolites to perform this much sought after conversion.
The proposal – for Cu/MOR – that the active site could be composed of three copper atoms is experimentally underpinned by two pieces of evidence. Firstly, the observation that the methanol yield obtained from these materials, up to a point, varied linearly with copper loading and with a gradient of 1/3. More recently, Pappas et al. (as shown above in Fig. 22) have derived a similar relation for their Cu/MOR systems, and obtained a gradient of 1/2.
In both cases, however, though these gradients describe a quantitative relation, they cannot determine the nuclearity of the active site. On their own these gradients tell us only – in any unambiguous sense – that in each case, the addition of three or two copper atoms respectively, results in the formation of one active site and one methanol molecule; the nuclearity of this site may therefore be one, two or three, or one or two depending on the example.
In both of these examples this indirect observation was then followed by X-ray spectroscopy: Grundner et al.29 using analysis of copper K-edge EXAFS and Pappas et al.31 using multivariate analysis of HERFD-XANES.
In the former case a standard analysis of the EXAFS could be fitted to the expectation of the DFT calculated [Cu3O3]2+ to a slightly better degree than a similarly DFT derived bis-μ-oxo dimer (Fig. 18). However, as has already been extensively discussed in Section 4.5 unequivocal “demonstration”29 of the presence of a single site that is trimeric in nature on this basis can be very much contested and cannot be taken as fact. The same can be said for the ability of EXAFS to distinguish between other types of copper–oxo speciation and that this spectroscopy has no really ability to distinguish between bis and mono μ-oxo di-copper motifs.80
In short, based upon all the experimental evidence that we have sifted through for a wide number of systems, we are lead to conclude that, thus far, no unequivocal evidence for either a single site Cu/MOR, a [Cu3O3]2+ trimeric copper site, or a reaction mechanism founded upon oxyl radical formation, has actually been presented in the literature.
By contrast, there is an abundance of evidence over a wide range of zeolite systems that points squarely to a (zeolite dependent) multiplicity of copper sites being present, and that this must be regarded as a central issue to the development of these materials for this application and for their fundamental comprehension. Practically speaking, the partitioning of the copper into numerous sites, that may or may not be active, reduces the atom efficiency and reactivity achievable. From a more fundamental viewpoint, such a multiplicity also makes isolation and detailed characterisation of the reactive species present, very challenging.
This brings us to a further unresolved issue: is the mono-μ-oxo di-copper motif the only species that can convert methane to methanol in these systems? Whilst the mono-μ-oxo di-copper species has been spectroscopically identified in Cu/MOR, there are reports of different types of Cu/MOR yielding very different speciation in UV-vis.110
Alternative suggestions, from DFT studies,79 have also been made regarding monomeric copper species and whether they too could mediate the activation of methane. This is especially the case of active small pore zeolites (such as CHA), wherein steric considerations might mitigate against copper centres of higher nuclearity.38 There is also the further possibility that within the same (sufficiently large) ring of a zeolite, two (proximal) monomers could associatively activate methane. This latter has recently been suggested by Sushkevich et al.50 on the basis of infrared studies using probe molecules (see Section 4.4). Fig. 24 illustrates the various possibilities for these active site motifs and how they might be thought of to activate methane in an overall sense.
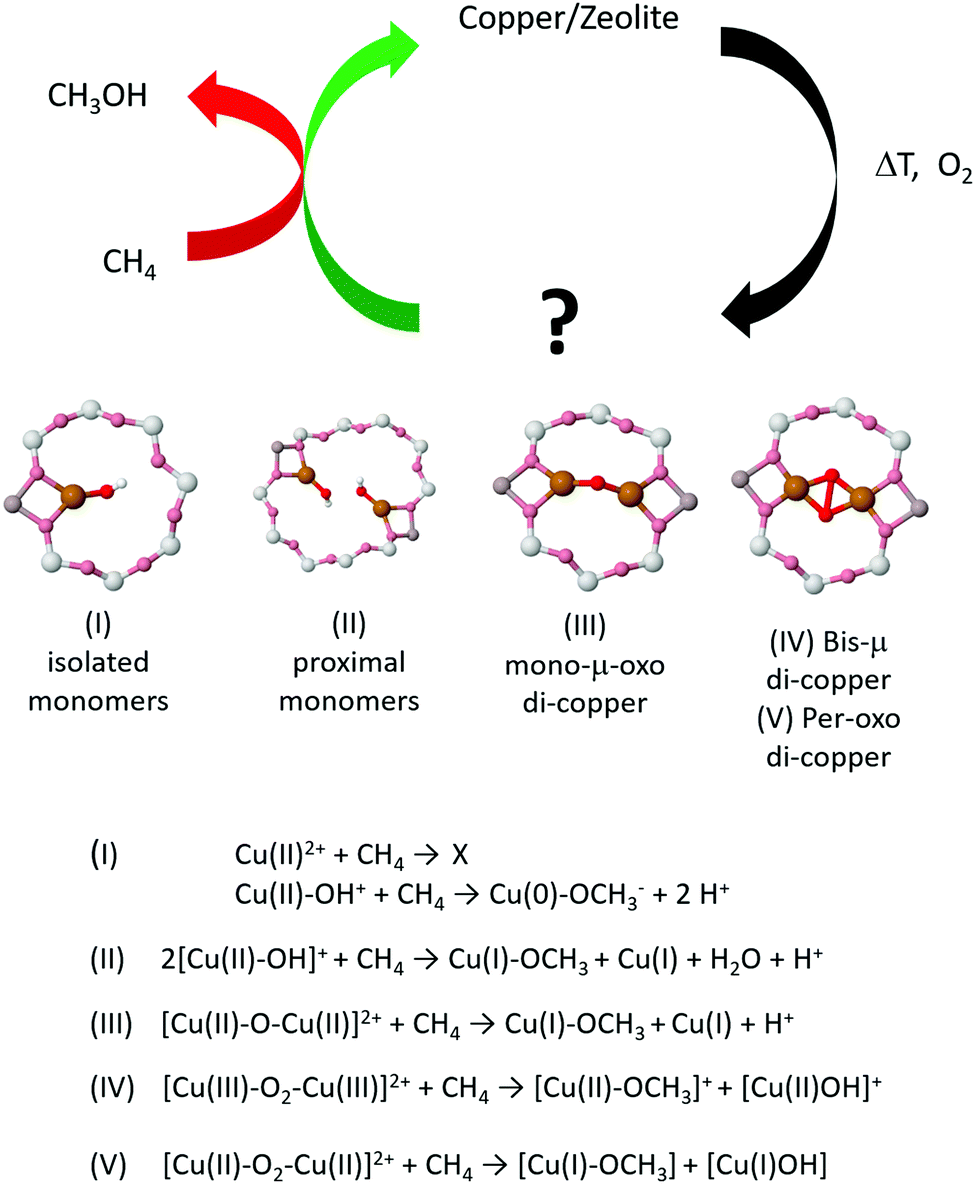 | ||
| Fig. 24 Schematic illustration of possible reactive schemes for the conversion of methane to methanol. The notional reactive consequences of stoichiometric methane activation in each case are also given. | ||
As previously delineated, we can consider the scheme by which the mono-moxo di-copper species acts ((III), Fig. 24) as the active species to have been demonstrated. We may also note that the proposition for two proximal monomers (eqn (II)) can be simply related to the mono-μ-oxo di-copper species (III) by addition of one water molecule, and that both reaction schemes are consistent with the expectation of recent studies.31,32,75,142 The proton that results from each of these mechanisms may be then involved in the formation of Brønsted acid sites as has also been observed.100
The reaction of a bis-μ-oxo di-copper or per-oxo di-copper species can achieve the same ends, the formation of two Cu(II) or Cu(I) species respectively, without any requirement for the involvement of other elements of the system (schemes IV and V, Fig. 24).
The last case, which arises from DFT studies,79 achieves methane activation and methoxy formation over isolated copper monomers (routes (I) Fig. 24), with any proton produced yielding a Brønsted acid site. However, such a route is at odds with the expectation of the two electron, two copper atom mechanism.
To summarise, whilst the presence and activity of the mono-μ-oxo di-copper species in Cu/MOR is clear, it is not yet the case that we can generalise that this is the universal motif for the high temperature, stepwise approach to the activation of copper zeolites. This is even more the case for the lower temperature isothermal/high pressure route to methane activation where the materials remain partially hydrated,75,163 and the temperatures used never reach those thought to be required for the generation of mono-μ-oxo di-copper species.28,31,80,164
Finally, to our last consideration: the role of aluminium in directing the selective conversion of methane to methanol in these systems. This parameter, along with topology of the zeolite, governs the structure of copper–oxo sites and, consequently, the activity of copper-exchanged samples in methane conversion to methanol. However, in contrast to the Cu/Al ratio, the activity of the materials does not scale linearly with the amount of aluminium in the framework. Firstly, Vanelderen et al.33 pointed to the existence of a certain maximum for the intensity of the UV band at 22700 cm−1, associated with the di-copper mono-μ-oxo sites. That was achieved for the materials with Si/Al ratio between 12 and 30, while (presumably) other Si/Al ratios lead to lower activity. This indicates that the certain distribution of aluminium atoms in the zeolite framework favours the formation of active copper oxo species.
High aluminium content, which one might consider as driving factor to introduce as much copper as possible per gram of material, does not necessarily lead to high methanol yield. In this respect, Ipek et al.106 showed that Cu/CHA (Cu–SSZ-13) with Si/Al of 5 is poorly active in spite of a high copper loading (Cu/Al = 0.39). Findings from PXRD indicate a high concentration of monomeric Cu(II) sites in 6 MR of Cu–SSZ-13. These sites are sterically restricted to such a degree that active sites for formation of methanol cannot be formed from them. In contrast, a higher concentration of Cu in 8 MR for the Si/Al = 12 sample was suggested to result in a higher concentration of copper–oxo species.
Similar trends have been observed for Cu/MOR, where the decrease of Si/Al ratio from 10 to 6 does not result in any significant change in methanol yield per gram of material. This, however, is not true for the methanol yield per copper, which increases with the increase of Si/Al. According to Newton et al.,142 Pappas et al.31 and Sushkevich et al.,50,100 this might be associated with the presence of different fractions of active and inactive copper species in the samples with different Si/Al ratio, as well as different selectivity towards methanol due to the over-oxidation in the materials with high Al content. We may note, however, that Cu/MAZ, a system for which high Si/Al ratios are intrinsic, has been demonstrated to be significantly more selective (for a single reactive cycle) than Cu/MOR.27 Thus, the question about the effect of Si/Al ratio falls into the more general problem of the quantifying and controlling the nature and quantity of copper sites hosted in zeolites. Equally, however, the precise role(s) that this parameter has in determining the efficacy of direct conversion of methane to methanol are yet to be fully understood.
Given the above, and the structural-reactive complexity that is emerging from the study of these materials, we might also ask whether there may be better ways to interrogate them than have generally been achieved to date. The next section addresses the question as to whether there are methods, or combinations of methods, that have thus far not been utilised in this arena, but that might shed more quantitative light into the behaviour of these elusive systems?
6. Gaps in our knowledge and how they might be closed
One of the issues that might be regarded as largely unresolved for methane to methanol conversion by copper hosted in zeolites is the detailed nature (nuclearity, local bonding geometry) of the active sites. A significant further question is whether the nature of these active sites is conserved across the range of active zeolite topologies, aluminium distributions, and the different (high temperature activation and isothermal/elevated pressure) cycling methodologies.As previously stated, the most robust assertions regarding the nature of the species present after high temperature activation are those derived on the basis of vibrational (Raman) spectroscopy and the symmetry based arguments that come with this approach.43,46,58,106 These studies uniformly come down in favour of di-copper mono-μ-oxo forms of copper being present in the activated Cu/zeolites independent (though the details do vary43,46,58,106) of the nature of the host i.e. ZSM-5, MOR, CHA. That said there are observations both from UV-vis and infrared,50 and theoretical studies79 that do not close the door to other structural-reactive motifs as potentially leading to activity in some circumstances.
A question therefore remains as to how to improve upon this situation and whether we can use other methods to further resolve active from inactive species, specify with greater precision their nature and, by extension, understand how they may be promoted or suppressed. In these respects we might point to a number of possibilities.
At this point we note that two recent publications165,166 have shown that nuclear magnetic resonance (1H, 13C, 27Al, 29Si) studies also have a significant potential for contributing to the understanding of this conversion. These studies have shed considerable light as to the range of products that result from reaction of activated CuII with methane, and where they may end up within the zeolite structure. For instance, in these investigations, the reaction with methane over a wide range of temperatures have shown that methoxy groups, the principle product of the reaction with methane, can be found at Brønsted acid sites rather than directly ligated to the copper centres.166 They have also shed light on the presence of species that hitherto had not been reported e.g. formaldehyde and the presence of both mono- and bi-dentate formates.165
Herein, however, we shall concentrate on those methods that have: firstly, the capacity to restore information that directly concerns the nature of the copper sites present and how they behave; secondly, that have a clear and present potential to be applied in an operando manner; but that, thirdly, as of yet, have not been applied to the selective conversion of methane to methanol using copper-containing zeolites.
6.1 Alternative probes of structure and function
In terms of other approaches that might yield more insight in terms of the systems and chemistry that are the subject of this review, we might reasonably point to four different methods that have either not been applied at all to these problems or have been but only in a relatively basic manner that belies their potential capacity. In the first case we might point to Valence to Core emission spectroscopy (V2C), total X-ray scattering/PDF analysis, and far infrared spectroscopy; in the second, electron paramagnetic resonance (EPR).Indeed, there have been examples of EPR applied to aspects of the methane to methanol conversion by Cu/containing zeolites for Cu/ZSM5,62 Cu/MOR,109 and Cu/CHA.59,136 These have been aimed at establishing the nature of the copper present after high temperature activation and/or cross-referencing changes in EPR post reaction with methanol to changes in UV-vis. The results of these exercises have tended therefore to indicate the presence of CuII monomers that do not react with methane and have therefore fulfilled an important role; that is of identifying some proportion of the copper present that is inactive for this conversion.
However, in the current context, it is the potential for EPR to be applied “in operando” and during all stages of the methane to methanol process that is of primary concern. That this can now be achieved has recently been demonstrated in application to Cu/CHA in the presence of gases of relevance to the catalytic reduction of NO.167Fig. 25 gives examples of the sorts of data achievable and how they may be used that are of direct relevance to the current discussion.
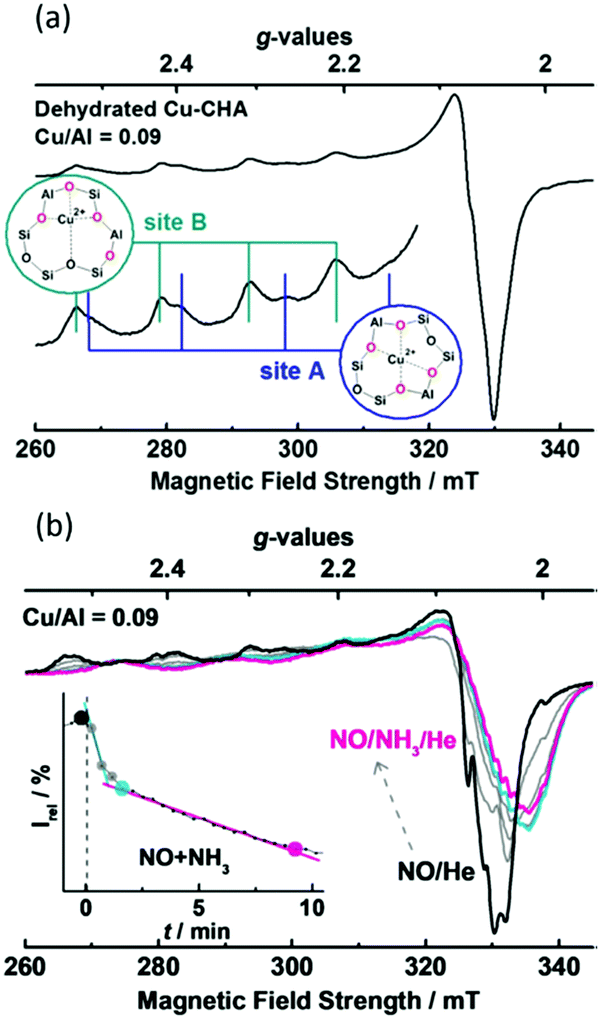 | ||
| Fig. 25 (a) In situ EPR spectra (473 K post dehydration at 523 K in 10% oxygen/helium) delineating the ability of EPR to identify different types of Cu(II) species residing in the 6 membered rings of CHA. The magnified area shows the parallel (g‖) region of the spectrum and the assignment of the features to geometrically distinct Cu(II) species. (b) First-derivative in situ EPR spectra of the reaction of Cu(II) species in the 6 membered rings in a Cu/Al = 0.09 sample during reaction with 1000 ppm NO + NH3 at 373 K. The inset shows the temporal progression of the reaction as observed by EPR and in terms of the % change in the intensity (I) of the total EPR signal. The net time resolution of the EPR was 45 seconds per spectrum. Reproduced from ref. 167 with permission from Wiley, copyright 2018. | ||
These results, Fig. 25(a), show firstly the discriminatory power of EPR, which clearly demonstrates the presence of two spectroscopically distinct types of Cu(II) residing in the 6-membered rings of CHA. Secondly, Fig. 25(b), the ability of in situ EPR to follow the progress if the chemistry is also shown in the removal of both starting features and their replacement with a single state that can be associated with the formation of [Cu(NH3)4]2+. The loss of intensity to the overall spectrum is associated with that portion of the Cu(II) initially present undergoing reduction to Cu(I). This process is also revealed to be composed of two (one fast, the second much slower) rates of reaction. This is ascribed to an inhibition effect due to an excess of NH3 and the initial product of reaction ([Cu(NH3)4]2+) being more stable toward reduction than its oxo antecedents.
These works clearly show that EPR has a significant, and relatively unexplored capacity, to extract considerable detail regarding site occupation, structure, and structural change in the Cu(II) species present in zeolitic systems in operando. As such, there seems no obvious reason why this method may not provide the same sort of insight into selective conversion of methane to methanol as well and given time.
Potential caveats to its application, however, do exist. The Kβ transitions are some orders of magnitude weaker than, for instance, the Kα fluorescence that is usually collected to obtain fluorescence yield Cu K-edge XANES and HERFD. As such, samples probed by V2C are required to withstand proportionately higher X-ray doses/power densities, often over considerable lengths of time for high quality data to be obtained. As we have already noted (Section 3) the possibility of unwanted and misleading reduction of CuII to CuI (or the reverse in the presence of water vapour applied during product extraction) is very much a possibility in the case of copper containing zeolites. This being so, protocols to ensure that what is eventually observed has more to do with changes to the material as a result of the chemistry, rather than the X-ray exposure, need to be in place and the intrinsic behavior of the system under such conditions should be fully understood before chemically relevant deductions can be made.
As XRD is founded upon the detailed analysis of Bragg reflections, it may only report on phases wherein sufficient order is present within the sample to generate such reflections; and even then the contributions from adsorbed atoms such as copper must be themselves sufficiently ordered to be revealed by diffraction analysis; and this is not always the case. Ordinarily, in a situation such as this then EXAFS would become the go-to method. However, as we have also delineated, such is the apparent multiplicity and disorder in many copper zeolite systems that this method is severely compromised in terms of elucidating the nature of the active sites present.
Total scattering (be it of X-rays or neutrons) combined with pair distribution function (PDF) analysis provides a means to restore structural data from X-ray or neutron scattering experiments that does not have any reliance upon the crystallinity of the sample.171–173 As such it has the potential to restore, in real-space rather than reciprocal space, short (as with EXAFS), mid, and long range (as with XRD) information and provide a very complete description of the behaviour of the material under study. This method heavily favours the use of high energy (>50 keV) X-rays, as it requires that a large range of Q (Å−1) space to be detected with high statistics, and high energy synchrotron beamlines equipped with state of the art flat panel detectors can be operated on the sub-millisecond timescale. The use of high energy X-rays (and minimal sample absorption) also means that the scattering observed is essentially kinematic in nature, the method can be applied under operationally relevant circumstances, and the likelihood of any beam induced effects is minimised. Most importantly, the X-ray scattering that underpins this method sees the sample under study in a different manner to, for instance, EXAFS. The photoelectron scattering that gives rise to the EXAFS is dominated by the core of the scattering element. In X-ray scattering the relative scattering intensities due to elements within the material vary with the square of the number of electrons surrounding the scattering atom. Moreover, and, to a first approximation, the scattering due to the presence of copper and the effects it has on the structure of the host zeolite may be revealed through a simple difference between copper loaded and copper free samples. As such, this method offers the prospect of a further way to see into the structure and bonding of the metal hosted within the zeolite as it is operating in a structurally direct manner. To date, however, no such study regarding the methane to methanol process using copper containing zeolites has appeared in the literature.
In these terms, the far infrared region of the IR spectrum is decidedly more attractive as methane is transparent to infrared radiation below ca. 900 cm−1, and even though water does yield some structure in the far infrared region it is notionally less obstructive that that found in the mid-range region.174 In the far IR, therefore, the possibility for in situ measurements using infrared spectroscopy arises. Moreover, it does so without the potential for probe-induced perturbation of the working system as is potentially the case for both Raman and X-rays.
The far-infrared region of the spectrum is also very rich in information arising from the framework vibrations of the zeolites. It is also where we might expect Cu–O bends and stretches to be found that can identify the nature of the copper species and how they change as a function of treatment, much as has been demonstrated from resonant Raman spectroscopy.43,58,81 In addition, the calculation of infrared spectra using methods such as DFT is tractable.175
Indeed, the very richness of this part of the IR spectrum necessarily means that a significant complexity is to be expected in the spectra and in their interpretation. That said, there do exist a limited number of studies of the nature of metallic cations in zeolite materials that suggest, at the very least, that this method may provide an additional route for deepening our understanding of the speciation of copper in these systems and how methane is converted to either methanol or over-oxidation products.
We might further note that third generation synchrotron are also very stable sources of far infrared radiation with brilliances that are orders of magnitude greater than those that can be obtained from a lab-based IR spectrometers; as a result, truly dynamic in situ study also seems possible using this approach.
6.2 Braiding theory and experiment; concurrence and deficits
Despite considerable progress in both theoretical investigations and experimental methods in application to identification of active sites of zeolites, the coherent interplay between the two approaches seems less than would ideally be desired, for instance, in respect of the mechanism of the methane to methanol conversion over the copper-exchanged zeolites. Theoretical investigations have recently been very vocal in suggesting a dominant radical mechanism of this reaction,67,176 a suggestion fuelled largely by a proposal of copper trimers as viable structures of active sites.29 Due to the presence of mixed valence Cu(II)/Cu(III) oxidation states of at least two copper atoms in such trimers, two oxygen atoms exhibit a radical oxyl character and are expected to activate the C–H bond of methane. This suggestion is in line with a similar radical mechanism widely assumed for the iron-exchanged zeolites.177 However, even for iron-exchanged zeolites there have been alternative mechanisms proposed,178 while for copper-exchanged zeolites recent experimental evidence has come down squarely in favour of a mechanism based on Cu(I)/Cu(II) pairs.31,32,142Similar difficulties hinder the attempts to identify a certain structure of an active site. While theoretical investigations often focus on the feasibility of individual configurations,29,68 many experimental methods appear to suggest the co-existence of multiple active sites.106
Moreover, if we consider the structures derived from DFT presented in Fig. 4 more closely we find that in all cases, the first shell of oxygen coordination is predicted to be split into pairs of longer (ca. >1.9 Å) and shorter (ca. <1.8 Å) Cu–O bonds. Whilst such a situation is no doubt plausible, techniques such as EXAFS have not reported such a partitioning of bond distances in the first coordination shell. Whilst we have seen that EXAFS does have severe limitations to it in its ability to determine copper-site structures in these materials, the first shell of coordination, that very much dominates the extracted EXAFS in these cases, should still give a reliable average view of this aspect of the structures present. As such, any partitioning of bond distances of this type should be resolvable.
An apparent discontinuity therefore seems to exist between the expectations of theory and the result of available experiment in these rather fundamental aspects of structure. It is, however, at present unclear as to why this may be the case. It is entirely possible that this could result simply from the mismatch that exists between the accuracy with which DFT might be able to specify bond distances (±0.1 Å) compared to the resolving power of EXAFS, where bond distances can be as precise as ca. ±1.5% (i.e. ∼0.05 Å). However, this does not fully account for why DFT partitions the first shell oxygen coordination as it does. As such, and though it might be very tempting to directly fit the DFT-derived geometries of the copper oxide active species in zeolites to the experimentally derived EXAFS, it may not currently be appropriate to do so.
We have also noted (vide supra Section 4.5) that, in general, the average coordination numbers returned from EXAFS for the first Cu–O shell of coordination in activated Cu(II) species are always significantly closer to 3 than to 4. Such a situation is only compatible with monomers (see for example the site structures derived from XRD and EPR for copper in the 6 MR of CHA and Fig. 4), mono-μ-oxo species, or mixtures thereof. Both bis-μ-oxo di-copper and trimeric copper oxo species are four coordinate and can only therefore be compatible with the experimental data if present as only minority species from this point of view.
However, although there do appear to be some apparent differences between experiment and theory that should be further examined, theoretical investigations are taking a leading role in certain areas. One of the big roles played by theory is understanding the role of water for stabilising certain reaction intermediates and aiding the reaction mechanism.30,68,179 Another theoretical breakthrough is associated with the dynamic behaviour of copper monomers to form multinuclear sites.64 In both cases, theory has been fundamental for the creation of the proposed ideas, and has inspired the corresponding experimental efforts.
Computational spectroscopy has generally become a widely used tool in zeolite chemistry research. For instance, such methods as IR, NMR, and UV-vis spectroscopies are implemented in majority of popular modelling suites, and have proven to provide reliable results in agreement with experimental evidence. We refer an interested reader to an excellent review of the recent developments of these methods by Van Speybroeck et al.,180 and only touch upon the body of literature relevant to the methane to methanol process.
One of the popular IR techniques experimentally is the use of probe molecules, such as for instance CO and NO to reveal information on the geometry of active sites present in the material. As CO will mostly probe Cu(I) sites, while NO will probe both Cu(I) and Cu(II) centers, the combination of theory and experiment can yield a very detailed structural information and even allow following the transformations of the adsorbates and/or active sites during the reaction. This field has recently seen a rapid growth and the reader is referred to extensive reviews on this topic.121,181
Nachtigall et al. demonstrated the applicability of theoretical infrared calculations to determine the configurations of the active sites based on CO adsorption,182–184 Hafner et al. have also contributed to a deeper theoretical understanding of CO adsorption in zeolites, for instance in MOR185,186 and CHA.187 Their simulations allowed distinguishing between high- and low-frequency bands in experimental spectra. The adsorption of NO has also been studied, mostly in the context of the NO decomposition processes, and important insights into metal cation coordination to the framework has been established.188
Computation of optical spectra is becoming increasingly more computationally accessible thanks to developments in theoretical algorithms, most importantly in time-dependent density functional theory (TD-DFT),189 as well as availability of extensive computational resources. Interestingly, already the seminal papers by Rice et al.190 and Woertink et al.43 made use of theory to identify the mono-μ-oxo di-copper active site in Cu–ZSM-5 zeolite. Furthermore, early investigations of the similar centres in enzymes, that later inspired extensive research of iron and copper sites in zeolites, also relied on theoretical methods.191,192
It therefore would not be an exaggeration to say that theoretical methods play a very important role both in the historical and recent developments of the zeolite catalysis. Specifically, both infrared and optical spectroscopies benefit greatly from theoretical collaborations. We hope that the remaining disparity of XAS and DFT results is therefore going to be solved soon as more groups turn their attention to the topic.
7. Conclusions
In this review, we have discussed various experimental and theoretical techniques that have been employed to understand the direct conversion of methane to methanol by copper containing zeolites; in doing so we have emphasized both the strengths and limitations of respective methods, and sought to clarify future routes to be explored. It is clear that the development of an efficient direct methane to methanol conversion process is a multi-faceted challenge, calling for extensive collaborations between fundamental chemistry, spectroscopy, process engineering and optimisation, and modelling, to name a few. Moreover, even a seemingly well-defined and isolated sub-problem of the geometrical configuration of active sites, and the reaction mechanisms compatible with them, is decidedly complex.Having said that, there has been a tremendous progress in the development and application of a range of powerful techniques over the years. High-resolution X-ray diffraction (XRD) has been successfully applied to identify the occupancy and location of copper ions in the pores of chabasite. UV-vis spectroscopy, having breathed life into the discussion of active site configurations in the first place, remains a powerful high throughput tool to study the copper-exchanged zeolites under in situ conditions. Resonant Raman has lent a specificity to UV-vis that has enabled the attribution of the vibrational bands to specific species as well as ruling out of other species by the same means. IR spectroscopy provides an almost unique opportunity to differentiate between various species of similar character, such as multiple Cu(I) and Cu(II) centers, and potentially also quantify the observed difference. X-ray absorption spectroscopy studies complete a tool set of necessary experimental techniques by providing means of observing the elemental specific structural and electronic changes the copper atoms undergo during the reaction and quantify these in time resolved fashion.
Theoretical methods serve a double duty, providing both the fundamental understanding of the experimentally observed phenomena by providing a refined structural model capturing essential chemistry of the system, and a means of potentially guiding the experimental efforts by screening the plethora of possible compositions, configurations, and reaction conditions before they enter the laboratory. However, they still suffer from the low level of accuracy and quantitative correlation with experiment and showing strong dependencies of the structural geometry and thermodynamic parameters that result from calculation on the initial guess and method used. Therefore, in an ideal case, a dynamical feedback system between theory and experiment should be built, where experimental evidence is initially used to build an appropriate theoretical model. Moreover, most of the theoretical investigations are limited to the use of DFT-based methods, while the redox chemistry of copper-exchanged zeolites is associated with the transfer of electrons, hence requiring the utilisation of high level theory. Strong demand of such studies should induce the new wave of interest to the development of fast and accurate theoretical methods for the modelling of redox sites and reactions in general.
Ultimately, two conclusions are apparent. Firstly, the necessity to employ a wide range of techniques concurrently, preferably in situ in the case of experimental methods, and incorporating state-of-the-art statistical thermodynamics and kinetic modelling to reach the relevant conditions in the case of theory, enabling comprehensive analysis of these systems under realistic conditions. Secondly, the goal of the characterisation is not only to understand the underlying chemistry, but also to enable rational design of the systems and processes relevant to real life applications.
On this front, accepting that a plurality of structures can react with methane, enables focusing on how to maximise their performance. Although the only definitive experimental derivation is that for the mono-μ-oxo di-copper center,81 there is also strong evidence for the formation of proximal monomers, that can become active through association.50 At the same time, clusters containing more than two copper atoms cannot be excluded, as one of the possible reactive configurations that may be present in the copper zeolites, while their sole formation in a form of single sites is doubtful.
The roles of the Si/Al and Cu/Al ratios in determining which species are present and with what fraction are starting to be unraveled, potentially opening an avenue to controlling and engineering of the properties of copper zeolites. What further complicates the issue is the fact that many experimental techniques used so far are inherently “bulk methods”, and thus yield a certain average measure of the active sites’ collective characteristics, whereas even within the same zeolite a multitude of active sites might co-exist and be responsible for the observed chemical behaviour. In this respect, the application and deep development of other methods, like EPR, PDF, and NMR becomes highly desirable and foreseen as an obvious step forward.
In combination with a multitude of reaction protocols, such a manifold of active structures could be translated into a range of available reaction mechanisms. This includes both the questions of the underlying redox properties and the activation/recovery procedures. The Cu(I)/Cu(II) redox mechanism, that requires the participation of two copper atoms per methane molecule activated, is now well founded experimentally,27,31,32,75,142 and more so than a mechanism involving Cu–O˙ oxyl radicals.29,67
Most importantly, alternative reaction protocols, such as the ones employing water as oxidant30 and eliminating temperature swings by making the process isothermal,48,49,193 should be further analysed and understood.
Notwithstanding extensive progress outlined in this review, a multitude of open questions relating to the structure of active sites and possible reaction mechanisms still remain, including but not limited to:
• What is the influence of water on the structure of active sites and the reaction mechanism?
• What is the dynamic behaviour of sites, and do active sites (inter)transform into various forms under the reaction conditions?
• Influence of the various zeolite frameworks and synthesis procedures on the configuration of copper oxide active sites.
• Influence of aluminium and copper loadings and aluminium distribution over T-sites on the configuration and chemical nature and reactivity of active sites.
We thus propose that the community should embrace the underlying structural and mechanistic complexity and work together towards understanding the ways this complexity yields relevant chemistry and with this find materials and process conditions, which give higher methanol yields.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
MAN would like to thanks Shell Global Solutions for the funding of his position. DP gratefully acknowledges generous CPU time allocation at the Swiss National Supercomputing Centre (CSCS) within the project s878. V. L. S. thank ESI platform of Paul Scherrer Institute for the financial support. The authors would like to thank and acknowledge all those beamline scientists (specifically at SNBL, DUBBLE, ID15, and ID26 at the ESRF, and SuperXAS at the Swiss Light Source), technical staff (ETH, ESRF, SLS), and co-workers who have made the work we do possible.Notes and references
- E. G. Nisbet, E. J. Dlugokencky and P. Bousquet, Science, 2014, 343, 493–496 CrossRef CAS PubMed
.
- D. Malakoff, Science, 2014, 344, 1465–1467 Search PubMed
- C. D. Elvidge, M. Zhizhin, K. Baugh, F. C. Hsu and T. Ghosh, Energies, 2016, 9, 14 CrossRef
- N. R. Foster, Appl. Catal., 1985, 19, 1–11 CrossRef CAS
- J.-P. Lange, K. P. De Jong, J. Ansorge and P. J. A. Tijm, Stud. Surf. Sci. Catal., 1997, 107, 81–86 CrossRef CAS
- J. H. Lunsford, Catal. Today, 2000, 63, 165–174 CrossRef CAS
- G. A. Olah, Angew. Chem., Int. Ed., 2005, 44, 2636–2639 CrossRef CAS PubMed
- C. Hammond, S. Conrad and I. Hermans, ChemSusChem, 2012, 5, 1668–1686 CrossRef CAS PubMed
- A. Caballero and P. J. Pérez, Chem. Soc. Rev., 2013, 42, 8809–8820 RSC
- P. V. L. Reddy, K. H. Kim and H. Song, Renewable Sustainable Energy Rev., 2013, 24, 578–585 CrossRef CAS
-
A. Mittasch, K. Winkler and M. Pier, DE Pat., 441433, 1923 Search PubMed
- G. C. Chinchen, P. J. Denny, J. R. Jennings, K. C. Waugh and P. Group, Appl. Catal., 1988, 36, 1–65 CrossRef CAS
- W. Wang, S. Wang, X. Ma and J. Gong, Chem. Soc. Rev., 2011, 40, 3703–3727 RSC
- M. Ahlquist, R. J. Nielsen, R. A. Periana and W. A. Goddard, J. Am. Chem. Soc., 2009, 131, 17110–17115 CrossRef CAS PubMed
- M. Ravi, M. Ranocchiari and J. A. van Bokhoven, Angew. Chem., Int. Ed., 2017, 56, 16464–16483 CrossRef CAS PubMed
- A. R. Kulkarni, Z. J. Zhao, S. Siahrostami, J. K. Nørskov and F. Studt, Catal. Sci. Technol., 2018, 8, 114–123 RSC
- M. Ravi, V. L. Sushkevich, A. J. Knorpp, M. A. Newton, D. Palagin, A. B. Pinar, M. Ranocchiari and J. A. van Bokhoven, Nat. Catal., 2019, 2, 485–494 CrossRef CAS
- V. C. C. Wang, S. Maji, P. P. Y. Chen, H. K. Lee, S. S. F. Yu and S. I. Chan, Chem. Rev., 2017, 117, 8574–8621 CrossRef CAS PubMed
- B. E. R. Snyder, M. L. Bols, R. A. Schoonheydt, B. F. Sels and E. I. Solomon, Chem. Rev., 2018, 118, 2718–2768 CrossRef CAS PubMed
-
D. Lance and H. S. Elworthy, FR Pat., 352687, 1905 Search PubMed
- R. B. Anderson, K. C. Stein, J. J. Feenan and L. J. E. Hofer, Ind. Eng. Chem., 1961, 53, 809–812 CrossRef CAS
- H. D. Gesser, N. R. Hunter and C. B. Prakash, Chem. Rev., 1985, 85, 235–244 CrossRef CAS
- C. Baerlocher and L. B. McCusker, Database of Zeolite Structures, http://www.iza-structure.org/databases/.
-
J. Cejka, H. V. Bekkum, A. Corma and F. Schueth, Introduction to Zeolite Science and Practice, Elsevier Science, Amsterdam, 3rd edn, 2007 Search PubMed
- P. Vanelderen, J. Vancauwenbergh, B. F. Sels and R. A. Schoonheydt, Coord. Chem. Rev., 2013, 257, 483–494 CrossRef CAS
- M. H. Groothaert, P. J. Smeets, B. F. Sels, P. A. Jacobs and R. A. Schoonheydt, J. Am. Chem. Soc., 2005, 127, 1394–1395 CrossRef CAS PubMed
- A. J. Knorpp, A. B. Pinar, M. Newton, V. Sushkevich and J. A. van Bokhoven, ChemCatChem, 2018, 10, 5593–5596 CrossRef CAS
- P. J. Smeets, M. H. Groothaert and R. A. Schoonheydt, Catal. Today, 2005, 110, 303–309 CrossRef CAS
- S. Grundner, M. A. C. Markovits, G. Li, M. Tromp, E. A. Pidko, E. J. M. Hensen, A. Jentys, M. Sanchez-Sanchez and J. A. Lercher, Nat. Commun., 2015, 6, 7546 CrossRef PubMed
- V. L. Sushkevich, D. Palagin, M. Ranocchiari and J. A. van Bokhoven, Science, 2017, 356, 523–527 CrossRef CAS PubMed
- D. K. Pappas, A. Martini, M. Dyballa, K. Kvande, S. Teketel, K. A. Lomachenko, R. Baran, P. Glatzel, B. Arstad, G. Berlier, C. Lamberti, S. Bordiga, U. Olsbye, S. Svelle, P. Beato and E. Borfecchia, J. Am. Chem. Soc., 2018, 140, 15270–15278 CrossRef CAS PubMed
- G. Brezicki, J. D. Kammert, T. B. Gunnoe, C. Paolucci and R. J. Davis, ACS Catal., 2019, 9, 5308–5319 CrossRef CAS
- P. Vanelderen, R. G. Hadt, P. J. Smeets, E. I. Solomon, R. A. Schoonheydt and B. F. Sels, J. Catal., 2011, 284, 157–164 CrossRef CAS PubMed
- E. M. C. Alayon, M. Nachtegaal, M. Ranocchiari and J. A. van Bokhoven, Chimia, 2012, 66, 668–674 CrossRef CAS PubMed
- S. E. Bozbag, E. M. C. Alayon, J. Pecháček, M. Nachtegaal, M. Ranocchiari and J. A. van Bokhoven, Catal. Sci. Technol., 2016, 6, 5011–5022 RSC
- K. Narsimhan, K. Iyoki, K. Dinh and Y. Román-Leshkov, ACS Cent. Sci., 2016, 2, 424–429 CrossRef CAS PubMed
- K. T. Dinh, M. M. Sullivan, K. Narsimhan, P. Serna, R. J. Meyer, M. Dinca and Y. Román-Leshkov, J. Am. Chem. Soc., 2019, 141, 11641–11650 CrossRef CAS PubMed
- B. Ipek and R. F. Lobo, Chem. Commun., 2016, 52, 13401–13404 RSC
- C. Hammond, M. M. Forde, M. H. Ab Rahim, A. Thetford, Q. He, R. L. Jenkins, N. Dimitratos, J. A. Lopez-Sanchez, N. F. Dummer, D. M. Murphy, A. F. Carley, S. H. Taylor, D. J. Willock, E. E. Stangland, J. Kang, H. Hagen, C. J. Kiely and G. J. Hutchings, Angew. Chem., Int. Ed., 2012, 51, 5129–5133 CrossRef CAS PubMed
- C. Hammond, R. L. Jenkins, D. J. Willock, N. Dimitratos, J. A. Lopez-Sanchez, H. Hagen, M. M. Forde, A. Thetford, M. H. ab Rahim, S. H. Taylor, D. M. Murphy, E. E. Stangland, J. M. Moulijn and G. J. Hutchings, Chem. – Eur. J., 2012, 18, 15735–15745 CrossRef CAS PubMed
- M. V. Parfenov, E. V. Starokon, L. V. Pirutko and G. I. Panov, J. Catal., 2014, 318, 14–21 CrossRef CAS
- B. E. R. Snyder, P. Vanelderen, M. L. Bols, S. D. Hallaert, L. H. Böttger, L. Ungur, K. Pierloot, R. A. Schoonheydt, B. F. Sels and E. I. Solomon, Nature, 2016, 536, 317–323 CrossRef CAS PubMed
- J. S. Woertink, P. J. Smeets, M. H. Groothaert, M. A. Vance, B. F. Sels, R. A. Schoonheydt and E. I. Solomon, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 18908–18913 CrossRef CAS PubMed
- E. M. Alayon, M. Nachtegaal, M. Ranocchiari and J. A. van Bokhoven, Chem. Commun., 2012, 48, 404–406 RSC
- M. J. Wulfers, S. Teketel, B. Ipek and R. F. Lobo, Chem. Commun., 2015, 51, 4447–4450 RSC
- D. K. Pappas, E. Borfecchia, M. Dyballa, I. A. Pankin, K. A. Lomachenko, A. Martini, M. Signorile, S. Teketel, B. Arstad, G. Berlier, C. Lamberti, S. Bordiga, U. Olsbye, K. P. Lillerud, S. Svelle and P. Beato, J. Am. Chem. Soc., 2017, 139, 14961–14975 CrossRef CAS PubMed
- T. Sheppard, C. D. Hamill, A. Goguet, D. W. Rooney and J. M. Thompson, Chem. Commun., 2014, 50, 11053–11055 RSC
- P. Tomkins, A. Mansouri, S. E. Bozbag, F. Krumeich, M. B. Park, E. M. C. Alayon, M. Ranocchiari and J. A. van Bokhoven, Angew. Chem., Int. Ed., 2016, 55, 5557–5561 CrossRef
- P. Tomkins, M. Ranocchiari and J. A. van Bokhoven, Acc. Chem. Res., 2017, 50, 418–425 CrossRef CAS PubMed
- V. L. Sushkevich, D. Palagin and J. A. van Bokhoven, Angew. Chem., Int. Ed., 2018, 57, 8906–8910 CrossRef CAS PubMed
- L. Artiglia, V. L. Sushkevich, D. Palagin, A. J. Knorpp, K. Roy and J. A. van Bokhoven, ACS Catal., 2019, 9, 6728–6737 CrossRef CAS
- D. Pappas, E. Borfecchia, M. Dyballa, K. A. Lomachenko, A. Martini, G. Berlier, B. Arstad, C. Lamberti, S. Bordiga, U. Olsbye, S. Svelle and P. Beato, ChemCatChem, 2019, 11, 621–627 CrossRef CAS
- M. B. Park, S. H. Ahn, A. Mansouri, M. Ranocchiari and J. A. van Bokhoven, ChemCatChem, 2017, 9, 3705–3713 CrossRef CAS
- S. E. Bozbag, P. Sot, M. Nachtegaal, M. Ranocchiari, J. A. van Bokhoven and C. Mesters, ACS Catal., 2018, 8, 5721–5731 CrossRef CAS
- J. Meyet, K. Searles, M. A. Newton, M. Wörle, A. P. van Bavel, A. D. Horton, J. A. van Bokhoven and C. Coperet, Angew. Chem., Int. Ed., 2019, 58, 9841–9845 CrossRef CAS PubMed
- V. L. Sushkevich and J. A. van Bokhoven, ACS Catal., 2019, 9, 6293–6304 CrossRef CAS
- D. W. Fickel and R. F. Lobo, J. Phys. Chem. C, 2010, 114, 1633–1640 CrossRef CAS
- P. J. Smeets, R. G. Hadt, J. S. Woertink, P. Vanelderen, R. A. Schoonheydt, B. F. Sels and E. I. Solomon, J. Am. Chem. Soc., 2010, 132, 14736–14738 CrossRef CAS PubMed
- A. Godiksen, F. N. Stappen, P. N. R. Vennestrøm, F. Giordanino, S. B. Rasmussen, L. F. Lundegaard and S. Mossin, J. Phys. Chem. C, 2014, 118, 23126–23138 CrossRef CAS
- H. V. Le, S. Parishan, A. Sagaltchik, H. Ahi, A. Trunschke, R. Schomäcker and A. Thomas, Chem. – Eur. J., 2018, 24, 12592–12599 CrossRef CAS PubMed
- M. H. Groothaert, J. A. van Bokhoven, A. A. Battiston, B. M. Weckhuysen and R. A. Schoonheydt, J. Am. Chem. Soc., 2003, 125, 7629–7640 CrossRef CAS PubMed
- M. H. Groothaert, K. Pierloot, A. Delabie and R. A. Schoonheydt, Phys. Chem. Chem. Phys., 2003, 5, 2135 RSC
- N. V. Beznis, B. M. Weckhuysen and J. H. Bitter, Catal. Lett., 2010, 138, 14–22 CrossRef CAS
- R. R. Jacobson, Z. Tyeklar, A. Farooq, K. D. Karlin, S. Liu and J. Zubieta, J. Am. Chem. Soc., 1988, 110, 3690–3692 CrossRef CAS
- K. Fujisawa, M. Tanaka, Y. Moro-oka and N. Kitajima, J. Am. Chem. Soc., 1994, 116, 12079–12080 CrossRef CAS
- E. I. Solomon, D. E. Heppner, E. M. Johnston, J. W. Ginsbach, J. Cirera, M. Qayyum, M. T. Kieber-Emmons, C. H. Kjaergaard, R. G. Hadt and L. Tian, Chem. Rev., 2014, 114, 3659–3853 CrossRef CAS PubMed
- G. Li, P. Vassilev, M. Sanchez-Sanchez, J. A. Lercher, E. J. M. Hensen and E. A. Pidko, J. Catal., 2016, 338, 305–312 CrossRef CAS
- M. H. Mahyuddin, T. Tanaka, Y. Shiota, A. Staykov and K. Yoshizawa, ACS Catal., 2018, 8, 1500–1509 CrossRef CAS
- S. Grundner, W. Luo, M. Sanchez-Sanchez and J. A. Lercher, Chem. Commun., 2016, 52, 2553–2556 RSC
- D. Palagin, A. J. Knorpp, A. B. Pinar, M. Ranocchiari and J. A. van Bokhoven, Nanoscale, 2017, 9, 1144–1153 RSC
- S. I. Chan, K. H. C. Chen, S. S. F. Yu, C. L. Chen and S. S. J. Kuo, Biochemistry, 2004, 43, 4421–4430 CrossRef CAS PubMed
- S. I. Chan, V. C. C. Wang, J. C. H. Lai, S. S. F. Yu, P. P. Y. Chen, K. H. C. Chen, C. L. Chen and M. K. Chan, Angew. Chem., Int. Ed., 2007, 46, 1992–1994 CrossRef CAS PubMed
- R. Balasubramanian, S. M. Smith, S. Rawat, L. A. Yatsunyk, T. L. Stemmler and A. C. Rosenzweig, Nature, 2010, 465, 115–119 CrossRef CAS PubMed
- M. O. Ross, F. MacMillan, J. Wang, A. Nisthal, T. J. Lawton, B. D. Olafson, S. L. Mayo, A. C. Rosenzweig and B. M. Hoffman, Science, 2019, 364, 566–570 CrossRef CAS PubMed
- A. J. Knorpp, M. A. Newton, A. B. Pinar and J. A. van Bokhoven, Ind. Eng. Chem. Res., 2018, 57, 12036–12039 CrossRef CAS
- A. J. Knorpp, M. A. Newton, S. C. M. Mizuno, J. Zhu, H. Mebrate, A. B. Pinar and J. A. van Bokhoven, Chem. Commun., 2019, 55, 11794–11797 RSC
- P. Tomkins, A. Mansouri, V. L. Sushkevich, L. I. Van der Wal, S. E. Bozbag, F. Krumeich, M. Ranocchiari and J. A. van Bokhoven, Chem. Sci., 2019, 10, 167–171 RSC
- A. A. Verma, S. A. Bates, T. Anggara, C. Paolucci, A. A. Parekh, K. Kamasamudram, A. Yezerets, J. T. Miller, W. N. Delgass, W. F. Schneider and F. H. Ribeiro, J. Catal., 2014, 312, 179–190 CrossRef CAS
- A. R. Kulkarni, Z. J. Zhao, S. Siahrostami, J. K. Nørskov and F. Studt, ACS Catal., 2016, 6, 6531–6536 CrossRef CAS
- E. M. C. Alayon, M. Nachtegaal, A. Bodi, M. Ranocchiari and J. A. van Bokhoven, Phys. Chem. Chem. Phys., 2015, 17, 7681–7693 RSC
- P. Vanelderen, B. E. R. Snyder, M. L. Tsai, R. G. Hadt, J. Vancauwenbergh, O. Coussens, R. A. Schoonheydt, B. F. Sels and E. I. Solomon, J. Am. Chem. Soc., 2015, 137, 6383–6392 CrossRef CAS PubMed
- A. M. Beale, I. Lezcano-Gonzalez, W. A. Slawinksi and D. S. Wragg, Chem. Commun., 2016, 52, 6170–6173 RSC
- C. Paolucci, I. Khurana, A. A. Parekh, S. Li, A. J. Shih, H. Li, J. R. Di Iorio, J. D. Albarracin-caballero, A. Yezerets, J. T. Miller, W. N. Delgass, F. H. Ribeiro, W. F. Schneider and R. Gounder, Science, 2017, 357, 898–903 CrossRef CAS PubMed
- C. Paolucci, A. A. Parekh, I. Khurana, J. R. Di Iorio, H. Li, J. D. Albarracin Caballero, A. J. Shih, T. Anggara, W. N. Delgass, J. T. Miller, F. H. Ribeiro, R. Gounder and W. F. Schneider, J. Am. Chem. Soc., 2016, 138, 6028–6048 CrossRef CAS PubMed
- D. Palagin, V. L. Sushkevich and J. A. van Bokhoven, ACS Catal., 2019, 9, 10365–10374 CrossRef CAS
- L. Vilella and F. Studt, Eur. J. Inorg. Chem., 2016, 1514–1520 CrossRef CAS
- Z.-J. Zhao, A. Kulkarni, L. Vilella, J. K. Nørskov and F. Studt, ACS Catal., 2016, 6, 3760–3766 CrossRef CAS
- S. A. Bates, A. A. Verma, C. Paolucci, A. A. Parekh, T. Anggara, A. Yezerets, W. F. Schneider, J. T. Miller, W. N. Delgass and F. H. Ribeiro, J. Catal., 2014, 312, 87–97 CrossRef CAS
- M. A. Newton, S. Checchia, A. J. Knorpp, D. Stoian, W. Van Beek, H. Emerich, A. Longo and J. A. van Bokhoven, Catal. Sci. Technol., 2019, 9, 3081–3089 RSC
- M. Sommerhalter, R. L. Lieberman and A. C. Rosenzweig, Inorg. Chem., 2005, 44, 770–778 CrossRef CAS PubMed
- J. G. Mesu, A. M. Beale, F. M. F. De Groot and B. M. Weckhuysen, J. Phys. Chem. B, 2006, 110, 17671–17677 CrossRef CAS PubMed
- Y. D. West, Internet J. Vib. Spectrosc., 1996, 1, 5 Search PubMed
- G. T. Palomino, P. Fisicaro, S. Bordiga, A. Zecchina, E. Giamello and C. Lamberti, J. Phys. Chem. B, 2000, 104, 4064–4073 CrossRef CAS PubMed
- V. L. Sushkevich and J. A. van Bokhoven, Chem. Commun., 2018, 54, 7447–7450 RSC
- V. L. Sushkevich, A. V. Smirnov and J. A. van Bokhoven, J. Phys. Chem. C, 2019, 123, 9926–9934 CrossRef CAS
- E. A. Pidko, ACS Catal., 2017, 7, 4230–4234 CrossRef CAS
- D. J. Wales and J. P. K. Doye, J. Phys. Chem. A, 1997, 101, 5111–5116 CrossRef CAS
- D. M. Deaven and K. M. Ho, Phys. Rev. Lett., 1995, 75, 288–291 CrossRef CAS PubMed
- J. A. van Bokhoven, T. L. Lee, M. Drakopoulos, C. Lamberti, S. Thie and J. Zegenhagen, Nat. Mater., 2008, 7, 551–555 CrossRef CAS PubMed
- V. L. Sushkevich and J. van Bokhoven, Catal. Sci. Technol., 2018, 8, 4141–4150 RSC
- C. J. Cramer and D. G. Truhlar, Phys. Chem. Chem. Phys., 2009, 11, 10757–10816 RSC
-
M. M. Treacy and J. B. Higgins, Collection of Simulated XRD Powder Patterns for Zeolites, Elsevier, Amsterdam, 4th edn, 2001 Search PubMed
- B. T. W. Lo, L. Ye and S. C. E. Tsang, Chem, 2018, 4, 1778–1808 CAS
- U. Deka, A. Juhin, E. A. Eilertsen, H. Emerich, M. A. Green, S. T. Korhonen, B. M. Weckhuysen and A. M. Beale, J. Phys. Chem. C, 2012, 116, 4809–4818 CrossRef CAS
- C. W. Andersen, M. Bremholm, P. N. R. Vennestrøm, A. B. Blichfeld, L. F. Lundegaard and B. B. Iversen, IUCrJ, 2014, 1, 382–386 CrossRef CAS PubMed
- B. Ipek, M. J. Wulfers, H. Kim, F. Göltl, I. Hermans, J. P. Smith, K. S. Booksh, C. M. Brown and R. F. Lobo, ACS Catal., 2017, 7, 4291–4303 CrossRef CAS
- B. F. Mentzen and G. Bergeret, J. Phys. Chem. C, 2007, 111, 12512–12516 CrossRef CAS
- A. J. Knorpp, M. A. Newton, V. L. Sushkevich, P. P. Zimmermann, A. B. Pinar and J. A. van Bokhoven, Catal. Sci. Technol., 2019, 9, 2806–2811 RSC
- P. Vanelderen, J. Vancauwenbergh, M. L. Tsai, R. G. Hadt, E. I. Solomon, R. A. Schoonheydt and B. F. Sels, ChemPhysChem, 2014, 15, 91–99 CrossRef CAS PubMed
- K. Narsimhan, V. K. Michaelis, G. Mathies, W. R. Gunther, R. G. Griffin and Y. Román-Leshkov, J. Am. Chem. Soc., 2015, 137, 1825–1832 CrossRef CAS PubMed
- P. J. Smeets, J. S. Woertink, B. F. Sels, E. I. Solomon and R. A. Schoonheydt, Inorg. Chem., 2010, 49, 3573–3583 CrossRef CAS PubMed
- Y. Kim, T. Y. Kim, H. Lee and J. Yi, Chem. Commun., 2017, 53, 4116–4119 RSC
- H. V. Le, S. Parishan, A. Sagaltchik, C. Göbel, C. Schlesiger, W. Malzer, A. Trunschke, R. Schomäcker and A. Thomas, ACS Catal., 2017, 7, 1403–1412 CrossRef CAS
- R. Oord, J. E. Schmidt and B. M. Weckhuysen, Catal. Sci. Technol., 2018, 8, 1028–1038 RSC
- T. Ikuno, S. Grundner, A. Jentys, G. Li, E. Pidko, J. Fulton, M. Sanchez-Sanchez and J. A. Lercher, J. Phys. Chem. C, 2019, 123, 8759–8769 CrossRef CAS
- B. E. R. Snyder, P. Vanelderen, R. A. Schoonheydt, B. F. Sels and E. I. Solomon, J. Am. Chem. Soc., 2018, 140, 9236–9243 CrossRef CAS PubMed
- M. J. Baldwin, D. E. Roo, J. E. Pate, K. Fujisawa, N. Kitajima and E. I. Solomon, J. Am. Chem. Soc., 1992, 114, 10421–10431 CrossRef CAS
- J. Ryczkowski, Catal. Today, 2001, 68, 263–381 CrossRef CAS
- C. Lamberti, E. Groppo, G. Spoto, S. Bordiga and A. Zecchina, Adv. Catal., 2007, 51, 1–74 CAS
- F. Zaera, Chem. Soc. Rev., 2014, 43, 7624–7663 RSC
- C. Lamberti, A. Zecchina, E. Groppo and S. Bordiga, Chem. Soc. Rev., 2010, 39, 4951–5001 RSC
- S. Bordiga, C. Lamberti, F. Bonino, A. Travert and F. Thibault-Starzyk, Chem. Soc. Rev., 2015, 44, 7262–7341 RSC
- M. Shelef, Chem. Rev., 1995, 95, 209–225 CrossRef CAS
- F. Kapteijn, J. Rodriguez-Mirasol and J. A. Moulijn, Appl. Catal., B, 1996, 9, 25–64 CrossRef CAS
- S. Brandenberger, O. Kröcher, A. Tissler and R. Althoff, Catal. Rev., 2008, 50, 492–531 CrossRef CAS
- F. Giordanino, E. Borfecchia, K. A. Lomachenko, A. Lazzarini, G. Agostini, E. Gallo, A. V. Soldatov, P. Beato, S. Bordiga and C. Lamberti, J. Phys. Chem. Lett., 2014, 5, 1552–1559 CrossRef CAS PubMed
- T. Günter, H. W. P. Carvalho, D. E. Doronkin, T. Sheppard, P. Glatzel, A. J. Atkins, J. Rudolph, C. R. Jacob, M. Casapu and J. D. Grunwaldt, Chem. Commun., 2015, 51, 9227–9230 RSC
- T. V. W. Janssens, H. Falsig, L. F. Lundegaard, P. N. R. Vennestrøm, S. B. Rasmussen, P. G. Moses, F. Giordanino, E. Borfecchia, K. A. Lomachenko, C. Lamberti, S. Bordiga, A. Godiksen, S. Mossin and P. Beato, ACS Catal., 2015, 5, 2832–2845 CrossRef CAS
- G. Spoto, S. Bordiga, D. Scarano and A. Zecchina, Catal. Lett., 1992, 13, 39–44 CrossRef CAS
- C. Lamberti, S. Bordiga, A. Zecchina, M. Salvalaggio, F. Geobaldo and C. Otero Areán, J. Chem. Soc., Faraday Trans., 1998, 94, 1519–1525 RSC
- A. Zecchina, S. Bordiga, M. Salvalaggio, G. Spoto, D. Scarano and C. Lamberti, J. Catal., 1998, 173, 540–542 CrossRef CAS
- A. Zecchina, S. Bordiga, G. T. Palomino, D. Scarano, C. Lamberti and M. Salvalaggio, J. Phys. Chem. B, 1999, 103, 3833–3844 CrossRef CAS
- G. Turnes Palomino, S. Bordiga, A. Zecchina, G. L. Marra and C. Lamberti, J. Phys. Chem. B, 2000, 104, 8641–8651 CrossRef CAS
- F. X. Llabrés i Xamena, P. Fisicaro, G. Berlier, A. Zecchina, G. T. Palomino, C. Prestipino, S. Bordiga, E. Giamello and C. Lamberti, J. Phys. Chem. B, 2003, 107, 7036–7044 CrossRef
- V. Bolis, A. Barbaglia, S. Bordiga, C. Lamberti and A. Zecchina, J. Phys. Chem. B, 2004, 108, 9970–9983 CrossRef CAS
- F. Giordanino, P. N. R. Vennestrøm, L. F. Lundegaard, F. N. Stappen, S. Mossin, P. Beato, S. Bordiga and C. Lamberti, Dalton Trans., 2013, 42, 12741–12761 RSC
- G. Spoto, A. Zecchina, S. Bordiga, G. Ricchiardi, G. Martra, G. Leofanti and G. Petrini, Appl. Catal., B, 1994, 3, 151–172 CrossRef CAS
- C. Lamberti, S. Bordiga, M. Salvalaggio, G. Spoto, A. Zecchina, F. Geobaldo, G. Vlaic and M. Bellatreccia, J. Phys. Chem. B, 1997, 101, 344–360 CrossRef CAS
- N. S. Nesterenko, F. Thibault-Starzyk, V. Montouillout, V. V. Yuschenko, C. Fernandez, J. P. Gilson, F. Fajula and I. I. Ivanova, Microporous Mesoporous Mater., 2004, 71, 157–166 CrossRef CAS
- E. M. C. Alayon, M. Nachtegaal, A. Bodi and J. A. van Bokhoven, ACS Catal., 2014, 4, 16–22 CrossRef CAS
- E. Borfecchia, D. K. Pappas, M. Dyballa, K. A. Lomachenko, C. Negri, M. Signorile and G. Berlier, Catal. Today, 2019, 333, 17–27 CrossRef CAS
- M. A. Newton, A. J. Knorpp, A. B. Pinar, V. L. Sushkevich, D. Palagin and J. A. van Bokhoven, J. Am. Chem. Soc., 2018, 140, 10090–10093 CrossRef CAS PubMed
-
J. A. van Bokhoven and C. Lamberti, Emission Spectroscopy X-Ray Absorption and X-Ray Theory and Applications, John Wiley & Sons, Chichester, UK, 2016 Search PubMed
-
J. A. van Bokhoven and C. Lamberti, XAFS Techniques for Catalysis, Nanomaterials, and Surfaces, Springer, 2017, pp. 299–315 Search PubMed
- F. W. Lytle, R. B. Greegor and A. J. Panson, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 1550–1563 CrossRef CAS PubMed
- J. L. DuBois, P. Mukherjee, T. D. P. Stack, B. Hedman, E. I. Solomon and K. O. Hodgson, J. Am. Chem. Soc., 2000, 122, 5775–5787 CrossRef CAS
- K.-I. Shimizu, H. Maeshima, H. Yoshida, A. Satsuma and T. Hattori, Chem. Lett., 2000, 862–866 Search PubMed
- E. M. C. Alayon, M. Nachtegaal, E. Kleymenov and J. A. van Bokhoven, Microporous Mesoporous Mater., 2013, 166, 131–136 CrossRef CAS
- J. M. Tranquada, S. M. Heald and A. R. Moodenbaugh, Phys. Rev. B: Condens. Matter Mater. Phys., 1987, 36, 5263–5274 CrossRef CAS PubMed
- Y. Li and W. K. Hall, J. Catal., 1991, 129, 202–215 CrossRef CAS
- W. Keith Hall and J. Valyon, Catal. Lett., 1992, 15, 311–315 CrossRef
- H. J. Jang, W. Keith Hall and J. L. D’Itri, J. Phys. Chem., 1996, 100, 9416–9420 CrossRef CAS
- M. Occhiuzzi, G. Fierro, G. Ferraris and G. Moretti, Chem. Mater., 2012, 24, 2022–2031 CrossRef CAS
- S. C. Larsen, A. Aylor, A. T. Bell and J. A. Reimer, J. Phys. Chem., 1994, 98, 11533–11540 CrossRef CAS
- J. Sárkány, J. Mol. Struct., 1997, 410–411, 137–140 CrossRef
- A. J. Dent, G. Cibin, S. Ramos, S. A. Parry, D. Gianolio, A. D. Smith, S. M. Scott, L. Varandas, S. Patel, M. R. Pearson, L. Hudson, N. A. Krumpa, A. S. Marsch and P. E. Robbins, J. Phys.: Conf. Ser., 2013, 430, 012023 CrossRef
- O. Müller, M. Nachtegaal, J. Just, D. Lützenkirchen-Hecht and R. Frahm, J. Synchrotron Radiat., 2016, 23, 260–266 CrossRef PubMed
- A. Voronov, A. Urakawa, W. van Beek, N. E. Tsakoumis, H. Emerich and M. Rønning, Anal. Chim. Acta, 2014, 840, 20–27 CrossRef CAS PubMed
- A. Rochet, B. Baubet, V. Moizan-baslé, C. Pichon, A. Rochet, B. Baubet, V. Moizan-baslé, C. Pichon and V. B. Co-k, C. R. Chim., 2016, 19, 1337–1351 CrossRef CAS
- A. Rochet, A. Ribeiro Passos, C. Legens and V. Briois, Catal., Struct. React., 2017, 3, 33–42 CrossRef CAS
- A. Martini, E. Borfecchia, K. A. Lomachenko, I. A. Pankin, C. Negri, G. Berlier, P. Beato, H. Falsig, S. Bordiga and C. Lamberti, Chem. Sci., 2017, 8, 6836–6851 RSC
- K. Hamalainen, D. P. Siddons, J. B. Hastings and L. E. Berman, Phys. Rev. Lett., 1991, 67, 2850–2853 CrossRef CAS PubMed
- V. L. Sushkevich and J. A. van Bokhoven, Catal. Sci. Technol., 2020, 10, 382–390 RSC
- M. H. Groothaert, P. J. Smeets, B. F. Sels, P. A. Jacobs and R. A. Schoonheydt, J. Am. Chem. Soc., 2005, 127, 1394–1395 CrossRef CAS
- V. L. Sushkevich, R. Verel and J. A. van Bokhoven, Angew. Chem., Int. Ed., 2020, 59, 910–918 CrossRef CAS PubMed
- M. Dyballa, K. Thorshaug, D. K. Pappas, E. Borfecchia, K. Kvande, S. Bordiga, G. Berlier, A. Lazzarini, U. Olsbye, P. Beato, S. Svelle and B. Arstad, ChemCatChem, 2019, 11, 5022–5026 CrossRef CAS
- A. Godiksen, O. L. Isaksen, S. B. Rasmussen, P. N. R. Vennestrøm and S. Mossin, ChemCatChem, 2018, 10, 366–370 CrossRef CAS
- C. J. Pollock and S. DeBeer, Acc. Chem. Res., 2015, 48, 2967–2975 CrossRef CAS
- K. A. Lomachenko, E. Borfecchia, C. Negri, G. Berlier, C. Lamberti, P. Beato, H. Falsig and S. Bordiga, J. Am. Chem. Soc., 2016, 138, 12025–12028 CrossRef CAS
- R. Zhang, H. Li and J. S. McEwen, J. Phys. Chem. C, 2017, 121, 25759–25767 CrossRef CAS
- V. Petkov, Mater. Today, 2008, 11, 28–38 CrossRef CAS
-
T. Egami and S. J. L. Billinge, Underneath the Bragg Peaks Structural Analysis of Complex Materials, Pergamon-Elsevier Science LTD, Oxford, 2nd edn, 2012 Search PubMed
-
K. W. Chapman and P. J. Chupas, in In-Situ Characterization of Heterogeneous Catalyst, ed. J. A. Rodriguez, J. C. Hanson and P. J. Chupas, Wiley, Hoboken, 2013, pp. 147–168 Search PubMed
- Y. Maréchal, J. Mol. Struct., 2011, 1004, 146–155 CrossRef
- F. Göltl and J. Hafner, J. Chem. Phys., 2012, 136, 064502 CrossRef PubMed
- K. D. Vogiatzis, G. Li, E. J. M. Hensen, L. Gagliardi and E. A. Pidko, J. Phys. Chem. C, 2017, 121, 22295–22302 CrossRef CAS PubMed
- K. A. Dubkov, N. S. Ovanesyan, A. A. Shteinman, E. V. Starokon and G. I. Panov, J. Catal., 2002, 207, 341–352 CrossRef CAS
- K. Yoshizawa, Acc. Chem. Res., 2006, 39, 375–382 CrossRef CAS PubMed
- T. Yumura, Y. Hirose, T. Wakasugi, Y. Kuroda and H. Kobayashi, ACS Catal., 2016, 6, 2487–2495 CrossRef CAS
- V. Van Speybroeck, K. Hemelsoet, L. Joos, M. Waroquier, R. G. Bell and C. R. A. Catlow, Chem. Soc. Rev., 2015, 44, 7044–7111 RSC
- E. Stavitski and B. M. Weckhuysen, Chem. Soc. Rev., 2010, 39, 4615–4625 RSC
- D. Nachtigallová, O. Bludský, C. Otero Areán, R. Bulánek and P. Nachtigall, Phys. Chem. Chem. Phys., 2006, 8, 4849–4852 RSC
- E. Garrone, R. Bulánek, K. Frolich, C. O. Areán, M. R. Delgado, G. T. Palomino, D. Nachtigallová and P. Nachtigall, J. Phys. Chem. B, 2006, 110, 22542–22550 CrossRef CAS PubMed
- P. Nachtigall, O. Bludský, L. Grajciar, D. Nachtigallová, M. R. Delgado and C. O. Areán, Phys. Chem. Chem. Phys., 2009, 11, 791–802 RSC
- L. Benco, T. Bucko, J. Hafner and H. Toulhoat, J. Phys. Chem. B, 2004, 108, 13656–13666 CrossRef CAS
- T. Bučko, J. Hafner and L. Benco, J. Phys. Chem. B, 2005, 109, 7345–7357 CrossRef PubMed
- F. Göltl and J. Hafner, J. Chem. Phys., 2012, 136, 064503 CrossRef PubMed
- A. Pulido and P. Nachtigall, ChemCatChem, 2009, 1, 449–453 CrossRef CAS
- M. A. L. Marques, C. A. Ullrich, F. Nogueria, A. Rubio, K. Burke and E. K. U. Gross, Time-Dependent Density Functional Theory, Springer-Verlag Berlin Heidelberg, 2006 Search PubMed.
- M. J. Rice, A. K. Chakraborty and A. T. Bell, J. Phys. Chem. B, 2000, 104, 9987–9992 CrossRef CAS
- M. J. Baldwin, P. K. Ross, J. E. Pate, Z. Tyeklàr, K. D. Karlin, E. I. Solomon, J. E. Pate, Z. Tyeklàr and K. D. Karlin, J. Am. Chem. Soc., 1991, 113, 8671–8679 CrossRef CAS
- P. Chen, K. Fujisawa and E. I. Solomon, J. Am. Chem. Soc., 2000, 122, 10177–10193 CrossRef CAS
- J.-P. Lange, V. L. Sushkevich, A. J. Knorpp and J. A. van Bokhoven, Ind. Eng. Chem. Res., 2019, 58, 8674–8680 CAS
| This journal is © The Royal Society of Chemistry 2020 |