
A tetraphenylethene-based hexacationic molecular cage with an open cavity†
Fan
Cao‡
,
Honghong
Duan‡
,
Qingfang
Li
and
Liping
Cao
 *
*
College of Chemistry and Materials Science, Northwest University, Xi’an, 710069, P. R. China. E-mail: chcaoliping@nwu.edu.cn
First published on 8th November 2022
Abstract
A tetraphenylethene-based hexacationic molecular cage (1) with an open cavity was synthesized. 1 exhibited 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 or 1:1 host–guest recognition for two nicotinamide adenine dinucleotide molecules (NADH and NAD+) with different CD and fluorescence responses in water.
:2 or 1:1 host–guest recognition for two nicotinamide adenine dinucleotide molecules (NADH and NAD+) with different CD and fluorescence responses in water.
Supramolecular chemistry has attracted extensive attention in scientific research as a multidisciplinary science composed of chemistry, biology, physics, and materials science.1 Specifically, supramolecular chemists constructed a variety of macrocyclic host molecules (e.g., cyclophane, cyclodextrin, calixarene, and cucurbituril) with selective recognition function for specific guests.2 Inspired from three-dimensional (3D) hydrophobic pockets of some functional proteins, furthermore, many molecular cages with 3D cavities of different sizes and shapes were designed and synthesized to achieve some specific functions, such as catalysis, separation, and drug delivery.3–5
Tetraphenylethene (TPE) has the advantages of convenient synthesis and modification,6 aggregation-induced emission (AIE),7 and dynamic rotational conformation.8 As a result, its derivatives show excellent photoelectric performance and biological applications.9–13 Given their good fluorescence and novel structure, TPEs have been successfully introduced into the supramolecular and material fields for the construction of macrocyclic compounds,8 metal–organic frameworks (MOFs) and covalent-organic frameworks (COFs).14,15 In previous reports, our group has developed a series of TPE-based monocyclophanes,16 dicyclophanes,17 3D cages,18 and supramolecular organic frameworks (SOFs)19 for molecular recognition and luminescence materials. Particularly, a TPE-based octacationic cage with four pillars, which has a closed cavity (Scheme 1a, left), has been constructed for host–guest recognition, fluorescent sensors, energy and electron transfer, and drug delivery.18 Here, we report a TPE-based hexacationic molecular cage (1) with only three pillars, which has an open cavity (Scheme 1a, right). Compared with the four-pillared cage, 1 with an open cavity exhibits 1:2 or 1:1 host–guest recognition for large nicotinamide adenine dinucleotide (NAD) molecules including NADH and NAD+ with dual responses of circular dichroism (CD) and fluorescence in water.
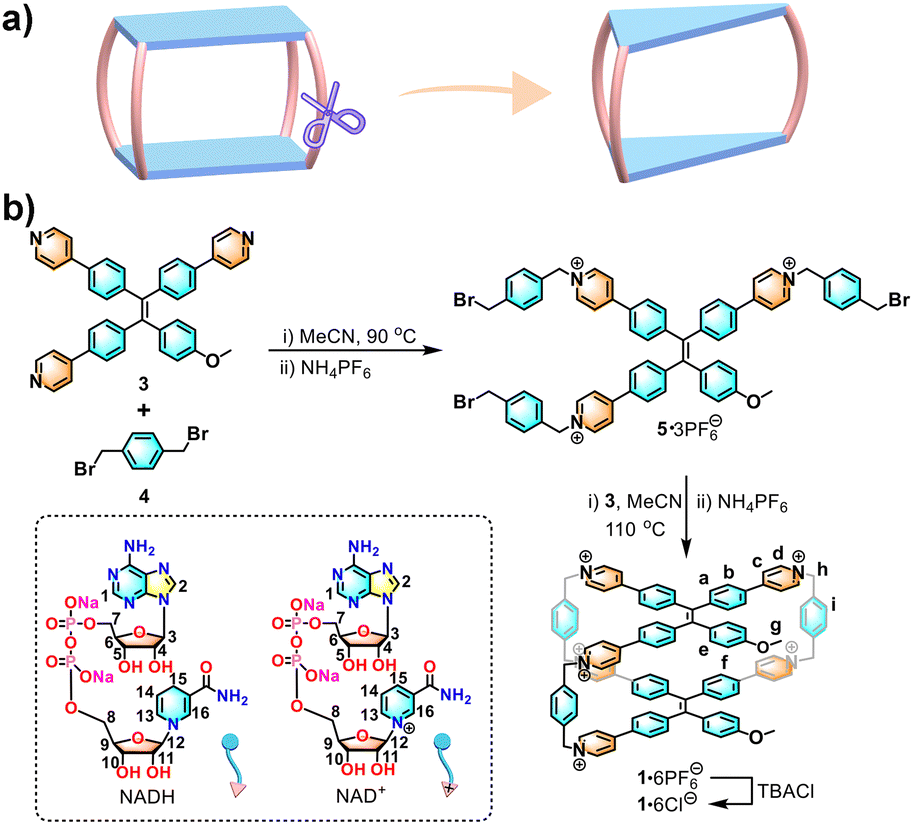 | ||
| Scheme 1 (a) Structural evolution of the three-pillared cage from a four-pillared cage. (b) Synthesis of 1 and chemical structures of NADH and NAD+. | ||
As shown in Scheme 1b, 1·6PF6− was synthesized by two-step SN2 reactions. Firstly, the tripyridyl TPE compound (3) was reacted with 1,4-bis(bromomethyl)benzene (4) to obtain an acyclic tripyridinium compound (5) in ∼53% yield. Next, 5 and 3 were mixed at 110 °C for 3 days to obtain an orange powder of crude 1·6PF6−. The crude product was purified via column chromatography to give pure 1·6PF6− in 4.3% yield. The water-soluble 1·6Cl− was obtained by adding an excess amount of tetrabutylammonium chloride in ∼75% yield (Fig. S1–S13, ESI†).
The X-ray-quality crystal of 1·6Cl− was obtained by slow vapour diffusion of isopropyl ether into a solution of 1·6Cl− in methanol at room temperature. Due to the lack of a pillar, the X-ray structure of 1·6Cl− shows that it has a large open portal with a distance of ∼20.4 Å (Fig. S14, ESI†). The two methoxy benzene rings on the two sides of the portal are stretched with a large distance of ∼14.5 Å. Furthermore, the inside room of the hydrophobic cavity with a triangular cube-like shape is about ∼16.4 Å × ∼13.3 Å × ∼6.7 Å (Fig. 1a). Additionally, there are multiple CH⋯π interactions (dH⋯π plane = ∼3.1 Å, ∼3.7 Å, and ∼2.8 Å, respectively) between the methoxy groups and benzene rings on the portals of the cavities, resulting in an intercrossing dimerization between two neighbouring cages (Fig. 1b and Fig. S15, ESI†). A solvent-dependent NMR experiment showed that 1·6Cl− exhibited broad peaks in D2O but sharp peaks in CD3OD, indicating that 1·6Cl− is in the self-aggregation state in water but in the individual state in methanol (Fig. S16, ESI†). In addition, a concentration-dependent NMR experiment in D2O confirmed that 1·6Cl− kept the aggregated state at low concentrations (50 μM–0.40 mM) (Fig. S17, ESI†). A dimerization constant of (5.89 ± 0.52) × 103 M−1 was calculated from a dilution process by the isothermal titration calorimetry (ITC) experiment (Fig. S18, ESI†).20 Diffusion-ordered spectroscopy (DOSY) of 1 in D2O gave a diffusion coefficient [DH = (1.617 ± 0.177) × 10−10 m2 s−1] with a hydrodynamic radius of ∼1.2 nm (the whole size is ∼2.4 nm; Fig. S19, ESI†), which is consistent with the size (1.9–2.8 nm) of the dimer (Fig. S20, ESI†). Therefore, the hydrophobic effect in the aqueous phase promotes the dimerization of 1·6Cl−, which is similar to that in the crystalline state.
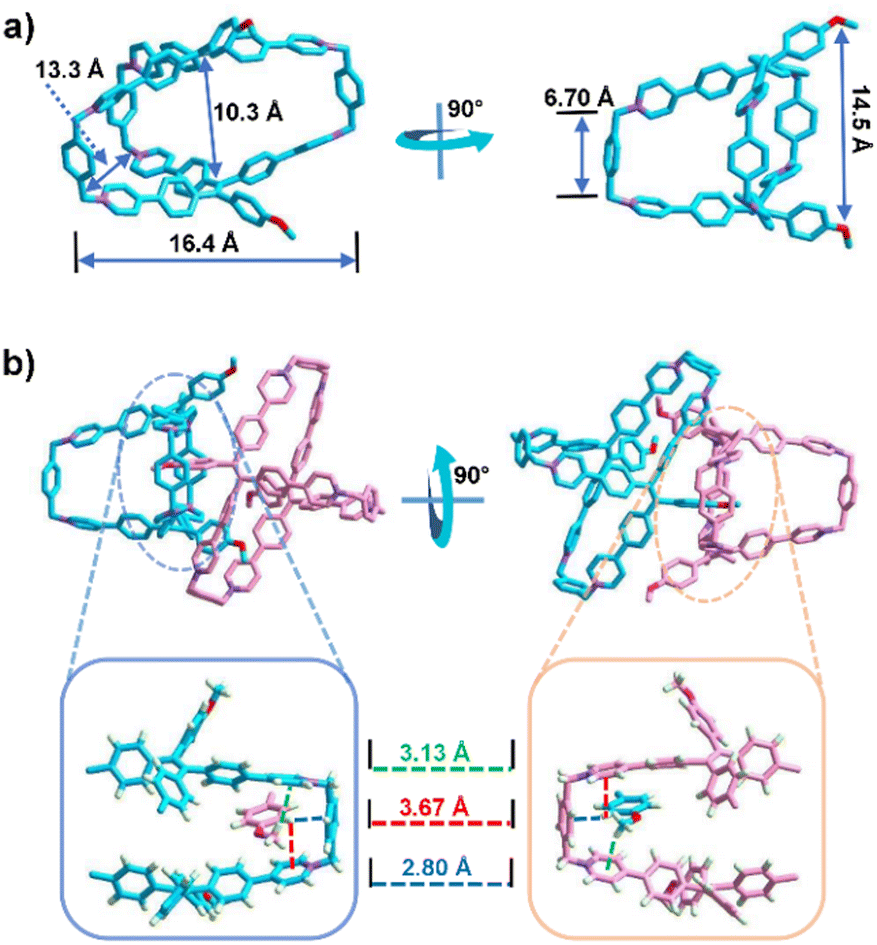 | ||
| Fig. 1 The X-ray structure of 1·6Cl−: (a) single molecule and (b) dimerization between two cages. Counter ions Cl− and hydrogen atoms are omitted for clarity. | ||
The host–guest capacity of 1 was firstly tested using polycyclic aromatic hydrocarbons (PAHs) including anthracene, triphenylene, pyrene, perylene, and coronene by 1H NMR experiments (Fig. S21–S27, ESI†). Compared with the four-pillared cage,18 the binding affinities of 1·6PF6− with PAHs were weaker due to the lack of the restriction in its open cavity (Table S2, ESI†). As a result of weak binding, the host–guest complexation of 1·6PF6− with PAHs could not exert any effects on the photophysical properties (Fig. S28–S32, ESI†). The single crystals of 1·6PF6−⊃coronene were obtained by slow vapor diffusion of isopropyl ether into a solution of 1·6PF6−⊃coronene in acetonitrile at room temperature (Fig. 2). In the sandwich-like structure of 1·6PF6−⊃coronene, coronene is not located at the center of the cavity, but close to one pillar benzene ring at the long side of the cavity, which results from the CH⋯π interaction (dH⋯π plane = ∼3.0 Å) between coronene and the pillar benzene ring. At the same time, the host–guest complexation changes the butterfly-like conformation of methoxy benzene rings on the two sides of the portal in free 1. For example, 1 in the host–guest complex has a compact cavity with a size of approximately ∼16.5 Å × ∼8.93 Å × ∼5.8 Å, which is smaller than the uncomplexed cage (Fig. 2a). This is a guest-induced conformational adaptation, resulting from the CH⋯π (dH⋯π plane = ∼2.8 Å) and π⋯π (dπ⋯π plane = ∼3.4 Å) interactions between coronene and two aromatic faces of 1 (Fig. S33, ESI†). In addition, the stack packing of the host–guest complexes from the b axis results in a 2D-layered structure via the CH⋯π interaction (dH⋯π plane = ∼3.2 Å) (Fig. 2b and Fig. S34, ESI†).
 | ||
| Fig. 2 The X-ray structure of 1·6PF6−⊃coronene: (a) the single complex; (b) the side view of the stack packing. The disordered guests, counter ions PF6−, and hydrogen atoms are omitted for clarity. | ||
Nicotinamide adenine dinucleotides (NADs) including the oxidized form NAD+ and reduced form NADH are important coenzymes in biological metabolic activities,21 which can participate in some important biological reactions.22,23 As a result, NAD+ and NADH can be markers or catalysts to indicate the physiological status or promote chemical reactions.24 Therefore, the selective recognition of NAD by artificial host molecules can provide an effective approach to detect NAD molecules.25 The host–guest chemistry of 1·6Cl− and NAD+/NADH in an aqueous solution were investigated by 1H NMR and ITC experiments. As shown in Fig. 3a, the 1H NMR spectrum of 1·6Cl− in D2O was broadened and unidentified owing to its self-aggregation. When mixing 1·6Cl− and NAD+ in a 1:1 molar ratio, the proton resonances (H1 and H2) of the adenine part of NAD+ disappeared, and the proton resonance (H3) exhibited an upfield shift, which is consistent with the nucleobase being encapsulated (Fig. 3a and Fig. S35, ESI†). At the same time, the proton resonances (H13–H16) of the nicotinamide unit of NAD+ were slightly shifted downfield, which indicated that the nicotinamide unit is located outside the cavity of 1·6Cl− owing to the charge repulsion between the positively-charged nicotinamide unit of the guest and the pyridinium rings of the host. In addition, the energy-minimized calculation and mass spectrometry of 1⊃NAD+ supported the possibility of the 1:1 host–guest complex (Fig. S36 and S37, ESI†).
 | ||
| Fig. 3 Partial 1H NMR spectra of 1·6Cl− (0.4 mM) titrated with (a) NAD+ in D2O and (b) NADH in CD3OD. ITC of (c) NAD+ and (d) NADH titrated with 1·6Cl− at 298 K in water. | ||
Due to the precipitation generated by the charge neutralization between 1·6Cl− and NADH in D2O, 1H NMR titration of 1 with NADH was performed in CD3OD (Fig. 3b and Fig. S38 and S39, ESI†). After 2.0 equiv. of NADH was added, the 1H NMR spectra of the host–guest complexes showed broad peaks for 1·6Cl−, and the peaks of NADH disappeared owing to rapid exchange. At the same time, the proton resonances (Ha, Hi and Hg) of the cage exhibited obvious splitting, indicating that chiral NADH can induce an asymmetric chemical environment of the inner cavity. At the same time, the proton resonance of the neutral nicotinamide unit and the ribose moiety of NADH were shifted upfield, which indicated that the nicotinamide unit is located inside the cavity of 1·6Cl−. Energy-minimized calculations showed that the tail-to-tail (tail is the nicotinamide unit) complex has the lowest energy when compared with the head-to-head and head-to-tail complexes (head is the adenine unit), which is consistent with the NMR results (Fig. S40, ESI†). The ITC experiment confirmed the 1:1 stoichiometry between 1·6Cl− and NAD+ with a binding constant of (3.47 ± 0.72) ×105 M−1 and the 1:2 stoichiometry between 1·6Cl− and NADH, with a binding constant of (2.29 ± 0.43) ×105 M−1 in water (Fig. 3c, d and Fig. S41 and S42, ESI†). The DOSY of 1⊃(NADH)2 and 1⊃NAD+ also exhibited a single band, which provided strong support for the formation of single complexes (Fig. S43 and S44, ESI†). In both cases, the main driving force is the hydrophobic effect for the cage to encapsulate NADH and NAD+ in water. Based on the above host–guest chemistry, we speculate that (1) the binding models between 1·6Cl− and NAD+ are in a 1:1 ratio, in which only the adenine unit of NAD+ is encapsulated inside the cavity of 1·6Cl− and the positively-charged nicotinamide unit is located outside the cavity of 1·6Cl− (Fig. 4, right); (2) the binding models between 1·6Cl− and NADH are in a 1:2 ratio, in which two nicotinamide units are located inside the cavity of 1·6Cl− (Fig. 4, left).
 | ||
| Fig. 4 Schematic representation of the possible binding models between 1·6Cl− and NAD+/NADH. | ||
The optical response of 1·6Cl− to NADH/NAD+ in the host–guest recognition was further studied by fluorescence and CD experiments (Fig. 5). In host–guest recognition, the chiral conformation of the host can be induced by chiral guests to exhibit chiroptical responses.26 Given the right-handed (P) and left-handed (M) rotational conformations of TPE units, achiral 1·6Cl− can be induced into chiral P- or M-conformation in water.8 A CD signal around 380 nm appeared when NADH was added into the solution of 1·6Cl− in water (Fig. 5a), indicating that the formation of a host–guest complex can cause a chirality transfer from NADH to 1·6Cl−. The negative CD signal in the 300–450 nm region contributes to the P-rotational conformation of the TPE units.8 Given their 1:2 host–guest complexation, two chiral NADH in the cavity could effectively restrict the free rotation of TPE units to induce an excess P-rotational conformation. On the contrary, NAD+ could not induce any CD response of 1·6Cl−, probably because only one guest is not sufficient to induce the chiral rotation of TPE (Fig. 5b).
 | ||
| Fig. 5 CD spectra of 1·6Cl− (10 μM) titrated with (a) NADH and (b) NAD+ (0–10.0 equiv.). Fluorescence spectra of 1·6Cl− titrated with (c) NADH and (d) NAD+ (0–5.0 equiv.). | ||
Fluorescence experiments showed different responses of 1·6Cl− as a fluorescent sensor to NADH and NAD+, respectively (Fig. 5c and d). The emission of 1·6Cl− in aqueous solution appeared at about 570 nm. With the addition of NADH and NAD+, NADH quenched the emission of 1·6Cl−, while NAD enhanced the emission of 1·6Cl−. When NADH enters the cavity of the cage as an electron donor, the formation of the host–guest complex enhances the charge transfer interaction between 1·6Cl− and NADH, resulting in the photoinduced electron transfer (PET). When the adenine unit of NAD+ enters the cavity of the cage, the free rotation of the TPE unit is restricted. As a result, the fluorescence intensity is significantly enhanced due to the restricted intramolecular rotation (RIR) of TPE units.18
In conclusion, we have designed and synthesized a TPE-based hexacationic cage (1) with an open cavity. Its host–guest chemistry shows that 1 can encapsulate NAD molecules inside the hydrophobic cavity to form a 1:1 complex for NAD+ and a 1:2 complex for NADH in water. Furthermore, given the fluorescence and dynamic rotational conformation of TPE units, 1 as a chiroptical and fluorescent sensor can recognize NADH and NAD+ with the dual responses of CD and fluorescence in aqueous solution. In future, this molecular cage could provide a host–guest approach to monitor and control NAD-related biological processes.
This work was supported by the National Natural Science Foundation of China (22122108 and 21971208).
Conflicts of interest
There are no conflicts to declare.Notes and references
- J.-M. Lehn, Chem. Soc. Rev., 2017, 46, 2378–2379 RSC.
- (a) Z. Liu, S. K. M. Nalluri and J. F. Stoddart, Chem. Soc. Rev., 2017, 46, 2459–2478 RSC; (b) G. Montà-González, F. Sancenón, R. Martínez-Máñez and V. Martí-Centelles, Chem. Rev., 2022, 122, 13636–13708 CrossRef PubMed.
- T. S. Koblenz, J. Wassenaar and J. N. H. Reek, Chem. Soc. Rev., 2008, 37, 247–262 RSC.
- M. A. Little and A. I. Cooper, Adv. Funct. Mater., 2020, 30, 1909842 CrossRef CAS.
- (a) J. Zhou, G. Yu and F. Huang, Chem. Soc. Rev., 2017, 46, 7021–7053 RSC; (b) D. Zhang, A. Martinez and J.-P. Dutasta, Chem. Rev., 2017, 117, 4900–4942 CrossRef CAS.
- J. Zhou, Z. Chang, Y. Jiang, B. He, M. Du, P. Lu, Y. Hong, H. S. Kwok, A. Qin, H. Qiu, Z. Zhao and B. Z. Tang, Chem. Commun., 2013, 49, 2491–2493 RSC.
- J. Li, J. Wang, H. Li, N. Song, D. Wang and B. Z. Tang, Chem. Soc. Rev., 2020, 49, 1144–1172 RSC.
- (a) H. Qu, Y. Wang, Z. Li, X. Wang, H. Fang, Z. Tian and X. Cao, J. Am. Chem. Soc., 2017, 139, 18142–18145 CrossRef CAS PubMed; (b) J. B. Xiong, H.-T. Feng, J. P. Sun, W. Z. Xie, D. Yang, M. H. Liu and Y.-S. Zheng, J. Am. Chem. Soc., 2016, 138, 11469–11472 CrossRef CAS PubMed.
- (a) X. Zheng, W. Zhu, C. Zhang, Y. Zhang, C. Zhong, H. Li, G. Xie, X. Wang and C. Yang, J. Am. Chem. Soc., 2019, 141, 4704–4710 CrossRef CAS PubMed; (b) Y. Huang, X. You, L. Wang, G. Zhang, S. Gui, Y. Jin, R. Zhao and D. Zhang, Angew. Chem., Int. Ed., 2020, 59, 10042–10051 CrossRef CAS PubMed.
- G. Huang, Q. Xia, W. Huang, J. Tian, Z. He, B. S. Li and B. Z. Tang, Angew. Chem., Int. Ed., 2019, 58, 17814–17819 CrossRef CAS.
- H. Shi, R. T. K. Kwok, J. Liu, B. Xing, B. Z. Tang and B. Liu, J. Am. Chem. Soc., 2012, 134, 17972–17981 CrossRef CAS.
- C. Liu, H. Bai, B. He, X. He, J. Zhang, C. Chen, Y. Qiu, R. Hu, F. Zhao, Y. Zhang, W. He, J. H. C. Chau, S. Chen, J. W. Y. Lam and B. Z. Tang, Angew. Chem., Int. Ed., 2021, 60, 12424–12430 CrossRef CAS.
- H.-T. Feng, Y. Li, X. Duan, X. Wang, C. Qi, J. W. Y. Lam, D. Ding and B. Z. Tang, J. Am. Chem. Soc., 2020, 142, 15966–15974 CrossRef CAS PubMed.
- Z. Wang, C.-Y. Zhu, J.-T. Mo, P.-Y. Fu, Y.-W. Zhao, S.-Y. Yin, J.-J. Jiang, M. Pan and C.-Y. Su, Angew. Chem., Int. Ed., 2019, 58, 9752–9757 CrossRef CAS PubMed.
- S. Dalapati, E. Jin, M. Addicoat, T. Heine and D. Jiang, J. Am. Chem. Soc., 2016, 138, 5797–5800 CrossRef CAS PubMed.
- H. Zhang, L. Cheng, H. Nian, J. Du, T. Chen and L. Cao, Chem. Commun., 2021, 57, 3135–3138 RSC.
- Y. Li, Y. Dong, L. Cheng, C. Qin, H. Nian, H. Zhang, Y. Yu and L. Cao, J. Am. Chem. Soc., 2019, 141, 8412–8415 CrossRef CAS PubMed.
- (a) H. Duan, Y. Li, Q. Li, P. Wang, X. Liu, L. Cheng, Y. Yu and L. Cao, Angew. Chem., Int. Ed., 2020, 59, 10101–10110 CrossRef CAS PubMed; (b) L. Cheng, K. Liu, Y. Duan, H. Duan, Y. Li, M. Gao and L. Cao, CCS Chem., 2020, 2, 2749–2751 Search PubMed; (c) L. Cheng, P. Tian, Q. Li, A. Li and L. Cao, CCS Chem., 2021, 3, 3608–3614 Search PubMed.
- Y. Li, Q. Li, X. Miao, C. Qin, D. Chu and L. Cao, Angew. Chem., Int. Ed., 2021, 60, 6744–6751 CrossRef CAS PubMed.
- S.-X. Nie, H. Guo, T.-Y. Huang, Y.-F. Ao, D.-X. Wang and Q.-Q. Wang, Nat. Commun., 2020, 11, 6257 CrossRef CAS PubMed.
- T. Saba, J. W. H. Burnett, J. Li, P. N. Kechagiopoulos and X. Wang, Chem. Commun., 2020, 56, 1231–1234 RSC.
- W. Ying, Antioxid. Redox Signaling, 2007, 10, 179–206 CrossRef PubMed.
- G. Sultani, A. F. Samsudeen, B. Osborne and N. Turner, J. Neuroendocrinol., 2017, 29, e12508 CrossRef PubMed.
- M. Li, K. H. Gebremedhin, D. Ma, Z. Pu, T. Xiong, Y. Xu, J. S. Kim and X. Peng, J. Am. Chem. Soc., 2022, 144, 163–173 CrossRef CAS PubMed.
- (a) C.-L. Kwok, S.-C. Cheng, P.-Y. Ho, S.-M. Yiu, W.-L. Man, V. K.-M. Au, P.-K. Tsang, C.-F. Leung, C.-C. Ko and M. Robert, Chem. Commun., 2020, 56, 7491–7494 RSC; (b) H. Sharma, N. K. Tan, N. Trinh, J. H. Yeo, E. J. New and F. M. Pfeffer, Chem. Commun., 2020, 56, 2240–2243 RSC.
- (a) C. Lopez-Leonardo, A. Saura-Sanmartin, M. Marin-Luna, M. Alajarin, A. Martinez-Cuezva and J. Berna, Angew. Chem., Int. Ed., 2022, 61, e202209904 CrossRef CAS PubMed; (b) J. Li, H. Y. Zhou, Y. Han and C. F. Chen, Angew. Chem., Int. Ed., 2021, 60, 21927–21933 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details including synthesis, ITC, NMR, UV/vis, fluorescence, and crystal data in the cif format. CCDC 2203594 and 2217483. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cc05153b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |