Polymer-assisted solution phase synthesis of the antihyperglycemic agent Rosiglitazone (Avandia™)†
Xin Lia, Chris Abella, Brian H. Warringtonb and Mark Ladlow*b
aDepartment of Chemistry, University Chemical Laboratories, Lensfield Road, Cambridge, UK CB2 1EW. Fax: +44 (0) 1223 336362; Tel: +44 (0) 1223 336405
bGlaxoSmithKline Cambridge Technology Centre, University Chemical Laboratory, Lensfield Road, Cambridge, UK CB2 1EW. E-mail: Mark.2.Ladlow@gsk.com; Fax: +44 (0) 1223 331532; Tel: +44 (0) 1223 336349
First published on 6th November 2003
Abstract
The commercially important antihyperglycemic agent Rosiglitazone 1 (Avandia™) has been prepared in high purity by a multi-step, polymer-assisted solution phase (PASP) synthesis without the need for the conventional chromatographic purification of any intermediates.
Introduction
Rosiglitazone (Avandia™) 1 is a recently-introduced antihyperglycemic thiazolidinedione (TZD) effective in the treatment of non-insulin-dependent diabetes mellitus (NIDDM), or type II diabetes as it is more commonly known.1 The disease is characterised by a combination of pancreatic β-cell dysfunction and insulin resistance which together lead to elevated blood glucose levels.2 Rosiglitazone 1 activates the peroxisome proliferator-activated nuclear receptor sub-type PPARγ.3 The PPARs are orphan receptors belonging to the nuclear hormone receptor super-family of lipid-activatable nuclear transcription factors, and there is increasing evidence that activation of the PPARγ isoform may be the basis for the antidiabetic effects of this class of thiazolidinedione.4 Although already prevalent throughout the world, it is estimated that currently, at least a third of all cases of NIDMM currently remain undiagnosed. Moreover, hyperglycemia is increasingly implicated in other metabolic disorders, such as hypertension, atherosclerosis and obesity, and therefore represents an important target for medicinal chemical intervention.5In an attempt to improve cost-efficiencies in the drug discovery process and at the same time increase the output of diverse compound collections to fuel high-throughput screening initiatives, an increasing amount of effort is being expended in identifying new paradigms for compound synthesis.6 Solid-phase organic synthesis (SPOS) has enjoyed considerable success, particularly in facilitating high-throughput combinatorial chemistry.7 However, this approach typically suffers from disadvantages such as long reaction development and optimisation times exasperated by difficulties associated with inconvenient on-resin reaction monitoring, the need to use large reagent excesses to drive reactions to completion, and an inability to perform purification of resin-bound intermediates. Consequently, in recent years, attention has re-focussed on identifying improved solution phase approaches. However, whilst solution phase chemistry is able to capitalise on the vast amount of validated information present in the literature, and reactions may be readily monitored using conventional methods, the need to perform routine reaction work-ups and column chromatographic purifications is both inconvenient and leads to reduced throughput.
One solution to this problem is the use of polymer-supported reagents and scavenger resins.8 These combine the advantages of solid-phase chemistry with the flexibility of solution phase synthesis (e.g. ease of work-up by simple filtration, use of reagent excesses to drive reactions to completion, and ready reaction monitoring). Moreover, polymer-assisted solution phase (PASP) synthesis techniques can afford both intermediate and final compounds in good yields and high purity without the need for further purification.9
Herein, to further demonstrate the utility of polymer-supported reagents in the efficient preparation of drug substances, we report a new multi-step PASP synthesis of the antihyperglycemic agent Rosiglitazone 1 starting from five core subunits (the 2,4-thiazolidinedione I, a substituted phenol II, an α-haloester III, a primary amine IV, and a pyridinyl moiety V) (Scheme 1). In particular, the route was devised to utilise supported reagents and in-line purification by ‘resin capture and release’10 using functionalised supports to simplify reaction work-up and afford all intermediates in high yield and purity without the need for conventional column chromatography at any stage.
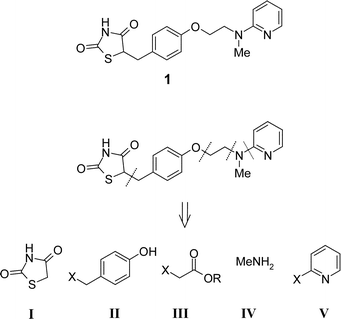 | ||
| Scheme 1 Retrosynthesis of Rosiglitazone 1 | ||
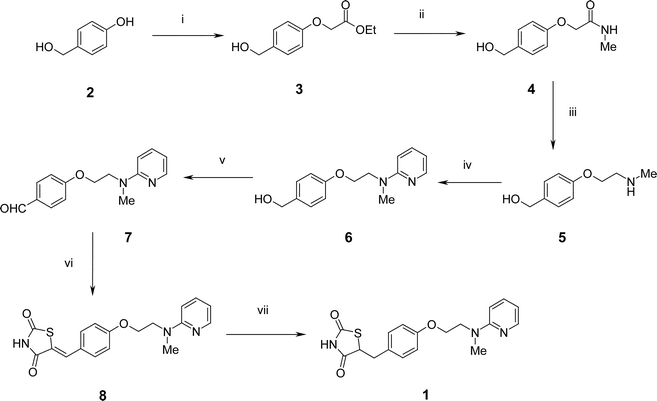 | ||
| Scheme 2 Reagents and conditions: i, ethyl iodoacetate, K2CO3, DMF, 90 °C, 96% or ethyl iodoacetate, PS-BEMP, DMF, rt, 93%; ii, MeNH2, H-15, THF, 100%; iii, (a) BH3·THF, 65 °C, (b) Et2NH, (c) SCX-2, 84% overall; iv, 2-fluoropyridine, 120 °C, 82%; v, PS–CrO3, THF, Δ, 86%; vi, (a) 2,4-thiazolidinedione, piperazinomethyl polystyrene, PhMe, 88 °C, (b) SCX-2, 99%; vii, H2, Pd(OH)2, 1,4-dioxane, 80 °C, 82%. | ||
Results and discussion
Our route to Rosiglitazone 1 began with 4-(hydroxymethyl)phenol112 which was alkylated under basic conditions in the presence of ethyl iodoacetate to afford the ether 3.12 This reaction was found to proceed smoothly in 2 h at room temperature utilising the polymer-supported Schwesinger base PS-BEMP13 and a slight excess of 4-(hydroxymethyl)phenol 2 to drive the reaction to completion. Excess phenol 2 was conveniently removed by performing the reaction in the presence of excess PS-BEMP, or by adding the macroporous basic scavenger MP-carbonate.14 However, although less convenient on a small scale, potassium carbonate could be used as an alternative more cost-effective base, particularly on multi-gram scale. Under these circumstances, the added inconvenience of an aqueous carbonate work-up was offset by a considerable cost advantage. Using either procedure, following solvent evaporation, 3 was obtained in high yield and >95% purity according to 1H NMR analysis without the need for any additional purification.Treatment of the ester 3 with methylamine in the presence of the supported sulfonic acid ion-exchange resin Amberlyst H-15 cleanly gave the carboxamide 4 after a simple filtration and evaporative work-up. Reduction of 4 was then performed by a two-step, one-pot procedure using borane–tetrahydrofuran complex at reflux. Decomposition of the resulting intermediate N-borane complexes with diethylamine cleanly gave the desired N-methylamine 5. On a small scale, 5 was readily purified by ‘catch and release’ using an Isolute™ SCX-2 sulfonic acid cartridge15 followed by elution with methanolic ammonia to release the amine product. No aqueous work-up was found to be necessary. However, on a larger scale, an aqueous work-up prior to solid phase extraction proved to be more efficient since it permitted a higher material loading onto the SCX-2 cartridge. Lithium aluminium hydride proved to be much less satisfactory than borane–tetrahydrofuran for this reduction and gave a more complex reaction mixture.
The amine 5 was treated with neat 2-fluoropyridine at 120 °C to give, after evaporation of excess 2-fluoropyridine, the aminopyridine 6 in high yield and excellent purity. We also attempted to perform this displacement reaction under microwave irradiation in DMF at temperatures up to 155 °C. However, a cleaner transformation was observed with conventional heating at 120 °C overnight.
Oxidation of 6 with polymer-supported chromic acid16 effected clean conversion to the aldehyde 7 with no evidence for formation of the corresponding carboxylic acid product of over-oxidation. In this case the use of the polymer-supported reagent was particularly advantageous and led to a selective transformation that was conveniently worked-up by a simple filtration.
The aldehyde 7 was heated with an excess of 2,4-thiazolidinedione in the presence of polymer-supported piperazine17 and the product purified by ‘catch and release’ using an SCX-2 cartridge to furnish the benzylidene 8 in excellent yield and purity. Finally, 8 was subjected to catalytic hydrogenation18 in the presence of Pearlmann's catalyst at 80 °C to afford Rosiglitazone 1 in 82% yield and high purity (>90% acccording to 1H NMR) with identical physical and spectroscopic properties to those reported.1
Conclusions
In summary, the antihyperglycemic agent Rosiglitazone (Avandia™) 1 has been prepared in 46% overall yield by a seven-step polymer-assisted solution phase synthesis. Although the present synthesis is longer than the previously reported route, the overall yield obtained is higher.19 Moreover, by incorporating in-line ‘catch and release’ purification steps, all compounds were obtained with excellent purities according to 1H NMR analysis20 (ESI†) without the need for conventional column chromatographic purification at any stage. We believe that the present synthesis further demonstrates the utility of polymer-assisted methods to rapidly prepare drug-like compounds in high purity by multi-step solution phase synthesis.Experimental
All moisture-sensitive reactions were carried out under a nitrogen atmosphere in oven-dried glassware. All solvents and reagents were used as supplied unless otherwise stated. Analytical thin layer chromatography (TLC) was carried out on Polygram™ SIL G/UV254 plates. Analytical high pressure liquid chromatography (HPLC) was performed using a Hewlett Packard Series 1050 instrument. Column: Supelcosil™ ABZ+PLUS 3.3 cm × 4.6 mm, 3 µm. Eluent A: water, 0.1% TFA, B: acetonitrile 95%, water 5%, TFA 0.05%. Flow rate: 1 mL min−1. Detection: UV (diode array: 215, 230, 254 nm). Method: gradient 10–95% B in A over 7 min. Melting points were measured on a Mettler FP5 automatic melting point apparatus in open capillaries and are uncorrected. Infrared spectra were collected on a Perkin-Elmer Spectrum One ATR FT-IR instrument. Liquid chromatography–mass spectra (LC–MS) were recorded on a Waters Alliance HT/Micromass ZQ under electrospray positive and negative ionisation conditions. Column: Supelcosil™ ABZ+PLUS 3.3 cm × 4.6 mm, 3 µm. Eluent A: 10 mM solution of ammonium acetate in water, 0.1% formic acid, B: acetonitrile 95%, water 5%, formic acid 0.05%. Flow rate: 1 mL min−1. Detection: UV (diode array: 215, 230, 254 nm). Method: gradient 0–100% B in A over 3.5 min. Accurate mass spectra were recorded on a VG Autospec mass spectrometer in positive electrospray mode. NMR spectra were recorded in the indicated solvent. Chemical shifts (δ) are reported in parts per million (ppm) relative to tetramethylsilane as an internal standard; the following abbreviations are used for multiplicities: s = singlet; d = doublet; t = triplet; m = multiplet; dd = doublet of doublets; br = broad; and coupling constant J-values are quoted in Hz. 1H NMR spectra were recorded on a Bruker AM 400 spectrometer at 400 MHz. 13C NMR spectra were recorded on a Bruker AM 400 at 100 MHz with proton decoupling.2-[4-(Hydroxymethyl)phenoxy]acetic acid ethyl ester (3)
To the solution of 4-(hydroxymethyl)phenol (13.6 g, 110 mmol) in DMF (100 mL), was added ethyl iodoacetate (8.0 mL, 67.6 mmol) and potassium carbonate (20.0 g; 145 mmol). The resulting suspension was stirred at 90 °C for 2 h and the solvent was evaporated in vacuo. The residue was partitioned between 2 M aqueous potassium carbonate solution (100 mL) and ethyl acetate (100 mL). The organic layer was washed with 2 M aqueous potassium carbonate solution, saturated brine, and dried (MgSO4). The solvent was removed in vacuo to afford the ethyl ester 3 as a colourless oil (13.6 g, 96%). IR νmax/cm−1: 3406 (νOH) and 1752 (νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O); 1H NMR (400 MHz, CDCl3): δ 1.27 (3H, t, J
= 7.2 Hz), 2.05 (1H, t, J
= 5.6 Hz), 4.24 (2H, q, J
= 7.2 Hz), 4.57 (2H, d, J
= 5.6 Hz), 4.58 (2H, s), 6.86 (2H, d, J
= 8.8 Hz), 7.25 (2H, d, J
= 8.8 Hz); 13C NMR (100 MHz, CDCl3): δ 168.3, 156.7, 133.6, 128.0 (2C), 114.0 (2C), 64.8, 64.1, 60.8, 13.5; LC–MS: tR
= 3.22 min, m/z
(ESI)
[M − OH]+
= 193.2. HR-MS (ESI): m/z calc. for C11H14O4Na 233.0790, found 233.0786 [M + Na]+.
O); 1H NMR (400 MHz, CDCl3): δ 1.27 (3H, t, J
= 7.2 Hz), 2.05 (1H, t, J
= 5.6 Hz), 4.24 (2H, q, J
= 7.2 Hz), 4.57 (2H, d, J
= 5.6 Hz), 4.58 (2H, s), 6.86 (2H, d, J
= 8.8 Hz), 7.25 (2H, d, J
= 8.8 Hz); 13C NMR (100 MHz, CDCl3): δ 168.3, 156.7, 133.6, 128.0 (2C), 114.0 (2C), 64.8, 64.1, 60.8, 13.5; LC–MS: tR
= 3.22 min, m/z
(ESI)
[M − OH]+
= 193.2. HR-MS (ESI): m/z calc. for C11H14O4Na 233.0790, found 233.0786 [M + Na]+.2-[4-(Hydroxymethyl)phenoxy]-N-methylacetamide (4)
To the ethyl ester 3 (7.00 g, 33.3 mmol) in a 2 M solution of methylamine in THF (100 mL), was added Amberlyst H-15 ion exchange resin (250 mg). The resulting suspension was stirred at room temperature for 48 h. After filtration, the resin was thoroughly washed with THF (4 × 5 mL) and the combined filtrates were concentrated in vacuo to give the amide 4 as white solid (6.55 g, 100%). Mp: 123–124 °C; IR νmax/cm−1: 3446 (νOH), 3331 (νN–H) and 1652 (νCO); 1H NMR (400 MHz, CDCl3): δ 1.80 (1H, br s), 2.90 (3H, d, J
= 4.8 Hz), 4.46 (2H, br s), 4.63 (2H, s), 6.59 (1H, br s), 6.88 (2H, d, J
= 8.8 Hz), 7.32 (2H, d, J
= 8.8 Hz); 13C NMR (100 MHz, CDCl3): δ 168.1, 156.0, 134.0, 128.2 (2C), 114.0 (2C), 66.7, 64.1, 25.1; LC–MS (ESI): tR
= 2.61 min, m/z
(ESI)
[M + H]+
= 196.2; HR-MS (ESI): m/z calc. for C10H13NO3Na 218.0793, found 218.0782 [M + Na]+.4-[2-(Methylamino)ethoxyphenyl]methanol (5)
A solution of borane–THF (1 M, 65 mL, 65.0 mmol) was added steadily to a stirring solution of the amide 4 (5.48 g, 28.1 mmol) in anhydrous THF (80 mL) at ambient temperature. The resulting mixture was stirred at 65 °C for 24 h and then allowed to cool to room temperature. Dilute hydrochloric acid (10%, ∼20 mL) was slowly added to the flask, and the mixture was stirred at room temperature for 1 h to give a white suspension. The suspension was treated with 2 M NaOH aqueous solution until a clear biphasic mixture was obtained (pH 12–14). Saturated brine (50 mL) was added, and the aqueous layer was extracted with diethyl ether (3 × 50 mL). The combined organic extracts were dried (MgSO4), and concentrated in vacuo. The residue obtained was treated with 10% diethylamine solution in THF (50 mL) at room temperature for 16 h and then concentrated in vacuo. This material was loaded equally onto 6 × 10 g SCX-2 cartridges. The cartridges were thoroughly washed with methanol, and the amine 5 was liberated by elution with 2 M methanolic ammonia as a pale yellow liquid (4.28 g, 84%). IR νmax/cm−1: 3310 (νN–H and OH); 1H NMR (400 MHz, CDCl3): δ 2.35 (3H, s), 2.81 (2H, t, J = 5.0 Hz), 3.36 (1H, br s), 3.88 (2H, t, J = 5.0 Hz), 4.47 (2H, s), 6.73 (2H, d, J = 8.8 Hz), 7.16 (2H, d, J = 8.8 Hz); 13C NMR (100 MHz, CDCl3): δ 157.3, 133.6, 127.6 (2C), 113.6 (2C), 65.9, 63.3, 49.9, 35.3; LC–MS: tR = 1.33 min, m/z (ESI) [M + H]+ = 182.2;. HR-MS (ESI): m/z calc. for C10H16NO2 182.1181, found 182.1179 [M + H]+.{4-[2-(Methylpyridin-2-ylamino)ethoxy]phenyl}methanol (6)
The amine 5 (3.71 g, 20.5 mmol) was dissolved in 2-fluoropyridine (40 mL) and the resulting solution was heated at 120 °C under nitrogen for 24 h. The mixture was concentrated in vacuo and the residue obtained was loaded onto a 10 g silica cartridge, and the cartridge was eluted with ethyl acetate (100 mL). The eluent was evaporated in vacuo to afford the tertiary amine 6 as an oil (4.34 g, 82%). IR νmax/cm−1: 3348 (νOH); 1H NMR (400 MHz, CDCl3): δ 2.05 (1H, br s), 3.13 (3H, s), 3.96 (2H, t, J = 5.6 Hz), 4.16 (2H, t, J = 5.6 Hz), 4.58 (2H, s), 6.52 (2H, m), 6.86 (2H, d, J = 8.8 Hz), 7.24 (2H, d, J = 8.8 Hz), 7.44 (1H, m), 8.13 (1H, m); 13C NMR (100 MHz, CDCl3): δ 157.8, 157.6, 147.2, 136.7, 132.6, 128.0 (2C), 113.8 (2C), 111.1, 105.1, 65.7, 64.3, 48.8, 37.2; LC–MS: tR = 2.54 min, m/z (ESI) [M + H]+ = 259.4; HR-MS (ESI): m/z calc. for C15H19N2O2 259.1446, found 259.1445 [M + H]+.4-[2-(Methylpyridin-2-ylamino)ethoxy]benzaldehyde (7)
To the solution of benzylic alcohol 6 (3.0 g, 11.6 mmol) in anhydrous THF (50 mL), was added polymer-supported chromic acid15 (2.5 mmol g−1, 9.0 g, 22.5 mmol). The resulting suspension was heated under reflux with stirring for 24 h. Additional polymer-supported chromic acid (2.5 mmol g−1, 4.5 g, 11.2 mmol) was added and the mixture was heated under reflux for a further 24 h. The suspension was cooled to room temperature, filtered, and the resin was thoroughly washed with ethyl acetate. The combined filtrates were evaporated in vacuo, and the residue was filtered through a short silica plug eluting with ethyl acetate (50 mL) to afford the aldehyde 7 as a gum (2.58 g, 86%). IR νmax/cm−1: 1690 (νCO) cm−1; 1H NMR (400 MHz, CDCl3): δ 3.11 (3H, s), 3.99 (2H, t, J
= 5.6 Hz), 4.24 (2H, t, J
= 5.6 Hz), 6.49 (1H, d, J
= 8.8 Hz), 6.54 (1H, m), 6.97 (2H, d, J
= 8.8 Hz), 7.43 (1H, m), 7.77 (2H, d, J
= 8.8 Hz), 8.13 (1H, m), 9.83 (1H, s); 13C NMR (100 MHz, CDCl3): δ 190.2, 163.3, 157.5, 147.2, 136.7, 131.3 (2C), 129.2, 114.1 (2C), 111.3, 105.1, 66.0, 48.6, 37.3. LC–MS: tR
= 2.72 min, m/z
(ESI)
[M + H]+
= 257.4; HR-MS (ESI): m/z calc. for C15H17N2O2 257.1290, found 257.1295 [M + H]+.(Z)-5-{4-[2-(Methylpyridin-2-ylamino)ethoxy]benzylidene}thiazolidine-2,4-dione (8)
To a solution of the aldehyde 7 (1.46 g, 5.70 mmol) and 2,4-thiazolidinedinone (3.33 g, 28.5 mmol) in toluene (50 mL), was added piperazinomethyl polystyrene16 (0.83 mmol g−1, 3.00 g, 2.49 mmol). The resulting mixture was heated at 88 °C for 24 h and then allowed to cool to room temperature. The suspension was filtered and the resin was washed thoroughly with CH2Cl2. The combined filtrates were evaporated in vacuo, and the residue was loaded onto 3 × 10 g SCX-2 cartridges. The cartridges were eluted first with ethyl acetate, and then with a mixture of dimethylethylamine and methanol (1 ∶ 1) to elute the benzylidene 8 as a yellow solid (2.02 g, 99%). Mp: 196–198 °C (from MeOH); IR νmax/cm−1: 1688 (νCO) cm−1; 1H NMR (400 MHz, d6-DMSO): δ 3.04 (3H, s), 3.90 (2H, t, J
= 5.6 Hz), 4.19 (2H, t, J
= 5.6 Hz), 6.54 (1H, m), 6.62 (1H, d, J
= 8.8 Hz), 7.07 (2H, d, J
= 8.8 Hz), 7.47 (1H, m), 7.50 (2H, d, J
= 8.8 Hz), 8.05 (1H, m); 13C NMR (100 MHz, d6-DMSO): δ 167.8, 167.4, 160.0, 157.8, 147.3, 137.1, 131.9 (2C), 131.5, 125.4, 120.3, 115.1 (2C), 111.4, 105.6, 65.6, 48.1, 36.8; LC–MS: tR
= 3.19 min, m/z
(ESI)
[M + H]+
= 356.2; HR-MS (ESI): m/z calc. for C18H18N3O3S 356.1069, found 356.1082 [M + H]+.5-{4-[2-(Methylpyridin-2-ylamino)ethoxy]benzyl}thiazolidine-2,4-dione (1)
Palladium hydroxide (20% on charcoal, 300 mg) was suspended in a solution of the benzylidene 8 (355 mg, 1.00 mmol) in 1,4-dioxane (40 mL). The suspension was heated at 80 °C with stirring under a hydrogen atmosphere (∼1 atm) for 24 h and then allowed to cool to room temperature. The mixture was filtered through a Celite pad and the pad was washed thoroughly with ethanol. The combined organic filtrates were evaporated in vacuo and the residue obtained was loaded onto a 5 g SCX-2 cartridge. The cartridge was first washed with methanol, and then eluted with 2 M methanolic ammonia. The methanolic ammonia fractions were collected and the solvent was evaporated in vacuo. The residue obtained was diluted with water (10 mL), adjusted to pH 7.5–8.0 with 10% hydrochloric acid, and extracted with CH2Cl2 (3 × 10 mL). The combined organic extracts were dried (MgSO4) and concentrated in vacuo to give Rosiglitazone 1 as a white solid (0.30 g, 82%) with physical properties identical to those previously reported.1 Mp: 153–154 °C (from MeOH); Found: C, 60.66; H, 5.42; N, 11.57; S, 8.68. C18H19N3O3S requires: C, 60.49; H, 5.36; N, 11.76; S, 8.97%; IR νmax/cm−1: 1695 (νCO); 1H NMR (400 MHz, CDCl3): δ 3.08 (1H, dd, J
= 9.2 and 14.0 Hz), 3.13 (3H, s), 3.40 (1H, dd, J
= 3.6 and 14.0 Hz), 3.94 (2H, m), 4.13 (2H, m), 4.46 (1H, dd, J
= 3.6 and 9.2 Hz), 6.50 (1H, d, J
= 8.8 Hz), 6.54 (1H, m), 6.82 (2H, d, J
= 8.8 Hz), 7.10 (2H, d, J
= 8.8 Hz), 7.44 (1H, m), 8.12 (1H, m); 1H NMR (400 MHz, d6-DMSO): δ 3.03 (1H, dd, J
= 9.2 and 14.0 Hz), 3.05 (3H, s), 3.26 (1H, dd, J
= 4.1 and 14.0 Hz), 3.86 (2H, t, J
= 6.0 Hz), 5.68 (2H, t, J
= 6.0 Hz), 4.83 (1H, dd, J
= 4.1 and 9.2 Hz), 6.54 (1H, dd, J
= 4.6 and 7.0 Hz), 6.61 (1H, d, J
= 9.0 Hz), 6.85 (2H, d, J
= 8.4 Hz), 7.11 (2H, d, J
= 8.4 Hz), 7.47 (1H, m), 8.05 (1H, m); 13C NMR (100 MHz, CDCl3): δ 174.4, 170.6, 157.6, 157.5, 146.9, 136.8, 129.7 (2C), 127.2, 114.0 (2C), 111.1, 105.3, 65.5, 53.1, 48.9 37.2, 37.1; LC–MS: tR
= 2.89 min, m/z
(ESI)
[M + H]+
= 358.2; HR-MS (ESI): m/z calc. for C18H20N3O3S 358.1225, found 358.1228 [M + H]+;Acknowledgements
We thank GlaxoSmithKline for financial support and a postdoctoral fellowship (to X. L.).References
-
(a) K. M. Brummond and J. Lu, J. Org. Chem., 1999, 64, 1723 CrossRef CAS
; (b) B. C. C. Cantello, M. A. Cawthorne, G. P. Cottam, P. T. Duff, D. Haigh, R. M. Hindley, C. A. Lister, S. A. Smith and P. L. Thurlby, J. Med. Chem., 1994, 37, 3977 CrossRef CAS
- R. A. DeFronzo, R. C. Bonnadonna and E. Ferrannini, Diabetes Care, 1992, 15, 318 Search PubMed
- J. M. Lehmann, L. B. Moore, T. A. Smith-Oliver, W. O. Wilkison, T. M. Willson and S. A. Kliewer, J. Biol. Chem., 1995, 270, 12953 CrossRef CAS
- T. M. Willson, P. J. Brown, D. D. Sternbach and B. R. Henke, J. Med. Chem., 2000, 43, 27 CrossRef CAS
-
(a) V. Shantaram, Clin. Exp. Hypertens., 1999, 21, 69 CAS
-
(a) S. V. Ley and I. R. Baxendale, Nat. Rev. Drug Discovery, 2002, 573 Search PubMed
-
(a) J. Yoshida and K. Itami, Chem. Rev., 2002, 102, 3693 CrossRef CAS
-
(a) S. V. Ley, I. R. Baxendale, R. N. Bream, P. S. Jackson, A. G. Leach, D. A. Longbottom, M. Nesi, J. S. Scott, R. I. Storer and S. J. Taylor, J. Chem. Soc., Perkin Trans. 1, 2000, 3815 RSC
- For example:
(a) I. R. Baxendale, A.-L. Lee and S. V. Ley, J. Chem. Soc., Perkin Trans. 1, 2002, 1850 RSC
- D. L. Flynn, Med. Res. Rev., 1999, 19, 408 CrossRef CAS
- An alternative, more advanced, starting material would have been 4-hydroxybenzaldehyde. However, in our hands, this material was found to undergo a Cannizzaro reaction under strongly basic conditions to afford a mixture of the corresponding alcohol and carboxylic acid products.
- C. P. Miller, M. D. Collini, B. D. Tran, H. A. Harris, Y. P. Kharode, J. T. Marzolf, R. A. Moran, R. A. Henderson, R. H. W. Bender, R. J. Unwalla, L. M. Greenberger, J. P. Yardley, M. A. Abou-Gharbia, C. R. Lyttle and B. S. Komm, J. Med. Chem., 2001, 44, 1654 CrossRef CAS
- PS-BEMP (2-tert-Butylimino-2-diethylamino-1,3-dimethyl-perhydro-1,3,2-diazaphosporine on polystyrene), 2.3 mmol g−1 was obtained from Fluka Cat. No. 20026.
- MP-carbonate was obtained from Argonaut Technologies Inc. Cat. No. 800268.
- Isolute™ SCX-2 (alkylsulfonic acid) cartridges were obtained from Jones Chromatography (e.g. Cat. No. 532-0100-C).
- Chromic acid on Amberlyst A-26 was obtained from Sigma-Aldrich Cat. No. 35,982-3..
- Piperazinomethyl polystyrene was obtained from NovaBiochem Cat. No. 01-64-0310; cf: J. Simpson, D. L. Rathbone and D. C. Billington, Tetrahedron Lett., 1999, 40, 7031 Search PubMed
- We found that this 1,4-reduction could also be mediated using aqueous sodium dithionite, however the procedure proved to be difficult to reproduce reliably and therefore catalytic hydrogenation under forcing conditions was preferred.
- According to the reported procedure in ref. 2b, Rosiglitazone was obtained in 31% overall yield as compared to 46% overall yield in the present synthesis.
- For the present synthesis, we found that 1H NMR spectroscopy was more indicative of the purity of synthetic intermediates than LC–MS since many of the compounds are quite polar and tend to elute close to the origin using conventional RP-HPLC methods and solvent gradients.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR and 13C NMR spectra for all reported compounds. See http://www.rsc.org/suppdata/ob/b3/b307549d/ |
| This journal is © The Royal Society of Chemistry 2003 |