Nitrophenyl derivatives of pyrrole 2,5-diamides: structural behaviour, anion binding and colour change signalled deprotonation†
Salvatore Camiolo, Philip A. Gale*, Michael B. Hursthouse and Mark E. Light
School of Chemistry, University of Southampton, Southampton, UK SO17 1BJ. E-mail: philip.gale@soton.ac.uk; Fax: 44-23-8059-6805; Tel: 44-23-8059-3332
First published on 28th January 2003
Abstract
Two new pyrrole 2,5-diamide clefts have been synthesized containing 4-nitrophenyl or 3,5-dinitrophenyl groups appended to the amide positions. The 3,5-dinitrophenyl derivative has been shown to deprotonate in the presence of fluoride, which in acetonitrile solution, gives rise to a deep blue colour.
Introduction
The selective recognition of anionic species by organic host molecules is a rapidly developing area of supramolecular chemistry.1 We have recently been investigating the anion complexation properties of 2,5-diamide-substituted pyrroles (e.g. 1a), simple receptors that show oxo-anion selectivity.2 We found that introduction of electron withdrawing chlorine substituents to the 3- and 4- positions of the pyrrole ring, significantly enhanced the affinity of the receptors (e.g.1b) for anions such as chloride,3 but more basic anions would deprotonate the pyrrole NH group leading to the formation of interlocked pyrrole anion dimers.3,4 We decided to introduce electron withdrawing groups to the amide positions in an attempt to enhance the affinity of the receptor without increasing the acidity of the pyrrole NH proton to such an extent that deprotonation would occur upon addition of anions. We therefore synthesized compounds 2 and 3 containing nitro-aromatic moieties and investigated their binding properties upon addition of anionic guests. In most cases, these receptors were found not to deprotonate and enhanced binding as compared to compound 1a was observed. However, addition of fluoride to receptor 3 did cause deprotonation, resulting (in acetonitrile) in the formation of a deep blue coloured solution. Colorimetric sensors for fluoride are particularly desirable as putative sensors for the nerve gas Sarin (GB) (isopropyl methylphosphonofluoridate) which loses a fluoride anion during hydrolysis.5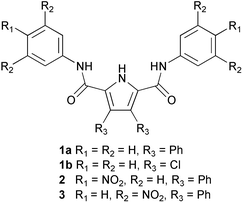
Results and discussion
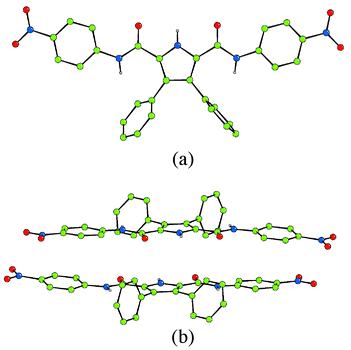
Compounds 2 and 3 were synthesised by reaction of either 4-nitro- or 3,5-dinitroaniline with 3,4-diphenyl-1H-pyrrole-2,5-dicarbonyl dichloride in dichloromethane in the presence of Et3N and DMAP which afforded the receptors in 43 and 11% yields respectively.Crystals of compound 2 suitable for analysis by single crystal X-ray diffraction were obtained by recrystallisation from hot acetonitrile.6 The crystals were very small and hence the diffraction weak, leading to a very high merging R factor. The asymmetric unit contains one and a half molecules. The crystal structure does not reveal the formation of dimers in the solid state which has been observed with a number of other similar molecules (Fig. 1).2 This may be due to extensive π-stacking in this crystal. Crystals of both compounds 2 and 3 were obtained from DMSO solutions of the receptors‡. The crystal structures reveal a DMSO bound to the pyrrole NH and one of the amide NH groups with the receptors both adopting a “semi-cleft conformation” as has been previously observed with compound 12 (Fig. 2).7,8 When crystallised from a dichloromethane solution, receptor 3 forms sheets that extend along the −1 0 1 plane via pyrrole NH–ON(nitro) hydrogen bonds (Fig. 3).9
 | ||
| Fig. 1 (a) The crystal structure of compound 2. Colour key: carbon = green, nitrogen = blue, oxygen = red. (b) Side view of the π-stacking interaction. | ||
 | ||
| Fig. 2 Crystal structures of the DMSO solvates of (a) compound 2 and (b) compound 3. Colour key: carbon = green, nitrogen = blue, oxygen = red, sulfur = yellow. | ||
 | ||
| Fig. 3 The crystal structure of compound 3 showing NH–ON hydrogen bonds (phenyl groups in the 3- and 4- positions of the pyrrole rings have been omitted for clarity). Colour key: carbon = green, nitrogen = blue, oxygen = red. | ||
Interestingly, in DMSO-d6–0.5% water solution, the amide NH proton resonances of 1, 2 and 3 are at 9.36, 10.19 and 11.29 ppm respectively indicating the increasing degree of de-shielding of this proton as the number of nitro groups increases. Proton NMR titrations were performed in order to assess the affinity of the receptors for anions.10 Addition of solutions of anions (as their tetrabutylammonium salts) to solutions of receptor 2 in DMSO-d6–0.5% water and fitting of the resultant titration curves gave association constants of 1245 M−1 for fluoride, 39 M−1 for chloride and 4150 M−1 for benzoate. The titration curve found with dihydrogen phosphate could not be fitted to a simple 1 ∶ 1 binding model. This may be due to oligomerisation of the phosphate anions in solution11 or alternatively simply to the formation of a variety of complexes with differing stoichiometries. Unfortunately, the pyrrole NH proton frequently disappears (presumably due to exchange processes) upon addition of anions to the solution for both receptors 2 and 3 making it impossible to calculate an association constant based on the shift of this proton resonance.
Under the same conditions, addition of anions to solutions of receptor 3 in DMSO-d6–0.5% water gave unusual results. Surprisingly, upon addition of fluoride and benzoate, no significant shift in the NH resonance was observed (with the resonance disappearing at higher anion concentrations). However the CH protons on the nitro-aromatic ring do shift downfield in what appears to be a three-stage process for fluoride (Fig. 4). Upon addition of the first two equivalents of anion a curve is obtained that is similar to that observed during the titration of compound 1b with fluoride in dichloromethane.2 Further addition of anions causes a downfield shift until the plateau is reached after the addition of three equivalents of fluoride. This behaviour seems to be consistent with a three step process that may be hypothesized as follows: the first equivalent of fluoride is coordinated by the receptor (causing a significant downfield shift); the second equivalent promotes the deprotonation process3,4 and the third equivalent of fluoride is coordinated by the deprotonated receptor with the participation of the phenyl CH groups that, in this receptor, are particularly acidic because of the presence of the electron withdrawing groups present in the phenyl ring.
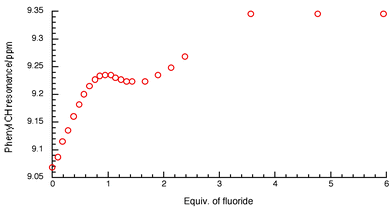 | ||
| Fig. 4 NMR titration curve of compound 3 with fluoride anions in DMSO-d6–0.5% water. | ||
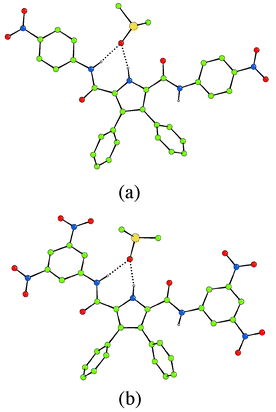
X-Ray crystallographic analysis of crystals of 3 grown by slow evaporation of a dichloromethane–methanol solution of the receptor in the presence of excess tetrabutylammonium fluoride, revealed the presence of an adventitious chloride anion coordinated to receptor 3.12 The crystals were very small and hence the diffraction weak, leading to a very high merging R factor. The structure confirms that the receptor has been deprotonated (two tetrabutylammonium counter cations are present) and reveals a chloride anion bound by two CH–Cl and two NH–Cl interactions in the plane of the three rings with C(H)–Cl hydrogen bonds being in the range 3.377(11)–3.397(12) Å and N(H)–Cl hydrogen bonds in the range 3.385(13)–3.403(12) Å. The chloride–pyrrolic nitrogen separation is 3.500(8) Å. We assume that if an amide had in fact deprotonated, the chloride would not be bound in its current position (Fig. 5).
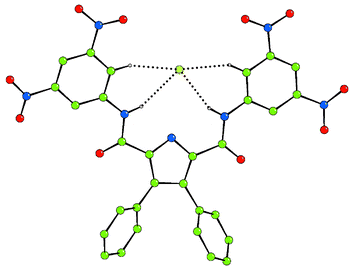 | ||
| Fig. 5 The crystal structure of 3-H+ binding chloride via NH and CH hydrogen bonds (two tetrabutylammonium counter cations have been omitted for clarity). Colour key: carbon = green, nitrogen = blue, oxygen = red, chloride = light green. | ||
For benzoate, the shift of the CH protons could be fitted to a 1 ∶ 1 binding model that corresponds to an association constant of 4200 M−1. With chloride, the amide NH protons do shift allowing an association constant of 53 M−1 to be calculated. Addition of dihydrogen phosphate gave a continuous downfield shift of the amide NH protons.
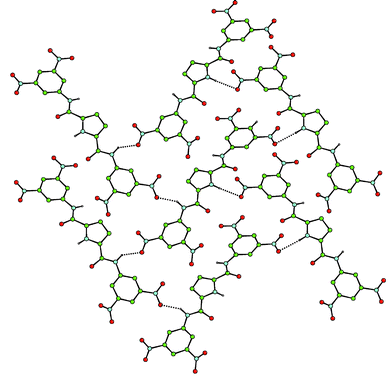
We also investigated the possibility of using these receptors as colorimetric sensors for anions in acetonitrile (Fig. 6). In this solvent, the receptors are not soluble, however addition of anions solubilises the host (in all cases except the addition of bromide to compound 2). In the case of receptor 2, a yellow colour appears with the anions that are bound most strongly (in DMSO-d6 solution) i.e. fluoride, benzoate and dihydrogen phosphate. However, with compound 3, an intense blue colour is observed upon addition of fluoride anions in acetonitrile. We believe this colour is due to a deprotonation process caused by fluoride acting as a base and subsequent charge transfer interaction between the deprotonated pyrrole and the nitroaromatic functionalities. In order to confirm this, receptor 3 was treated with 20 equivalents of TBAOH and, after removing the excess base, a dark red material was crystallised from a mixture of diethyl ether–dichloromethane. The crystal structure of this material reveals that it is indeed the deprotonated ligand, crystallised as the tetrabutylammonium salt (Fig. 7).13 The crystallographic asymmetric unit contains two half pyrrole anions and a single tetrabutylammonium cation. The pyrrole 2,5-diamide anions interlock as has been observed previously in 3,4-dichloropyrrole 2,5-diamides with NH⋯N− distances in the range 2.97–3.08 Å.3,4 Interestingly when this material is dissolved in dichloromethane it maintains the dark red colour in solution. However a variety of colours are observed when the compound is dissolved in more polar solvents: blue in acetonitrile or acetone, purple in methanol and violet in water. UV/VIS analysis carried out on both an acetonitrile solution of this material and compound 3 in the presence of excess fluoride, revealed identical UV/VIS spectra (with a maximum at 598 nm). Consequently, we believe that the deep blue colour observed upon addition of an excess of fluoride anions to acetonitrile solutions of compound 3 is due to deprotonation of the receptor by the fluoride anion.
 | ||
| Fig. 6 Solutions of (a) receptor 2 and (b) receptor 3 (2 mM) in acetonitrile with various anionic guests (added as their tetrabutylammonium salt at a concentration of 20 mM). In the absence of an anion, the receptors are not soluble in this solvent but are solubilised upon addition of the anion (with the exception of receptor 2 and bromide). | ||
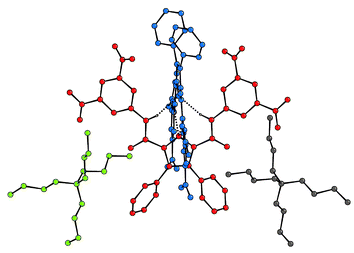 | ||
| Fig. 7 The X-ray crystal structure of the tetrabutylammonium salt of 3-H+. Colours represent individual components of the assembly. Certain hydrogen atoms have been omitted for clarity. | ||
The strategy of attaching electron withdrawing groups3,14 to an anion receptor to enhance anion affinity has worked in this case with the affinity for benzoate increasing from 560 M−1 for compound 1a to 4150 M−1 for compound 2 and 4200 M−1 for compound 3. With our previous 3,4-dichloropyrrole-based systems, benzoate and dihydrogen phosphate were shown to deprotonate the receptors3 however in this case only receptor 3 undergoes a deprotonation process upon anion addition and then only with fluoride. A number of research groups are currently synthesizing new colorimetric anion sensors.15 We have shown that deprotonation of a functionalised pyrrole provides a selective colorimetric method of sensing fluoride in acetonitrile solution.
Experimental
J-values are given in Hz.N,N′-Bis(4-nitrophenyl)-3,4-diphenyl-1H-pyrrole-2,5-dicarboxamide (2):
3,4-Diphenyl-1H-pyrrole-2,5-dicarboxylic acid16 (1.0 g, 3.3 mmol) was suspended in SOCl2 (20 cm3) and refluxed overnight. The reaction was allowed to cool and the excess of SOCl2 was removed in vacuo. A brown solid formed that was dissolved in CH2Cl2 (30 cm3) and Et3N (0.74 g, 7.3 mmol) and DMAP (5 mg, 0.04 mmol) were added. After addition of 4-nitroaniline (0.98 g, 7.15 mmol), the mixture was stirred at room temperature for 72 hours. The solvent was removed in vacuo and acetonitrile (20 cm3) was added to the red solid. A pale yellow compound crystallised, that was collected and washed with acetonitrile (2 × 5 cm3). After drying, 770 mg of the desired compound was obtained (43%). δH (300 MHz; DMSO-d6; Me4Si) 7.26–7.35 (10H, m, Arom.), 7.85 (4H, d, J 9.1, NHCCH), 8.33 (4H, d, J 9.1, NO2CCH), 10.19 (2H, s, CONH), 12.91 (1H, s, NH). δC (75 MHz; DMSO-d6; Me4Si) 119.1, 125.0, 126.9, 127.8, 128.1, 130.5, 133.2, 142.4, 144.8, 159.3. m/z (ES+) 1095 (M + M+). HRMS (ES−): 546 (M−), Δ = 0.7 ppm. Found: C, 64.51; H, 3.75; N, 12.10%. M+ 0.2 DCM requires C, 64.26; H, 3.82; N, 12.41%. Mp >320 °C.N,N′-Bis(3,5-dinitrophenyl)-3,4-diphenyl-1H-pyrrole-2,5-dicarboxamide (3):
3,4-Diphenyl-1H-pyrrole-2,5-dicarboxylic acid16 (1.5 g, 4.9 mmol) was suspended in SOCl2 (25 cm3) and refluxed overnight. The reaction was allowed to cool and the excess of SOCl2 was removed in vacuo. A brown solid formed that was dissolved in CH2Cl2 (50 cm3) and Et3N (1.21 g, 12 mmol) and DMAP (5 mg, 0.04 mmol) were added. After addition of 3,5-dinitroaniline (1.88 g, 10 mmol), the mixture was stirred at room temperature for 72 hours. The solvent was removed in vacuo and acetonitrile (100 cm3) was added. A white solid corresponding to the desired compound slowly crystallised from the hot acetonitrile solution. The solid was collected, washed with acetonitrile (2 × 10 cm3) and dried in vacuo (348 mg, 11%). δH (300 MHz; DMSO-d6; Me4Si) 7.26–7.33 (10H, m, Arom.), 8.63 (2H, s, Arom.), 9.13 (4H, s, Arom.), 11.29 (2H, s, CONH), 13.34 (1H, s, NH). δC (75 MHz; DMSO-d6; Me4Si) 112.5, 118.8, 126.7, 127.5, 130.1, 130.7, 133.4, 141.4, 148.2, 159.2. m/z (ES−) 636 (M−), 672 (M + Cl−), 749 (M− + TFA). HRMS (ES−): 636 (M−), Δ = 2.1 ppm. Found C, 56.20; H, 2.62; N, 15.35%. M requires C, 56.52; H, 3.00; N, 15.38%. Mp >320 °C.Acknowledgements
We would like to thank the EPSRC for a project studentship (to S. C.) and for use of the X-ray facilities at the University of Southampton. P. A. G. would like to thank the Royal Society for a University Research Fellowship.We would like to thank Christopher J. Woods for the excellent cover illustration.
References
- P. A. Gale, Coord. Chem. Rev., 2003, in press Search PubMed; P. A. Gale, Coord. Chem. Rev., 2001, 213, 79 Search PubMed; P. A. Gale, Coord. Chem. Rev., 2000, 199, 181 CrossRef CAS; P. D. Beer and P. A. Gale, Angew. Chem., Int. Ed., 2001, 40, 486 CrossRef CAS; J. L. Sessler and W. E. Allen, CHEMTECH, 1999, 29, 16 CrossRef; F. P. Schmidtchen and M. Berger, Chem. Rev., 1997, 97, 1609 Search PubMed; P. D. Beer and D. K. Smith, Prog. Inorg. Chem., 1997, 46, 1 CrossRef CAS; J. L. Atwood, K. T. Holman and J. W. Steed, Chem. Commun., 1996, 1401 Search PubMed; K. Kavallieratos, S. R. de Gala, D. J. Austin and R. H. Crabtree, J. Am. Chem. Soc., 1997, 119, 2325 RSC; A. P. Davis, J. F. Gilmer and J. J. Perry, Angew. Chem., Int. Ed., 1996, 35, 1312 CrossRef CAS ; Supramolecular Chemistry of Anions, eds. A. Bianchi, K. Bowman-James and E. García-España, Wiley, New York, 1997 CrossRef CAS.
- P. A. Gale, S. Camiolo, C. P. Chapman, M. E. Light and M. B. Hursthouse, Tetrahedron Lett., 2001, 42, 5095 CrossRef CAS; P. A. Gale, S. Camiolo, G. J. Tizzard, C. P. Chapman, M. E. Light, S. J. Coles and M. B. Hursthouse, J. Org. Chem., 2001, 66, 7849 CrossRef CAS; S. Camiolo, P. A. Gale, M. B. Hursthouse and M. E. Light, Tetrahedron Lett., 2002, 43, 6995 CrossRef CAS.
- S. Camiolo, P.A. Gale, M. B. Hursthouse, M. E. Light and A. J. Shi, Chem. Commun., 2002, 758 RSC.
- P. A. Gale, K. Navakhun, S. Camiolo, M. E. Light and M. B. Hursthouse, J. Am. Chem. Soc., 2002, 124, 11228 CrossRef CAS.
- S. Franke, Manual of Military Chemistry, Vol. 1 Chemistry of Chemical Warfare Agents, Deutscher Militirverlag, East Berlin, 1967, Search PubMedtranslated from German by U. S. Department of Commerce, National Bureau of Statistics, Institute for Applied Technology, NTIS no. ASD-849866, p. 247, 252.
- Crystal data for 2 colourless plate, C30H21N5O6, Mr = 547.52, T = 120(2) K, orthorhombic, space group Pbcn, a = 32.285(2), b = 11.8982(6), c = 19.8475(11) Å, V = 7624.2(8) Å3, ρcalc = 1.431 g cm −3, µ = 0.102 mm−1, Z = 12, reflections collected: 40433, independent reflections: 5946 (Rint = 0.4075), final R indices [I > 2σI]: R1 = 0.0941, wR2 = 0.1043, R indices (all data): R1 = 0.3726. wR2 = 0.1634.
- Crystal data for 2–3(DMSO) yellow plate, C36H36N5O9S3, Mr
= 778.88, T
= 120(2) K, triclinic, space group P
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a
= 11.0659(2), b
= 12.0400(2), c
= 15.9608(4)
Å, α
= 84.462(1), β
= 81.333(1), γ
= 64.306(1)°, V
= 1893.21(7)
Å3, ρcalc
= 1.366 g cm −3, µ
= 0.256 mm−1, Z
= 2, reflections collected: 19454, independent reflections: 6390 (Rint
= 0.0621), final R indices [I > 2σI]: R1 = 0.0452, wR2 = 0.1118, R indices (all data): R1 = 0.0675. wR2 = 0.1222.
, a
= 11.0659(2), b
= 12.0400(2), c
= 15.9608(4)
Å, α
= 84.462(1), β
= 81.333(1), γ
= 64.306(1)°, V
= 1893.21(7)
Å3, ρcalc
= 1.366 g cm −3, µ
= 0.256 mm−1, Z
= 2, reflections collected: 19454, independent reflections: 6390 (Rint
= 0.0621), final R indices [I > 2σI]: R1 = 0.0452, wR2 = 0.1118, R indices (all data): R1 = 0.0675. wR2 = 0.1222. - Crystal data for 3–2DMSO colourless plate, C34H31N7O12S2, Mr
= 793.78, T
= 120(2) K, triclinic, space group P, a
= 8.608(5), b
= 15.212(5), c
= 15.257(5)
Å, α
= 67.786(5), β
= 84.475(5), γ
= 73.886(5)°, V
= 1776.8(13)
Å3, ρcalc
= 1.484 g cm −3, µ
= 0.225 mm−1, Z
= 2, reflections collected: 19499, independent reflections: 6028 (Rint
= 0.1126), final R indices [I > 2σI]: R1 = 0.0559, wR2 = 0.0998, R indices (all data): R1 = 0.1424. wR2 = 0.1242.
- Crystal data for 3 colourless prism, C30H19N7O10, Mr = 637.52, T = 120(2) K, monoclinic, space group P21/c, a = 20.3034(11), b = 10.1636(5), c = 13.4570(6) Å, β = 104.057(2)°, V = 2693.8(2) Å3, ρcalc = 1.572 g cm −3, µ = 0.122 mm−1, Z = 4, reflections collected: 17978, independent reflections: 4716 (Rint = 0.0965), final R indices [I > 2σI]: R1 = 0.0550, wR2 = 0.1074, R indices (all data): R1 = 0.1256. wR2 = 0.1308. Close contacts exist between both O7⋯N7(i) (2.763(7) Å) and O6⋯O10(i) (2.760(7) Å). Presumably the former interaction contains an electrostatic component, and this combined with a potential CH⋯O interaction (C14⋯O10(i) = 3.34(5) Å) bring O6 and O10I into close proximity. (i) = x, 0.5 − y, 0.5 + z.
- M. J. Hynes, J. Chem. Soc., Dalton Trans., 1993, 311 RSC.
- M. E. Light, S. Camiolo, P. A. Gale and M. B. Hursthouse, Acta Crystallogr. Sect. E: Struct. Rep. Online, 2001, 57, o727 Search PubMed.
- Crystal data for bistetrabutylammonium (3–H+) Cl− colourless block, C62H90N9O10Cl, Mr
= 1156.88, T
= 120(2) K, triclinic, space group P, a
= 13.408(5), b
= 15.847(5), c
= 17.023(5)
Å, α
= 73.042(5), β
= 74.733(5), γ
= 68.046(5)°, V
= 3159.9(18)
Å3, ρcalc
= 1.216 g cm −3, µ
= 0.123 mm−1, Z
= 2, reflections collected: 18298, independent reflections: 7903 (Rint
= 0.2876), final R indices [I > 2σI]: R1 = 0.1330, wR2 = 0.1941, R indices (all data): R1 = 0.4834. wR2 = 0.2967.
- Crystal data for tetrabutylammonium salt of 3- H+, red block, C46H54N8O10, Mr = 878.97, T = 120(2) K, monoclinic, space group C2/c, a = 33.3632(3), b = 15.2635(3), c = 23.1686(3) Å, β = 130.061(1), V = 9029.98(19) Å3, ρcalc = 1.293 g cm −3, µ = 0.093 mm−1, Z = 8, reflections collected: 42281, independent reflections: 7954 (Rint = 0.0497), final R indices [I > 2σI]: R1 = 0.0382, wR2 = 0.0903, R indices (all data): R1 = 0.0498. wR2 = 0.097.
- P. Anzenbacher Jr., A. C. Try, H. Miyaji, K. Jursíková, V. M. Lynch, M. Marquez and J. L. Sessler, J. Am. Chem. Soc., 2000, 122, 10268 CrossRef; J. L. Sessler, P. Anzenbacher Jr., J. A. Shriver, K. Jursíková, V. M. Lynch and M. Marquez, J. Am. Chem. Soc., 2000, 122, 12061 CrossRef CAS; P. A. Gale, J. L. Sessler, W. E. Allen, N. A. Tvermoes and V. Lynch, Chem. Commun., 1997, 665 RSC.
- S. C. McCleskey, A. Metzger, C. S. Simmons and E. V. Anslyn, Tetrahedron, 2002, 58, 621 CrossRef CAS; P. Piatek and J. Jurczak, Chem. Commun., 2002, 2450 RSC; P. Anzenbacher Jr., T. P. Bombuwala, K. Jursíková and K. Bombuwala, Abstr. Pap. Am. Chem. Soc., 2001, 222 Search PubMed , Part 2, ORGN-544; H. Miyaji and J. L. Sessler, Angew. Chem., Int. Ed., 2001, 40, 154 Search PubMed; C. Lee, D. H. Lee and J. I. Hong, Tetrahedron Lett., 2001, 42, 8665 CrossRef CAS; S. Yamaguchi, S. Akiyama and K. Tamao, J. Am. Chem. Soc., 2001, 123, 11372 CrossRef CAS; D. H. Lee, H. Y. Lee, K. H. Lee and J. I. Hong, Chem. Commun., 2001, 1188 CrossRef CAS; S. L. Wiskur and E. V. Anslyn, J. Am. Chem. Soc., 2001, 123, 10109 RSC; H. Ait-Haddou, S. L. Wiskur, V. M. Lynch and E. V. Anslyn, J. Am. Chem. Soc., 2001, 123, 11296 CrossRef CAS; F. Sancenón, A. B. Descalzo, R. Martínez-Máñez, M. A. Miranda and J. Soto, Angew. Chem., Int. Ed., 2001, 40, 2640 CrossRef CAS; H. Miyaji, W. Sato, J. L. Sessler and V. M. Lynch, Tetrahedron Lett., 2000, 41, 1369 CrossRef CAS; H. Miyaji, W. Sato and J. L. Sessler, Angew. Chem., Int. Ed., 2000, 39, 1777 CrossRef CAS; C. B. Black, B. Andrioletti, A. C. Try, C. Ruiperez and J. L. Sessler, J. Am. Chem. Soc., 1999, 121, 10483 CrossRef CAS; P.A. Gale, L. J. Twyman, C. I. Handlin and J. L. Sessler, Chem. Commun., 1999, 1851; A. Metzger and E. V. Anslyn, J. Am. Chem. Soc., 1998, 120, 8533 RSC.
- M. Friedman, J. Org. Chem., 1965, 30, 859 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H NMR, 13C NMR and mass spectra for compounds 2 and 3, NMR anion titration profiles in DMSO-d6–0.5% water. See http://www.rsc.org/suppdata/ob/b2/b210848h/ |
| ‡ CCDC reference number(s) 195412-195416. See http://www.rsc.org/suppdata/ob/b2/b210848h/ for crystallographic files in .cif or other electronic format. |
| This journal is © The Royal Society of Chemistry 2003 |