Metabolic engineering—a genetic toolbox for small molecule organic synthesis†
Michael D.
Burkart
Department of Chemistry and Biochemistry, University of California, San Diego, 9500 Gilman Dr., La Jolla, CA 92093-0358, USA
First published on 6th December 2002
Abstract
Metabolic engineering is an emerging field that exploits biosynthetic machinery as a means to genetically design small molecule production within heterologous host organisms. Molecular design and synthesis with biological tools has lagged behind total synthesis technology for about seventy-five years and owes its existence to relatively new molecular biology techniques. Here the field of metabolic engineering is explained as a comparison to total organic synthesis, including the sequence of scientific events leading up to successful implementation and future goals in the field. It is expected that metabolic engineering will take a place alongside traditional organic synthesis as a powerful means to design and create small organic molecules.
Introduction
It was in late February of 1993 in the jungle of Costa Rica when I had an epiphany. I was performing natural product isolation for a Central American biodiversity institute while on leave from undergraduate school, and at that moment I had just gotten my boot stuck in a foot of sticky mud. Why, I reasoned, couldn't we isolate the genes responsible for the biosynthesis of natural products at the same time as we are isolating the metabolites and subsequently using the DNA-coded enzymes as catalysts to make the molecules in vivo? This seemed to be a valid alternative to total synthesis, and I vowed to spend my future education studying enzyme catalysis and natural product biosynthesis as a means to construct small molecules. What I didn't know was that the defining experiments of this very discipline, metabolic engineering, were being conducted at that moment in a couple of labs in the US and UK. This work would redefine the relationship between natural product biosynthesis and synthetic organic chemistry and reposition their roles in molecular synthesis. After a moment of inspiration, I redirected my attention to freeing my foot from its muddy confinement.Synthetic organic chemistry has a rich history of borrowing from Nature.1 Indeed, the triumphs of total synthesis lie within the construction of natural molecules by chemists whose schemes often compete with those of the producing organism. The great historical power behind organic synthesis is the ability to produce molecules on scales exceeding those available from natural sources. Moreover, the knowledge gained from total synthetic programs has provided a wealth of tools for the creation and modification of non-natural organic molecules.
In contrast, fermentation systems, while complicated to develop, often provide the most efficient means to obtain large quantities of complex molecules. At least one-quarter of pharmaceuticals prescribed today are produced from natural sources, and greater than ninety-five percent of antibiotics are made by microorganism fermentation. These molecules come from producer organisms that naturally compete in the environment with their neighbors using these antibiotics as chemical warfare agents. The molecules are often larger and more complex than most synthetic drugs, and as a result, the only cost-effective means of production is microbial fermentation. Natural product chemists have long dreamed about genetically designing and programming small molecule biosynthesis in laboratory organisms, which could be used as factories for the synthesis of novel molecules. Yet only traditional synthetic organic chemists have been able to synthesize small molecules by design, thanks to the huge technological advances made by organic chemists in the first half of the last century.
An abridged history of metabolic engineering
Up until the end of the 20th century, natural product biosynthesis was limited mostly to the phenotypic study of culturable organisms and screening of mutant strains. The elucidation of reaction pathways from a chemical perspective was mostly relegated to intellectual pursuit. It is largely microbiological techniques such as strain isolation and improvement that were responsible for the introduction of antibiotics and their influence on modern health. Not until the invention of polymerase chain reaction (PCR) in 1985 were the genes involved in natural product biosynthesis able to be sequenced, cloned, and manipulated.2 A revolution in the biological and medicinal sciences has occurred since the advent of PCR, and the study of natural product biosynthesis has blossomed accordingly. The list of fully sequenced organisms grows rapidly each year, and individual natural product biosynthetic pathways are sequenced and elucidated at an astonishing pace. (Current rate = 25 molecules per year). Within these DNA sequences lies an inherent ability to manipulate small molecules in an exquisitely selective and specific manner with enzymes coded by the genes. The promise held within these sequences is the ability to produce selectively complex small molecules in vivo.A telling example is that of erythromycin, a macrolide antibiotic discovered in 1952 from the bacterium Saccharopolyspora erythraea. The structure of erythromycin was elucidated in 1957 followed by an X-ray structure in 1965. From a chemical synthesis standpoint, the first total synthesis of erythromycin B was published in 1981 by Woodward, et al.3 Macrolides had been of great synthetic interest for decades, and simpler macrolides, such as methymycin, had been previously synthesized.4 From a biosynthesis standpoint, most of what was understood about erythromycin biosynthesis up until 1991 came from radioisotope labeling studies and mutant strains of S. erythraea. These studies demonstrated that its biosynthesis arose from three carbon units through a pathway that bore a resemblance to fatty acid biosynthesis by its dependence upon coenzyme A. From mutation studies a resistance gene was identified, and the study of mutant strains had been found to not produce metabolites or intermediates, indicative of processive biosynthetic machinery.5 In 1990, Leadlay, et al. published the sequence of 6-deoxyerythronolide B synthase, three enormous genes found adjoining the resistance gene ermE, named EraAI, II, and III, which coded for three megasynthases called DEBS 1, DEBS 2, and DEBS 3.6 These proteins assemble the erythromycin aglycone from one propionyl-CoA and six molecules of methylmalonyl-CoA. At this point, the floodgates were opened, and a stream of publications involving the DEBS system followed. In 1994, Khosla, et al. demonstrated the genetic engineering and heterologous production of the DEBS macrolactone in Streptomyces coelicolor, a familiar and manipulable strain that is amenable to large-scale fermentation.7
Metabolic engineering (ME), the directed manipulation of metabolite formation within an organism, is a relatively new field (i.e., the journal Metabolic Engineering began in 1999). Already there are sub-categories of ME that emphasize the microbiological, mathematical, and chemical importance of the discipline. Nevertheless, there exists a basic ordering of disciplines leading up to the successful implementation of an ME cycle.
The metabolic engineering cycle—a hierarchy of disciplines
First natural molecules with biological activity are isolated and identified. This research has been ongoing for decades and is responsible for the discovery of all bioactive natural products today. Current research mainly focuses on marine organisms and involves organism collection, natural product isolation, bioassay screening, and structure elucidation. This field is a rapidly developing and vibrant course of research indispensable to the future discovery of novel molecular structure.Next is the elucidation of biosynthetic pathways from the producer organisms. This stage entails the isolation and sequencing of the biosynthetic genes involved in the natural biosynthesis of one molecule. Usually, in order to publish sequencing information, researchers must in addition definitively demonstrate activity of one enzyme in the biosynthetic cluster, either through knockout experiments which alter molecular structure or through in vitro proof of activity. Complicating this research is the abundance of non-culturable microorganisms producing highly interesting bioactive molecules. For instance, many natural products isolated from marine organisms such as sea cucumbers are believed to be produced by unculturable symbiotic bacteria living within the macroorganism.8 On a different but similar issue, many organismic strains producing a molecule of interest are often proprietary or carefully guarded by their discoverers.
Once a biosynthetic pathway has been fully sequenced, the activity, order, and timing of each enzyme in its pathway must be determined. Each gene product usually corresponds to an enzymatic step in the biosynthesis, and these must be determined and demonstrated. Here, one can often draw analogies from previously studied enzymes through protein sequence similarity, or homology, and parallels with other known secondary metabolism pathways. In many cases, each enzyme is produced individually and activity studies are performed in vitro to validate a proposed secondary metabolism pathway. Alternatively, the pathway may be studied genetically by generating mutants of the producer organism in which an individual gene has been inactivated, thereby producing pathway intermediates that may be correlated to the missing enzyme. Often combinations of these techniques lead to a complete understanding of enzyme activity. Gene sequence within a given pathway does not necessarily correspond to sequential enzyme activity, and the order of events must also be correlated to enzyme function in order to fully understand metabolite construction.
At this point the metabolic engineer enters the scene, assembling enzymes from known biosynthetic pathways into heterologous hosts. Here there are very few rules laid down, and most natural product pathways are being pursued. Non-ribosomal peptide (NRP), polyketide (PK), carbohydrate, terpene, sterol, shikimic acid, and fatty acid pathways are all of interest to current researchers. Most heterologous host organisms to date have been chosen from a set of easily manipulable bacteria, often E. coli. Once a new pathway has been created, mathematical models of metabolite flux are studied to determine optimum fermentative output and minimum growth requirements. New genetic tools, including gene promoters, repressors, and signaling pathways, are continually being developed and optimized for applications to ME.
Approaches to metabolic engineering
There are two basic schools of thought for the practice of ME. The first involves engineering of an entire biosynthetic cluster into an organism more suitable to laboratory manipulation. This is only necessary if the natural producer is difficult to culture and ferment or if it expresses the natural product at low levels. From a microbiological standpoint, engineering the biosynthetic enzymes in an organism with known culture conditions and promoter elements is much more desirable than the enormous effort necessary to study and manipulate each producer organism individually. Take the example of epothilone B, produced by the myxobacterium Sorangium cellulosum, which grows very slowly, doubling only once every 16 hours, and produces about 20 mg per liter of the metabolite. Because the cost of fermenting the natural producer was prohibitive even to produce material for clinical trials, Julien et al. successfully engineered the entire epothilone cluster into both S. coelicolor and Myxococcus xanthus, both of which are better-studied organisms with faster growth and known genetic switches for increased protein expression.9The second and more promising effort lies in the design and biosynthesis of novel metabolites through selective construction of new pathways drawing from different biosynthetic systems. For example, many bioactive natural products are of hybrid biomolecules, including NRP–PK (hybrid megasynthase products, e.g. epothilone), NRP–carbohydrate (glycopeptides, e.g. vancomycin), and PK–carbohydrate (glycoketides, e.g. daunomycin), to name just a few. An excellent example of ME with hybrid subunits is the recent engineering of the deoxysugar pathway of oleandromycin by Salas et al.10 The researchers produced novel elloramycin glycoketides through manipulation of modular deoxysugar biosynthetic pathways and a “sugar flexible” glycosyltransferase. This methodology could uncover novel molecules that are hybrids of metabolites with known metabolic pathways and, perhaps, display new bioactivities.
Industrial utility
ME has two major avenues of importance to chemical and pharmaceutical industries. Inherently, ME holds the promise of an inexpensive vector for bulk production of fine chemicals. Once an organism has been engineered with a small molecule-producing pathway, the cost of growth and isolation of the molecule is the only subsequent investment. Costs may be further reduced by proper choice of host organism growth conditions and addition of available carbon sources into feedstocks. Another major advantage lies in the avoidance of toxic waste streams often produced by non-renewable transformations. It is these advantages that have drawn attention in research and production. Both of these sectors have shown great interest in shifting to renewable resources and incorporating enzyme or fermentative science into their production streams. With the inevitable increase in petroleum prices and further tightening of environmental regulations, ME will only grow in importance as a logical and inexpensive addition to large-scale chemical synthesis.The second important influence of ME arises in the arena of discovery. As evidenced by the biosynthetic machinery of NRP, PK, and carbohydrate systems, a clear theme of modularity appears to be one of nature's tricks for the assemblage of diverse molecular species. Taking advantage of this modularity should enable diversity of molecular structure in much the same way that combinatorial chemistry imparts molecular diversity. Combinatorial biosynthesis, genetic reprogramming of secondary metabolites through high-throughput shuffling of biosynthetic enzymes, is a major goal in natural product genetic engineering.11 All natural product categories could theoretically be developed into a combinatorial biosynthetic system of drug discovery. Of particular interest in this arena are NRP and PK systems. Often referred to as assembly lines, PK and NRP synthases are incredibly large multifunctional proteins that are naturally organized into directional modules that load and transfer metabolite subunits in a linear fashion, with one molecular subunit added per module. The theoretical simplicity of their modular architecture coupled with near-infinite possible variations in molecular structure of each product has inspired researchers to investigate module connectivity and synthase procession. Incremental achievements have been made toward modular shuffling, particularly in PK systems, but a general impediment lies in the fact that time has evolved individual modules within megasynthase systems to communicate with their nearest neighbors, thereby imparting specificity. Random shuffling of modules may only lead to an inoperable assortment of mismatched units (i.e. neighbors that don't speak a common language). Unfortunately, basic principles such as quaternary molecular structure and intermodule kinetics remain largely unknown due to the sheer size and complexity of these systems. A more complete understanding of the basic enzymology of their modularity is needed before combinatorial biosynthesis will become a functional tool.
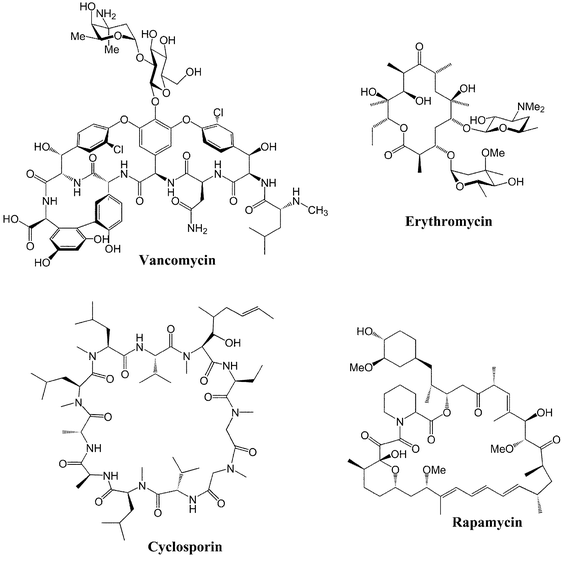 | ||
| Fig. 1 Elegant structures. Current and future PK and NRP molecules of interest to metabolic engineering. | ||
Right now systems are being created to overcome fundamental barriers to combinatorial biosynthesis. The creation of highly iterative combinatorial genetics, in which billions of permutations in modular organization may be created and selected for based upon successful metabolite formation, will be needed to select for the random coupling of enzymatic domains that fortuitously match (i.e. neighbors that speak related languages and can thus communicate). An undertaking such as this exceeds the current abilities of molecular biology in scale and screening technology, and new tools toward this end will certainly prove useful. Directed evolution will also be used to optimize and select modules or sub-modular domains that are more promiscuous with regard to their neighboring groups. In this way, assemblies of flexible modular enzymes could be engineered directionally or combinatorially for the production of new metabolites. Finally, molecular diversity may also be generated by chemo-enzymatic applications of biosynthetic enzymes in vitro. By utilizing enzymes with broad substrate specificity, chemists may capitalize upon both the strengths of synthetic chemistry (e.g., unusual precursors and facile library construction) and those of enzymatic catalysis (e.g., regio-/stereo-specificity and aqueous media).12
Conclusion
The use of biological tools in synthetic organic chemistry has only recently attained wide acceptance in the chemical community. Enzymes as in vitro chemical tools have proven quite useful in process chemical applications.13 The introduction of ME comes as a natural outgrowth of the use of enzymes in chemistry, bringing reactivity in vivo to capitalize on the metabolic power of living organisms. This move, however, brings with it the complexity of biological systems, and as a result requires knowledge of metabolism, molecular biology, and microbial cell culture, and new tools bridging these disciplines must be acquired to make the potential of ME a reality.It has been almost a decade since I successfully released my foot from the mud and got back to work, and during this time ME has blossomed into an exciting new area of molecular design and synthesis. We are presently at a pivotal moment in time where scientific discovery of natural product biosynthesis and organic synthesis have the ability to overlap. I am thrilled to be taking an active part in the accomplishments of the next ten years, bringing these tools to bear in synthetic applications from materials to pharmaceutical chemistry.
References
- K. C. Nicolaou and E. J. Sorensen, Classics in Total Synthesis: Targets, Stategies, Methods, John Wiley & Son Ltd., New York, 1996 Search PubMed.
- D. A. Hopwood and D. H. Sherman, Annu. Rev. Genet., 1990, 24, 37–66 CrossRef CAS.
- R. B. Woodward, et al., J. Am. Chem. Soc., 1981, 103, 3210–3216 CrossRef CAS.
- S. Masamune, C. U. Kim, K. E. Wilson, G. O. Spessard, P. E. Georghiou and G. S. Bates, J. Am. Chem. Soc., 1975, 97, 3512–3513 CrossRef CAS.
- J. Staunton and B. Wilkinson, Chem. Rev., 1997, 97, 2611–2629 CrossRef CAS.
- J. Cortés, S. F. Haydock, G. A. Roberts, D. J. Bevitt and P. F. Leadlay, Nature, 1990, 348, 176–178 CrossRef CAS.
- C. M. Kao, L. Katz and C. Khosla, Science, 1994, 265, 509–512 CAS.
- P. Young, ASM News, 1997, 63, 417–421 Search PubMed.
- L. Tang, S. Shah, L. Chung, J. Carney, L. Katz, C. Khosla and B. Julien, Science, 2000, 287, 640–642 CrossRef CAS; B. Julien and S. Shah, Antimicrob. Agents Chemother., 2002, 46, 2772–2778 CrossRef CAS.
- L. Rodríguez, I. Aguirrezabalaga, N. Allende, A. F. Braña, C. Méndez and J. A. Salas, Chem. Biol., 2002, 9, 721–729 CrossRef CAS.
- E. Rodriguez and R. McDaniel, Curr. Opin. Microbiol., 2001, 4, 526–534 CrossRef CAS.
- R. M. Kohli, C. T. Walsh and M. D. Burkart, Nature, 2002, 418, 658–661 CrossRef CAS.
- G. M. Whitesides and C.-H. Wong, Enzymes in Synthetic Organic Chemistry, Pergamon Press, Oxford, 1994 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Figure: The metabolic engineering and synthetic organic cycles. See http://www.rsc.org/suppdata/ob/b2/b210173d/ |
| This journal is © The Royal Society of Chemistry 2003 |