A novel asymmetric synthesis of highly enantiomerically enriched norbornane-type diamine derivatives
Carsten
Bolm
*,
Ingo
Schiffers
,
Iuliana
Atodiresei
,
Salih
Ozcubukcu
and
Gerhard
Raabe
Institut für Organische Chemie der RWTH Aachen, Prof.-Pirlet-Str. 1, 52056, Aachen, Germany. E-mail: Carsten.Bolm@oc.rwth-aachen.de; Fax: +49 241 809 2391
First published on 24th October 2002
Abstract
The simple and highly enantioselective methanolysis of norbornene dicarboxylic acid anhydride mediated by quinidine leads to the corresponding cis-monomethyl ester with 98% ee. By means of selective ester epimerization, followed by Curtius degradation of the intermediate trans-diacyl azide, two optically active norbornane-type diamines are obtained as their hydrochloric salts. Liberating the amine with an excess of triethylamine in situ and subsequent derivatization affords potential C1-symmetric ligands for asymmetric catalysis in excellent yields.
In recent years, the interest in the development of stereocontrolled syntheses of chiral 1,2-diamines has increased considerably.1 Vicinal diamines have become important synthetic targets, in particular due to their use as intermediates in the preparation of important biologically active compounds such as biotin,2 balanol,3 a broad range of κ-opioid receptor agonists,4 which are pharmacologically used as analgesic agents without the clinically side effects that characterize morphine, and 1,2-diamino platinum complexes, which serve as cytostatic remedies in chemotherapy with increased antitumoral activity and less toxicity as compared to cisplatin.5 Furthermore, vicinal diamines have found applications in the resolution of racemates,6 as chiral auxiliaries in enantio- and diastereoselective syntheses,1 and in the preparation of a large number of ligands for asymmetric catalysis, including epoxidation,7 alkene aziridination,8 epoxide ring-opening,9 Diels–Alder reactions,10 and transfer hydrogenation.11 In the light of their synthetic significance it is surprising that most of these studies have been focussed on the use of only a very few diamines. The most widely applied ones are trans-1,2-diphenylethylene diamine (1) and trans-1,2-diaminocyclohexane (2). They are readily accessible, and their large-scale preparations have been described in full detail.12 Interestingly, the related trans-diamine 3, having a more rigid norbornane backbone, has remained almost unnoticed. Since in contrast to 1 and 2, diamine 3 is C1-symmetric, special effects could result when ligands stemming from 3 are used in asymmetric catalysis. Only a few years ago, enantiopure 3 was first isolated by resolution of its racemate with O,O′-dibenzoyltartaric acid.13 Unsatisfyingly, however, this approach afforded the tartaric salt of ent-3 in only 16% yield, based on the racemat. So, a more efficient preparation, which would allow a broader application of this interesting 1,2-diamine, appeared desirable.

Recently, we described a simple and highly enantioselective methanolysis of meso-anhydrides such as 4, mediated by cinchona alkaloids to give optically active monomethylesters of type 5. Either enantiomer of ester 5 can be prepared in excellent yield with up to 99% ee by using inexpensive and quantitatively recoverable quinine or quinidine as the directing additive.14 As an extension of our work in this field, we reported on the synthesis and polymerization of both cis- and trans-β-amino acid methylesters starting from the same monomethylester 5.15 On the basis of these findings, we wish to demonstrate herein that 5 is also a valuable intermediate for the preparation of highly enantiomerically enriched norbornane diamine derivatives.
As shown in Scheme 1, the overall reaction sequence involves the asymmetric desymmetrization of meso-anhydride 4, followed by selective epimerization of the cis-hemiester 5 to its trans-isomer,16 which is then converted to the corresponding saturated diacid by saponification and hydrogenation.17 For the synthesis of the key intermediate 6, the diacid is activated by treatment with ethyl chloroformate and triethylamine. Addition of an aqueous solution of sodium azide affords dicarboxylic diazide 6, which is subjected to thermal rearrangement without further purification. Curtius degradation of the crude diazide in benzene, followed by acidic hydrolysis of the resulting isocyanate, leads to the dihydrochloric salt of diamine 3 in 70% yield. Direct alcoholysis of the intermediate isocyanate by performing the rearrangement in ethanol delivers the corresponding dicarbamate 7, which is converted into the dihydrochloric salt of diamine 8 by reduction and acidic work-up in 71% yield. The absolute configuration of 3·2HCl was determined by comparison of its sense of optical rotation with that reported in the literature13 and found to be in accordance with the proposed structure in Scheme 1, which was based on the uniform stereochemical outcome of the asymmetric anhydride opening. Since the value of the optical rotation differed strongly from the published data, the enantiomer ratio of 99∶1 was confirmed by HPLC analysis of the corresponding ditosylate 9, using a racemic sample of this compound as reference. As a consequence of the stereospecificity of the epimerization and the Curtius degradation, the enantiomeric excess was identical with that of the starting material 5. In addition, our findings were verified by the X-ray crystal structure analysis of 8·2HCl, shown in Fig. 1.18
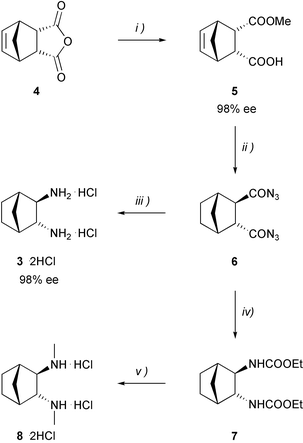 | ||
Scheme 1 Synthesis of the diamine dihydrochloric salts 3·2HCl and 8·2HCl. Reagents and conditions: (i) quinidine, methanol, −55![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) °C, 98%; (ii)
(a) LDA, THF, −78°C, 81%; (b) 1 N NaOH, methanol; (c) H2, Pd/C, THF, 88%
(2 steps); (d) ClCOOEt, Et3N, THF, −20°C; (e) aq. NaN3, −10°C to r.t.; (iii)
(a) benzene, 80°C; (b) 8 N HCl, 70%
(3 steps); (iv) ethanol, 79°C, 65%
(3 steps); (v) LiAlH4, THF, 71%. °C, 98%; (ii)
(a) LDA, THF, −78°C, 81%; (b) 1 N NaOH, methanol; (c) H2, Pd/C, THF, 88%
(2 steps); (d) ClCOOEt, Et3N, THF, −20°C; (e) aq. NaN3, −10°C to r.t.; (iii)
(a) benzene, 80°C; (b) 8 N HCl, 70%
(3 steps); (iv) ethanol, 79°C, 65%
(3 steps); (v) LiAlH4, THF, 71%. | ||
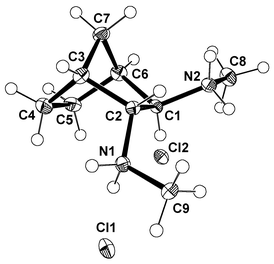 | ||
| Fig. 1 Molecular structure of 8·2HCl in the solid state as determined by X-ray crystal structure analysis. | ||
During the course of our investigations, Trost et al. reported on the application of the diphenylphosphinobenzoyl substituted diamine 3 as ligand in asymmetric allylic alkylations.20 Their synthesis involved a Diels–Alder reaction between (+)-dimenthyl fumarate and cyclopentadiene,21 followed by saponification of the resulting diester and subsequent degradation similar to that shown in Scheme 1. A direct comparison of the two methods in view of their preparative advantages on a large scale remains difficult at the present time. However, the need for an efficient synthetic access to enantiopure diamine 3 is further emphasized by this catalysis work.
In contrast to the free diamines,22 the corresponding dihydrochloric salts are stable in air and easy to handle. Since treatment of 3·2HCl with KOH and distillation under reduced pressure led to the free diamine in only moderate yield,13 the preferred method was to liberate 3in situ by addition of an excess of triethylamine before further derivatization. As shown in Scheme 2, this protocol afforded ditosylate 9 as well as salenes 10 and 11 in excellent yields. Selective mono-N-tosylation by adding dropwise one equivalent of tosylchloride to a methylene chloride solution of 3·2HCl and triethylamine failed, even at low temperature. However, enantiopure mono-N-substituted cis- and trans-norbornane diamines could finally be prepared by sequential introduction of the two amino moieties.23 Details of these syntheses will be described in due course.
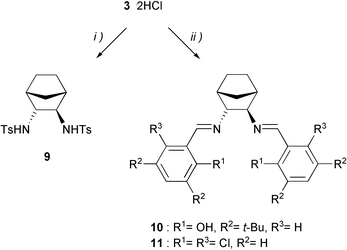 | ||
| Scheme 2 Synthesis of the ditosylate 9 and the salenes 10 and 11. Reagents and conditions: (i) TsCl, Et3N, CH2Cl2, 92%; (ii) methanol, Et3N, for salene 10: 3,5-di-tert-butyl-2-hydroxybenzaldehyde, for salene 11: 2,6-dichlorobenzaldehyde. | ||
In conclusion, we have described the transformation of a norbornane-type cis-dicarboxylic acid monoester, which is readily obtained from a quinidine mediated methanolysis of the anhydride, into optically active trans-norbornane diamine. All synthetic steps are easy to perform, giving highly enantiomerically enriched diamine derivatives in good yields. Currently, we are focusing our efforts on the application of these new compounds as ligands in asymmetric catalysis.
Experimental
Unless otherwise specified all reagents were purchased from commercial suppliers and used without further purification. All reactions were performed under argon using standard Schlenk and vacuum line techniques. THF was distilled from sodium/benzophenone ketyl radical, CH2Cl2 from CaH2, ethanol from sodium and stored over 4 Å molecular sieves. All other solvents were reagent grade and used as received. 1H NMR and 13NMR spectra were obtained on an Inova 400 spectrometer and were recorded relative to TMS as internal standard. HPLC analysis was performed using a Chiracel OD-H column, 4.6×250 mm, λ=254 nm. Mass spectra were measured on a Finnigan SSQ 7000 instrument or on a Hewlett Packard GCMS apparatus-system (column HP-5 MS, 30 m×0.25 mm×0.25 µm; Mass Selective Detector 5973). Melting points were measured in open glass capillaries with a Büchi apparatus and are uncorrected. Optical rotation values were determined on a Perkin–Elmer P241 instrument at room temperature using solvents of Merck UVASOL-quality. Infrared spectra were recorded on a Perkin–Elmer 1760 FT apparatus. All microanalyses were obtained with a Heraeus CHN-O-RAPID element analyzer at the Institut für Organische Chemie der RWTH Aachen. Preparative column chromatography: Merck silica gel 60, particle size 0.040–0.063 mm (230–400 mesh, flash). Analytical TLC: silica gel 60 F254 plates, Merck, Darmstadt.
Syntheses
°C and the reaction mixture was stirred at this temperature for 1 h. A solution of NaN3
(3.9 g, 60.0 mmol, 6.0 equiv) in H2O (24 ml) was added at −10°C. The temperature was gradually increased to r.t. and stirring continued for 2 h. The mixture was diluted with water, extracted with EtOAc and the organic phase was washed with NaHCO3, dried over MgSO4, filtered and concentrated to provide the coresponding acyl azide 6, which was dissolved in benzene (16 ml) and heated under reflux for 2 h. To the cooled isocyanate solution conc. aq. HCl (30 ml) was added and the resulting mixture was refluxed for 4 h. The reaction mixture was extracted with Et2O and the aqueous layer was concentrated to give a solid that was washed with THF and dried under high vacuum to give 1.4 g (7.05 mmol, 70%) of the title compound as a colorless solid. Mp: >250°C (rac, en), lit.22 mp: >350°C (rac). [α]D=−9.3 (c=1.52, MeOH), [α]D=−16.4 (c=1.18, H2O), lit.13 for ent-3·2HCl: [α]D=+26.0 (c=2.0, MeOH). 1H NMR (DMSO-d6): δ=1.22 (d, J=10.2 Hz, 1H), 1.30–1.39 (m, 2H), 1.49–1.59 (m, 2H), 1.98 (d, J=10.2 Hz, 1H), 2.3 (s, 1H), 2.95 (s, 1H), 3.30 (s, 1H), 3.46 (s, 1H), 8.54 (s, 3H), 8.59 (s, 3H). 13C NMR: δ=21.0, 27.5, 35.3, 39.8, 40.9, 57.2, 58.0. MS (EI, 70 eV): m/z=126 (44), 108 (16), 97 (20), 92 (18), 85 (25), 70 (43), 56 (100). IR (KBr): ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) =2970, 2614, 1604, 1565, 1496, 1120, 1061 cm−1. Anal. calcd. for C7H16Cl2N2
(199.12): C, 42.22%; H, 8.10%; N, 14.07%; found: C, 42.22%; H, 8.19%; N, 14.00%.
°C (en). [α]D=0 (c=1.07, CHCl3), [α]D=0 (c=1.28, MeOH). 1H NMR (CDCl3): δ=1.22 (t, J=7.1 Hz, 6H), 1.29–1.42 (m, 3H), 1.45–1.61 (m, 3H), 2.19 (d, J=3.3 Hz, 1H), 2.47 (s, 1H), 3.18 (s, 1H), 3.65 (s, 1H), 4.11 (q, J=7.1 Hz, 4H), 5.35 (s, 1H), 5.43 (s, 1H). 13C NMR: δ=14.6, 20.5, 27.4, 35.0, 40.2, 43.1, 60.8, 61.2, 61.8, 156.3, 156.7. MS (EI, 70 eV): m/z=225 (3), 181 (37), 153 (100), 128 (5), 108 (6), 81 (7). IR (KBr): =3293, 2976, 1681, 1552, 1325, 1308, 1279, 1235, 1110 cm−1. Anal. calcd. for C13H22N2O4
(270.32): C, 57.76%; H, 8.20%; N, 10.36%; found: C, 57.42%; H, 8.27%; N, 10.58%.
°C. After being stirred for 16 h at r.t. the reaction mixture was refluxed for 3 h, hydrolyzed with 1 N aq. NaOH, filtered; the filtrate was washed with Et2O. The organic phase was extracted with 1 N aq. HCl and the resulting aqueous layer was concentrated and dried in vacuo to give a solid that was recrystallized from methanol to provide 895.0 mg (3.94 mmol, 71%) of the title compound as colorless crystals. Mp: >250°C (en). [α]D=−23.2 (c=1.62, MeOH). 1H NMR (CD3OD): δ=1.46–1.72 (m, 5H), 1.8–1.93 (m, 2H), 2.65 (d, J=4.7 Hz, 1H), 2.79 (s, 3H), 2.81 (s, 3H), 3.18 (dd, J=2.2, 3.6 Hz, 1H), 3.67 (t, J=3.6 Hz, 1H), 4.83 (br s, 4H). 13C NMR: δ=20.2, 26.1, 32.0, 32.1, 34.8, 38.3, 39.5, 64.6, 65.2. MS (EI, 70 eV): m/z=139 (5), 124 (100), 113 (6), 96 (10), 84 (21), 70 (76), 57 (9). IR (KBr): =2965, 2887, 2805, 2672, 2561, 2483, 2455, 2397, 2344, 1586, 1478, 1427, 1057 cm−1. Anal. calcd. for C9H20Cl2N2
(227.17): C, 47.58%; H, 8.87%; N, 12.33%; found: C, 47.25%; H, 9.09%; N, 12.16%.
°C (en). [α]D=+41.3 (c=1.57, CHCl3). ee=98%
[HPLC analysis: Chiracel OD-H, n-Heptane–2-Propanol=95∶5, 0.7 ml min−1, 254 nm, t1=45.5 (major), t2=56.9 (minor)]. 1H NMR (CDCl3): δ=1.00–1.09 (m,1H), 1.16–1.32 (m, 3H), 1.41–1.52 (m, 2H), 2.13 (d, J=3.8 Hz, 1H), 2.23 (s, 1H), 2.42 (s, 6H), 2.61 (s, 1H), 3.06 (d, J=3.3 Hz, 1H), 5.02 (d, J=4.1 Hz, 1H), 5.21 (d, J=4.4 Hz, 1H), 7.29 (d, J=8.0 Hz, 4H), 7.72 (t, J=8.5 Hz, 4H). 13C NMR: δ=20.4, 21.6, 27.1, 34.9, 40.3, 41.9, 64.1, 64.6, 127.2, 127.3, 129.8, 136.5, 136.7, 143.7. MS (EI, 70 eV): m/z=279 (100), 182 (4), 155 (18), 108 (24), 91 (57), 81 (15). IR (KBr): =3272, 2959, 1446, 1325, 1305, 1290, 1161, 1093 cm−1. Anal. calcd. for C21H26N2O4S2
(434.57): C, 58.04%; H, 6.03%; N, 6.45%; found: C, 57.79%; H, 5.95%; N, 6.27%.
°C (rac), 196.5°C (en). [α]D=−356.0 (c=1.51, CHCl3). 1H NMR (CDCl3): δ=1.28 (s, 18H), 1.46 (s, 9H), 1.47 (s, 9H), 1.50–1.57 (m, 3H), 1.73–1.78 (m, 1H), 1.81–2.05 (m, 1H), 2.14 (d, J=10.2 Hz, 1H), 2.32 (d, J=4.1 Hz, 1H), 2.44 (s, 1H), 3.17 (s, 1H), 3.57 (s, 1H), 7.06 (t, J=2.5 Hz, 2H), 7.38 (s, 2H), 8.26 (s, 1H), 8.34 (s, 1H), 13.63 (s, 1H), 13.69 (s, 1H). 13C NMR: δ=22.5, 27.7, 29.8, 31.9, 34.5, 35.4, 36.7, 43.7, 45.6, 80.2, 80.7, 118.0, 118.2, 126.3, 127.1, 127.2, 136.8, 140.4, 158.0, 158.1, 164.4, 166.2. MS (EI, 70 eV): m/z=558 (M+, 19), 325 (100), 296 (14), 264 (32), 244 (12), 219 (7), 84 (7). IR (KBr): =2955, 2873, 1624, 1595, 1473, 1440, 1362, 1248, 1175, 1057 cm−1. Anal. calcd. for C37H54N2O2
(558.84): C, 79.52%; H, 9.74%; N, 5.01%; found: C, 79.47%; H, 9.92%; N, 5.13%.
°C (en). [α]D=−128.0 (c=1.0, CHCl3). 1H NMR (CDCl3): δ=1.48–1.60 (m, 3H), 1.70–1.79 (m, 1H), 2.16–2.23 (m, 2H), 2.40 (d, J=4.1 Hz, 1H), 2.51 (br s, 1H), 3.41 (t, J=2.2 Hz, 1H), 3.75 (br s, 1H), 7.20 (dd, J=7.4, 8.5 Hz, 2H), 7.31 (d, J=3.1 Hz, 2H), 7.33 (d, J=3.0 Hz, 2H), 8.32 (s, 1H), 8.48 (s, 1H). 13C NMR: δ=22.3, 28.0, 36.8, 44.3, 45.2, 80.8, 81.6, 128.7, 128.8, 130.3, 130.4, 133.6, 133.8, 134.8, 134.8, 155.3, 156.9. MS (EI, 70 eV): m/z=267 (100), 239 (40), 174 (39), 156 (24), 123 (24), 66 (22). IR (KBr): =2954, 2870, 1642, 1579, 1557, 1429, 1375, 1189, 1094, 781 cm−1. Anal. calcd. for C21H18N2Cl4
(440.19): C, 57.30%; H, 4.12%; N, 6.36%; found: C, 57.26%; H, 3.91%; N, 6.35%.
=2970, 2614, 1604, 1565, 1496, 1120, 1061 cm−1. Anal. calcd. for C7H16Cl2N2
(199.12): C, 42.22%; H, 8.10%; N, 14.07%; found: C, 42.22%; H, 8.19%; N, 14.00%.
°C (en). [α]D=0 (c=1.07, CHCl3), [α]D=0 (c=1.28, MeOH). 1H NMR (CDCl3): δ=1.22 (t, J=7.1 Hz, 6H), 1.29–1.42 (m, 3H), 1.45–1.61 (m, 3H), 2.19 (d, J=3.3 Hz, 1H), 2.47 (s, 1H), 3.18 (s, 1H), 3.65 (s, 1H), 4.11 (q, J=7.1 Hz, 4H), 5.35 (s, 1H), 5.43 (s, 1H). 13C NMR: δ=14.6, 20.5, 27.4, 35.0, 40.2, 43.1, 60.8, 61.2, 61.8, 156.3, 156.7. MS (EI, 70 eV): m/z=225 (3), 181 (37), 153 (100), 128 (5), 108 (6), 81 (7). IR (KBr): =3293, 2976, 1681, 1552, 1325, 1308, 1279, 1235, 1110 cm−1. Anal. calcd. for C13H22N2O4
(270.32): C, 57.76%; H, 8.20%; N, 10.36%; found: C, 57.42%; H, 8.27%; N, 10.58%.
°C. After being stirred for 16 h at r.t. the reaction mixture was refluxed for 3 h, hydrolyzed with 1 N aq. NaOH, filtered; the filtrate was washed with Et2O. The organic phase was extracted with 1 N aq. HCl and the resulting aqueous layer was concentrated and dried in vacuo to give a solid that was recrystallized from methanol to provide 895.0 mg (3.94 mmol, 71%) of the title compound as colorless crystals. Mp: >250°C (en). [α]D=−23.2 (c=1.62, MeOH). 1H NMR (CD3OD): δ=1.46–1.72 (m, 5H), 1.8–1.93 (m, 2H), 2.65 (d, J=4.7 Hz, 1H), 2.79 (s, 3H), 2.81 (s, 3H), 3.18 (dd, J=2.2, 3.6 Hz, 1H), 3.67 (t, J=3.6 Hz, 1H), 4.83 (br s, 4H). 13C NMR: δ=20.2, 26.1, 32.0, 32.1, 34.8, 38.3, 39.5, 64.6, 65.2. MS (EI, 70 eV): m/z=139 (5), 124 (100), 113 (6), 96 (10), 84 (21), 70 (76), 57 (9). IR (KBr): =2965, 2887, 2805, 2672, 2561, 2483, 2455, 2397, 2344, 1586, 1478, 1427, 1057 cm−1. Anal. calcd. for C9H20Cl2N2
(227.17): C, 47.58%; H, 8.87%; N, 12.33%; found: C, 47.25%; H, 9.09%; N, 12.16%.
°C (en). [α]D=+41.3 (c=1.57, CHCl3). ee=98%
[HPLC analysis: Chiracel OD-H, n-Heptane–2-Propanol=95∶5, 0.7 ml min−1, 254 nm, t1=45.5 (major), t2=56.9 (minor)]. 1H NMR (CDCl3): δ=1.00–1.09 (m,1H), 1.16–1.32 (m, 3H), 1.41–1.52 (m, 2H), 2.13 (d, J=3.8 Hz, 1H), 2.23 (s, 1H), 2.42 (s, 6H), 2.61 (s, 1H), 3.06 (d, J=3.3 Hz, 1H), 5.02 (d, J=4.1 Hz, 1H), 5.21 (d, J=4.4 Hz, 1H), 7.29 (d, J=8.0 Hz, 4H), 7.72 (t, J=8.5 Hz, 4H). 13C NMR: δ=20.4, 21.6, 27.1, 34.9, 40.3, 41.9, 64.1, 64.6, 127.2, 127.3, 129.8, 136.5, 136.7, 143.7. MS (EI, 70 eV): m/z=279 (100), 182 (4), 155 (18), 108 (24), 91 (57), 81 (15). IR (KBr): =3272, 2959, 1446, 1325, 1305, 1290, 1161, 1093 cm−1. Anal. calcd. for C21H26N2O4S2
(434.57): C, 58.04%; H, 6.03%; N, 6.45%; found: C, 57.79%; H, 5.95%; N, 6.27%.
°C (rac), 196.5°C (en). [α]D=−356.0 (c=1.51, CHCl3). 1H NMR (CDCl3): δ=1.28 (s, 18H), 1.46 (s, 9H), 1.47 (s, 9H), 1.50–1.57 (m, 3H), 1.73–1.78 (m, 1H), 1.81–2.05 (m, 1H), 2.14 (d, J=10.2 Hz, 1H), 2.32 (d, J=4.1 Hz, 1H), 2.44 (s, 1H), 3.17 (s, 1H), 3.57 (s, 1H), 7.06 (t, J=2.5 Hz, 2H), 7.38 (s, 2H), 8.26 (s, 1H), 8.34 (s, 1H), 13.63 (s, 1H), 13.69 (s, 1H). 13C NMR: δ=22.5, 27.7, 29.8, 31.9, 34.5, 35.4, 36.7, 43.7, 45.6, 80.2, 80.7, 118.0, 118.2, 126.3, 127.1, 127.2, 136.8, 140.4, 158.0, 158.1, 164.4, 166.2. MS (EI, 70 eV): m/z=558 (M+, 19), 325 (100), 296 (14), 264 (32), 244 (12), 219 (7), 84 (7). IR (KBr): =2955, 2873, 1624, 1595, 1473, 1440, 1362, 1248, 1175, 1057 cm−1. Anal. calcd. for C37H54N2O2
(558.84): C, 79.52%; H, 9.74%; N, 5.01%; found: C, 79.47%; H, 9.92%; N, 5.13%.
°C (en). [α]D=−128.0 (c=1.0, CHCl3). 1H NMR (CDCl3): δ=1.48–1.60 (m, 3H), 1.70–1.79 (m, 1H), 2.16–2.23 (m, 2H), 2.40 (d, J=4.1 Hz, 1H), 2.51 (br s, 1H), 3.41 (t, J=2.2 Hz, 1H), 3.75 (br s, 1H), 7.20 (dd, J=7.4, 8.5 Hz, 2H), 7.31 (d, J=3.1 Hz, 2H), 7.33 (d, J=3.0 Hz, 2H), 8.32 (s, 1H), 8.48 (s, 1H). 13C NMR: δ=22.3, 28.0, 36.8, 44.3, 45.2, 80.8, 81.6, 128.7, 128.8, 130.3, 130.4, 133.6, 133.8, 134.8, 134.8, 155.3, 156.9. MS (EI, 70 eV): m/z=267 (100), 239 (40), 174 (39), 156 (24), 123 (24), 66 (22). IR (KBr): =2954, 2870, 1642, 1579, 1557, 1429, 1375, 1189, 1094, 781 cm−1. Anal. calcd. for C21H18N2Cl4
(440.19): C, 57.30%; H, 4.12%; N, 6.36%; found: C, 57.26%; H, 3.91%; N, 6.35%.
Acknowledgements
We are grateful to the Deutsche Forschungsgemeinschaft (DFG) within the Collaborative Research Center (SFB) 380 ‘Asymmetric Synthesis by Chemical and Biological Methods’ and the Fonds der Chemischen Industrie for financial support. We also thank N. Brendgen and S. Grünebaum for experimental contributions, as well as Dr. C. W. Lehmann (MPI für Kohlenforschung, Mülheim) for data collection.References
- Review: D. Lucet, T. Le Gall and C. Mioskowski, Angew. Chem., 1998, 110, 2724 Search PubMed; D. Lucet, T. Le Gall and C. Mioskowski, Angew. Chem., Int. Ed., 1998, 37, 2580 CrossRef.
- (a) M. A. Eisenberg, in Escherichia coli Salmonella typhimurium, ed. F. C. Neidhardt, American Society for Microbiology, Washington, DC, 1987, vol. 1, p. 544 Search PubMed; (b) A. Marquet, Pure Appl. Chem., 1993, 65, 1249 CAS.
- P. Kulanthaivel, Y. F. Hallock, C. Boros, S. M. Hamilton, W. P. Janzen, L. M. Ballas, C. R. Loomis, J. B. Jiang, B. Katz, J. R. Steiner and J. Clardy, J. Am. Chem. Soc., 1993, 115, 6452 CrossRef CAS.
- (a) J. A. Lord, A. A. Waterfield, J. Hughes and H. W. Kosterlitz, Nature, 1977, 267, 495 CAS; (b) S. J. Paterson, L. E. Robson and H. W. Kosterlitz, Br. Med. Bull., 1983, 39, 31 Search PubMed; (c) J. Szmuszkovicz and P. F. VonVoigtlander, J. Med. Chem., 1982, 25, 1125 CrossRef CAS.
- (a) B. Rosenberg, L. VanCamp, J. E. Trosko and V. H. Mansour, Nature, 1969, 222, 385 CAS; (b) A. Pasini and F. Zunino, Angew. Chem., 1987, 99, 632 CAS; A. Pasini and F. Zunino, Angew. Chem., Int. Ed. Engl., 1987, 26, 615 CrossRef; (c) H. Brunner, P. Hankofer, U. Holzinger, B. Treittinger and H. Schönenberg, Eur. J. Med. Chem., 1990, 25, 35 CrossRef CAS; (d) H. Brunner, P. Hankofer, U. Holzinger and B. Treittinger, Chem. Ber., 1990, 123, 1029 CAS.
- (a) M. Kawashima and A. Hirayama, Chem. Lett., 1990, 2299 CAS; (b) M. Kawashima and A. Hirayama, Chem. Lett., 1991, 763 CAS; (c) M. Kawashima and R. Hirata, Bull. Chem. Soc. Jpn., 1993, 66, 2002 CAS; (d) H. Brunner and H. Schliessling, Angew. Chem., 1994, 106, 130 CAS; H. Brunner and H. Schliessling, Angew. Chem., Int. Ed. Engl., 1994, 33, 125 CrossRef; (e) P. Mangeney, A. Alexakis and J. F. Normant, Tetrahedron Lett., 1988, 29, 2677 CrossRef CAS; (f) A. Alexakis, P. Mangeney, I. Marek, F. Rose-Munch, E. Rose, A. Semra and F. Robert, J. Am. Chem. Soc., 1992, 114, 8288 CrossRef CAS; (g) A. Alexakis, P. Mangeney, N. Lensen, J.-P. Tranchier, R. Gosmini and S. Raussou, Pure Appl. Chem., 1996, 68, 531 CAS; (h) Y. L. Bennati and S. Hanessian, Chem. Rev., 1997, 97, 3161 CrossRef CAS.
- (a) Recent reports: E. N. Jacobsen and M. H. Wu, in Comprehensive Asymetric Catalysis, eds. E. N. Jacobsen, A. Pfaltz and H. Yamamoto, Springer, New York, 1999, ch. 18.2, pp. 649–677 Search PubMed; (b) T. Katsuki, in Catalytic Asymmetric Synthesis, ed. I. Ojima, Wiley-VCH, New York, 2nd edn., 2000, ch.6B, pp. 287–325 Search PubMed; (c) C. P. O'Mahony, E. M. McGarrigle, M. F. Renehan, K. M. Ryan, N. J. Kerrigan, C. Bosquet and D. G. Gilheany, Org. Lett., 2001, 22, 3435 CrossRef CAS.
- (a) Z. Li, K. R. Konser and E. N. Jacobsen, J. Am. Chem. Soc., 1993, 115, 5326 CrossRef CAS; (b) D. A. Evans, M. M. Faul and M. T. Bilodeau, J. Am. Chem. Soc., 1994, 116, 2742 CrossRef CAS; (c) Z. Li, R. W. Quan and E. N. Jacobsen, J. Am. Chem. Soc., 1995, 117, 5889 CrossRef CAS.
- (a) E. N. Jacobsen, Acc. Chem. Res., 2000, 33, 421 CrossRef CAS; (b) F. Fache, E. Schulz, M. L. Tommasino and M. Lemaire, Chem. Rev., 2000, 100, 2159 CrossRef CAS; (c) J. M. Ready and E. N. Jacobsen, J. Am. Chem. Soc., 2001, 123, 2687 CrossRef CAS.
- (a) D. A. Evans, T. Lectka and S. T. Miller, Tetrahedron Lett., 1993, 34, 7027 CrossRef CAS; (b) S. E. Schaus, J. Brånalt and E. N. Jacobsen, J. Org. Chem., 1998, 62, 403 CrossRef CAS.
- (a) Reviews: R. Noyori and S. Hashiguchi, Acc. Chem. Res., 1997, 30, 97 Search PubMed; (b) M. J. Palmer and M. Wills, Tetrahedron: Asymmetry, 1999, 10, 2045 CrossRef CAS; (c) M. Wills, M. J. Palmer, A. Smith, J. Kenny and T. Walsgrove, Molecules, 2000, 5, 4 Search PubMed.
- (a) For the diphenylethylenediamine see: D. Pini, A. Iuliano, C. Rosini and P. Salvadori, Synthesis, 1990, 1023 Search PubMed; (b) B. B. Lohray and J. R. Ahuja, J. Chem. Soc., Chem. Commun., 1991, 95 RSC; (c) R. Oi and K. B. Sharpless, Tetrahedron Lett., 1991, 32, 999 CrossRef CAS; (d) E. J. Corey, R. Imwinkelried, S. Pikul and Y. B. Xiang, J. Am. Chem. Soc., 1989, 111, 4486 CrossRef; (e) M. Shimizu, M. Kamei and T. Fujisawa, Tetrahedron Lett., 1995, 36, 8607 CrossRef CAS. For the diaminocyclohexane see: (f) A. Wieland, O. Schlichtung and W. V. Z. Langsdorf, Phys. Chem., 1926, 161, 74 Search PubMed; (g) G. Swift and D. Swern, J. Org. Chem., 1967, 32, 511 CrossRef CAS; (h) T. A. Whitney, J. Org. Chem., 1980, 45, 4214 CrossRef CAS.
- K. Hatano, T. Takeda and R. Saito, J. Chem. Soc., Perkin Trans. 2, 1994, 579 RSC.
- (a) C. Bolm, A. Gerlach and C. L. Dinter, Synlett, 1999, 195 CAS; (b) C. Bolm, I. Schiffers, C. L. Dinter and A. Gerlach, J. Org. Chem., 2000, 65, 6984 CrossRef CAS.
- (a) C. Bolm, I. Schiffers, C. L. Dinter, L. Defrère, A. Gerlach and G. Raabe, Synthesis, 2001, 1719 CrossRef CAS; (b) C. Bolm, C. L. Dinter, I. Schiffers and L. Defrère, Synlett, 2001, 1875 CrossRef CAS.
- For recent examples see: (a) K. Maruoka, M. Akakura, S. Saito, T. Ooi and H. Yamamoto, J. Am. Chem. Soc., 1994, 116, 6153 CrossRef CAS; (b) R. M. Borzilleri and S. M. Weinreb, J. Am. Chem. Soc., 1994, 116, 9789 CrossRef CAS; (c) J. T. Starr, G. Koch and E. M. Carreira, J. Am. Chem. Soc., 2000, 122, 8793 CrossRef CAS; (d) A. Bernardi, D. Arosio, D. Dellavecchia and F. Micheli, Tetrahedron: Asymmetry, 1999, 10, 3403 CrossRef CAS.
- For the experimental procedures, see the following literature: (a) alkaloid-mediated anhydride opening, see ref. 14b; (b) selective epimerization, see ref. 16a; (c) hydrogenation and saponification, see ref. 15a. Analytical data of all known intermediates were in accordance with the literature.
- X-Ray crystal structure analysis of 8·2HCl: formula C9H20N2Cl2; M=227.18, colorless crystal, 0.06×0.06×0.17 mm3, a=6.9118(2), b=8.1756(2), c=21.0256(5)
Å, V=1188.12 Å3, ρcalcd=1.270 g cm−3, μ=0.509 mm−1, Z=4, orthorhombic, space group P212121
(No. 19), λ=0.71073 Å, T=120 K, ω scans, 18496 reflections collected, 4544 independent and 3827 observed reflections [I
![[greater than or equal, slant]](https://www.rsc.org/images/entities/char_2a7e.gif) 4σ(I)], 198 refined parameters, R=0.043, wR=0.048, (w=[σ2(F)+0.0004·F2]−1), S=1.076, residual electron density −1.12/0.76 e Å−3, hydrogens located and refined isotropically. The structure has been solved by direct methods as implemented in the Xtal3.7 set of crystallographic routines,19a employing GENSIN19b for the generation of structure invariant relationships and GENTAN19c for the general tangent phasing procedure. CCDC reference number 189661. See http://www.rsc.org/suppdata/nj/b2/b206943c/ for crystallographic files in CIF or other electronic format.
4σ(I)], 198 refined parameters, R=0.043, wR=0.048, (w=[σ2(F)+0.0004·F2]−1), S=1.076, residual electron density −1.12/0.76 e Å−3, hydrogens located and refined isotropically. The structure has been solved by direct methods as implemented in the Xtal3.7 set of crystallographic routines,19a employing GENSIN19b for the generation of structure invariant relationships and GENTAN19c for the general tangent phasing procedure. CCDC reference number 189661. See http://www.rsc.org/suppdata/nj/b2/b206943c/ for crystallographic files in CIF or other electronic format. - (a) Xtal3.7 System, eds. S. R. Hall, S. J. du Boulay and R. Olthof-Hazekamp, University of Western Australia, 2000 Search PubMed; (b) V. Subramanian and S. R. Hall, in Xtal3.7 System, eds. S. R. Hall, D. J. du Boulay and R. Olthof-Hazekamp, University of Western Australia, 2000 Search PubMed; (c) S. R. Hall, in Xtal3.7 System, eds. S. R. Hall, D. J. du Boulay and R. Olthof-Hazekamp, University of Western Australia, 2000 Search PubMed.
- B. M. Trost, J. L. Zambrano and W. Richter, Synlett, 2001, 907 CrossRef CAS.
- K. Furuta, K. Iwanaga and H. Yamamoto, Tetrahedron Lett., 1986, 27, 4507 CrossRef CAS.
- K. Alder and G. Stein, Liebigs Ann. Chem., 1934, 514, 211 Search PubMed.
- I. Schiffers, Ph.D. Dissertation, RWTH Aachen University, 2002.
- H. Waldmann, Liebigs Ann. Chem., 1990, 671 Search PubMed.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2003 |