Focus. Dealing with ‘real’ samples: sample pre-treatment in microfluidic systems
Andrew J. de Mello and Nigel Beard review issues related to the analysis of ‘real’ samples using microfluidic chip technology
First published on 19th February 2003
Analytical chemistry is an expansive field, encompassing a myriad of methods and techniques employed to provide discrimination of an analyte of interest from its surroundings. A generic analytical procedure can be broken down into three broad categories (Fig. 1): the analytical principle on which the measurement is based, the analytical method (i.e. the concept of optimising the conditions for the analytical principle chosen), and finally the analytical procedure (that encompasses all considerations from analyte to analytical result). | ||
| Fig. 1 Schematic of a generic analytical process. | ||
Over the past decade, the concepts of miniaturization (and in particular the development of microfluidic sciences) have been seriously applied to chemical and biological problems.1 However, to date, most research has focused on the downsizing of the analytical principle, with many of the other necessary analytical procedures (such as reagent sampling and sample pre-treatment) still performed off-chip. This is perhaps unsurprising, since many of the primary benefits afforded through miniaturisation lie in improved performance characteristics of the analytical principle. For example, downsizing of capillary electrophoresis (CE) has repeatedly been shown to yield distinct advantages when compared to conventional capillary and slab-gel formats (such as reduced analysis times2 and extremely high separation efficiencies3). In addition, the unique environment provided by microfluidic systems allows for rapid, efficient and controllable chemical and biological synthesis (due to the scale dependence of thermal and mass transfer).4,5 These fundamental performance gains have done much to stimulate interest in the field and drive the development of microsystems for a wide range of unit applications. However, the ability to extract essential information from a chemical or biological system almost always involves performing a number of distinct analytical operations in sequence. Consequently, much recent focus has centred on the integration of functional components within monolithic systems. Lithographic printing techniques are well-suited to the fabrication of integrated analytical systems, and indeed many examples of multistep analytical procedures have been reported.6,7 A cursory survey of the literature shows that most examples of integrated processing within microfabricated devices have been directed at linking analytical principles (for example chemical or biological reactors with separation modules) rather that integration with front-end functions (such as sample extraction and filtration). Nevertheless, the ability to efficiently process raw sample (from the laboratory, the body or the field) and subsequently perform the required analytical operations ‘on-chip’ will be key in defining the eventual success and an application of microfluidic systems.
Sample processing and pre-treatment can take a number forms depending on the nature of the system to be sampled. Often an analyte of interest is accommodated within an extremely complex matrix (for example blood). Thus the isolation and ‘clean-up’ of a particular analyte or set of analytes is desirable under most circumstances. Typical processes may include sample filtration, centrifugation, distillation, dilution, target amplification and extraction. Successful execution of these processes is required to ensure that the analyte is present in a form compatible with the analytical principle. In addition, small volumes of sample and reagent (pL–nL) are representative of most miniaturized systems. This characteristic has clear advantages associated with cost and analytical throughput, but does pose constraints on appropriate or available detection methods. Consequently, much research has focused on the development of miniaturised and sensitive detection techniques.8 An alternative approach to increasing the sensitivity of analyte detection methods is to pre-concentrate the sample prior to analysis, thus indirectly yielding superior limits of detection. Finally, molecular species which may be difficult to detect using standard detection methods may be derivatized prior to analysis with an appropriate label or tag. Subsequent detection of the label can then be used to indirectly infer the presence of the target analyte. The array of sample pre-treatment techniques is vast, and it is therefore the aim of this mini-review to highlight a few of the major developments in sample pre-treatment techniques that have successfully been integrated into chip-based systems. More extensive details of sample pre-treatment techniques in microfabricated analytical systems can be found in an excellent review article by Jan Lichtenberg.9
It is often quite necessary to tailor the sample pre-treatment methodology to both the analyte of interest and the analytical technique employed; nonetheless, there are distinct generic techniques that have been successfully downsized in to the microchip environment. As stated earlier there are three main sub-categories of sample pre-treatment (isolation/clean-up, sample pre-concentration and sample derivatization), and for simplicity a selection of chip-based techniques within each category have been addressed below.
The isolation/clean-up of analyte of interest from the sample matrix
A challenging task faced by the analytical chemist when dealing with raw samples is extracting/isolating the analyte of interest from the sample matrix. It is fair to say that the majority of ‘real’ samples arrive in a format incompatible with most analytical instrumentation, and require some degree of clean-up. Even well-defined samples (e.g. aqueous solutions) require basic filtration prior to analysis. Other desirable techniques may include dilution, cell lysis, liquid/liquid extraction and solid phase extraction.Filtration
Possibly the most essential step when performing analysis in microfluidic systems is the filtration of sample prior to processing. Due to the small dimensions typical in microstructures, particulates can cause serious operational problems, providing sites for nucleation or blockage. The simplest solution is to filter all reagents and sample prior to introduction. Unfortunately, most conventional filtration methods require fluidic volumes far greater than those required for analytical processing. Consequently, it is desirable to integrate sample filtration on-chip prior to analysis.A number of microfabricated filters have been described for both collection of particles and clean-up of sample for downstream processing. A popular approach has been to create microfabricated frits, pillar structures or flow restrictions within fluidic channels to mimic conventional filters. Particulates can be ‘trapped’ within the structure as long as the diameter of the particulate is larger than the feature dimensions of the microstructure, and solution flow is unimpeded. However, such designs have typically been used as bead traps, or stationary phase reactors, in which a chemical reaction can be performed without causing blockage to the rest of the fluidic network. Göran Stemme and co-workers at Royal Institute of Technology, Stockholm have described a number of microfabricated filters based on this concept.10–13 An example of such a device for stationary phase trapping is illustrated in Fig. 2.12 Here filter pillars (3 μm wide and 50 μm high) define a square reaction chamber for stationary phase bead collection. The authors report reaction chambers with volumes as small as 500 pL and limiting flow rates of 2 μL min−1 when the chamber is completely packed.12 The integration of such filter array elements with passive valves has subsequently been applied to solid-phase biochemical assays, including single nucleotide polymorphism analysis.13 Importantly, the combination of filter and passive elements in a flow-through system affords facile filter regeneration (or bead removal) by reversal of the flow direction.
 | ||
| Fig. 2 SEM image of a microdevice for stationary phase trapping. (Adapted with permission. Copyright 2000, Elsevier Science B.V.) | ||
Fred Regnier and colleagues at Purdue University have proposed an alternative solution to the problem of filtering within microfluidic systems. Their in situ solvent and reagent filters are based on the concept of lateral percolation.14 In their lateral percolation filters (Fig. 3), the sample penetrates a filter bed of posts along the plane/face of the microstructure, continuing perpendicular through the filter bed (lateral to the point of entry). Particles are then retained in the bed as fluid flows laterally through the structure. First generation lateral percolation filters were fabricated in quartz using deep reactive ion etching techniques, with the filter element comprising a network of intersecting channels (1.5 × 10 μm) situated at the bottom of the fluidic reservoirs. Particulates with dimensions larger than the minimum filter features (1.5 μm) were successfully restricted, and the robustness of the device was examined with a variety of particulates (including dust particles and bacterial cells).
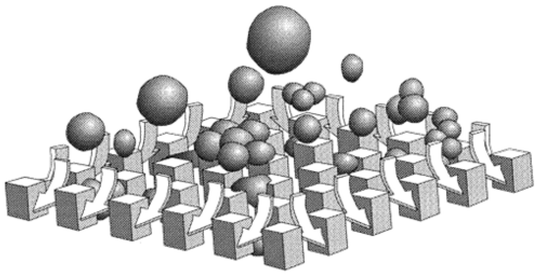 | ||
| Fig. 3 Lateral percolation in a microfabricated chamber. Liquid vertically enters an array of microfabricated cubes attached to an underlying substrate and is drawn laterally to the sides of the array through an interconnecting channel network. Particles larger than the channels separating the cubes are excluded, similar to axial filters. (Adapted with permission. Copyright 1999, The American Chemical Society.) | ||
In both previous studies the efficacy and application of the filter structure is determined by the resolution limits of the manufacturing process. Consequently, filtering of sub-micron sized particulates using physical structures puts stringent demands on device fabrication. To this end sacrificial layer technology has been used to create flow filters for particles as small as 10 microns.15,16 A detailed discussion of such approaches may be found elsewhere.9
Material transport in micron-sized vessels normally falls into the laminar flow regime (Reynold’s numbers are typically very low) where viscous forces dominate over inertia and dampen out irregularities in flow patterns. This means turbulence is often unattainable and mass transport can only occur via molecular diffusion.17 Interestingly, this property can be exploited to allow for the discrimination (or filtering) of molecular or particular species. Simply put, species of low molecular mass have greater mobility (larger diffusion coefficients) than large molecular species. Consequently, filtration (or spatial discrimination) can be induced by allowing analytes of interest to migrate across a laminar boundary (between a sample and solvent stream) whilst retaining unwanted heavier particulates in the original fluid stream. The process can be further controlled by altering the time in which the two fluids come into contact. Importantly, this approach addresses many of the problems associated with structurally-based filters, since its operation is reliant on the control of molecular diffusion rather that the resolution of the manufacturing process.
Microstructures based on this concept were first demonstrated by James Brody, Paul Yager and co-workers at the University of Washington in the mid-nineties.18 Initial devices incorporated both structural (etched barriers) and diffusion based filtration, and were successful at discriminating between and isolating 16 and 2.6 μm spheres. Due to the relative complexity of the fabrication process (a three mask process incorporating anisotropic etching of silicon) the authors developed their ideas to create microfabricated filters based solely on diffusional discrimination.19,20 Such devices (termed H-filters) function by bringing two laminar flows (a carrier stream and diluant stream) together in a central channel. Molecules (in the carrier stream) may then diffuse across the fluid barrier created at the boundary between the two streams. Highly mobile analyte molecules can cross between streams quickly, whilst heavier particles remain in the carrier stream. Consequently, only particles which have crossed the boundary before reaching the output channels will end up in the filtered output. Importantly, the approach is highly configurable since the time allowed for diffusional transfer between streams is directly controlled by fluid velocity and the length of the channel.
Recent developments in diffusional filtration have included the use of H-filter devices in complex preparative and detection processes. For example, combination of the H-filter and other microfluidic components within a monolithic chip device has been used to perform protein extraction and quantification from Gram-negative bacterial cells.21
Liquid/liquid extraction (LLE)
Liquid/liquid extraction (sometimes referred to as solvent/solvent extraction) is a technique widely used in conventional sample pre-treatment methodologies and describes the physical process by which a compound (or a mixture of compounds) is transferred from one liquid phase to another. The high surface-to-volume ratios and short diffusion distances typical within microfluidic environments, combined with laminar flow conditions, offer the possibility of performing LLE within microchannels without the need for stirring or agitation. However, to date, reports of LLE within chip-based systems have been occasional due to the difficulty of inducing electroosmotic flow in common organic solvents (e.g. chloroform and toluene). Takehiko Kitamori and colleagues at the University of Tokyo and Kanagawa Academy of Science and Technology were early in realising the merits of the microscale for LLE and have reported several microfabricated devices for LLE. The first microfluidic system for solvent extraction was a variation of a H-filter design, fabricated in quartz and comprising 250 μm wide channels. Introduction of Fe(II) in an aqueous stream and tri-octylmethylammonium chloride in an organic (chloroform) stream allowed extraction of the ion-pair product in the organic phase. Extraction was shown to occur in less than 45 s, representing an order of magnitude improvement over conventional extraction times in separation funnels.22 The authors have also demonstrated the extraction of Ni(II) complexes in microchannels,23 integration of neutral ionophore-based ion-pair extraction on-chip,24 and sequential ion-sensing via ‘slug’ flow in microchannel environments.25 The latter is of particular interest since the approach allows the determination of multiple ions in a single sample by pumping aqueous and organic phases intermittently through a fluidic network.A potential drawback when using microfluidic systems for solvent extraction is the low unit throughput (normally between 1 and 100 μL min−1). This problem can be obviated by operating arrays of parallel channels concurrently. To this end, researchers at AstraZeneca and CRL UK have reported the fabrication of silicon/glass micro-contactor arrays for the extraction of single feeds at rates of 250 mL h−1.26 LLE is achieved by contacting fluidic streams at constricted openings between distinct channels. The approach is attractive since flows can be separated naturally as the channels diverge.
Solid phase extraction (SPE)
Solid phase extraction is a broad technique in which a target molecule is retained by a chromatographic stationary phase material and subsequently eluted in an appropriate (and selective) solvent. SPE functions as both a sample clean-up method and a pre-concentration method.† This is due to the fact that as the target analyte is retained within the stationary phase and the unwanted components of the sample matrix flow to waste, retention is accompanied by pre-concentration. A brief survey of the literature indicates two popular methods for performing SPE in microfluidic systems. The first involves coating channel walls with a high affinity stationary phase. This coating interacts with the target analyte whilst unwanted components and matrix flow to waste. Although many early examples of SPE on-chip have incorporated this approach, the capacity of the SPE column is dependent on the surface area available for interaction. Accordingly, an alternative tactic is to fill or pack microchannels with a stationary phase material.Michael Ramsey and associates at Oak Ridge National Laboratory described one of the first examples of SPE on a microfluidic device.27 SPE of a neutral coumarin dye was achieved by coating specific channel walls with octadecyltrimethoxysilane. A simple fluidic network allowed for both enrichment (80 fold increase in concentration) and elution of the dye within 4 min. The problem of limited surface areas in open-channel devices can be ameliorated to some degree by utilising sophisticated fabrication techniques. For example, Alan Northrup and co-workers have reported successful SPE of DNA by flowing sample through a regular array of high aspect ratio silicon posts. Although the authors report a ten-fold increase in concentration and a 50% capture efficiency, the use of deep reactive ion etching methods for chip manufacture makes the approach rather complex.28
A simpler way to increase surface area is to pack microchannels with stationary phase material. For example, Jed Harrison and co-workers at the University of Alberta have reported the fabrication and testing of a 330 pL chromatographic bed integrated within an electroosmotically pumped microsystem.29 The authors utilize weirs within a microfabricated channel to trap coated silica beads (1.5–4 μm diameter). These are then used to perform both solid phase extraction and electrochromatography of small molecules. Concentration enhancements of up to 500 times were demonstrated for two fluorescent dyes. More recent studies on the same device have demonstrated improved packing and bed stabilisation, and the efficient detection of fluorescent dyes at concentrations below 100 fM.30
Similar in-stream SPE microdevices have been proposed by researchers at Lund University and AstraZeneca for sample clean-up and enrichment of protein and peptide samples prior to MALDI-TOF MS analysis.31 Silicon/glass devices incorporating a ‘weir’ structure facilitate the packing of reverse-phase chromatographic beads. These beads are then used to successfully purify and enrich a 10 nM peptide mixture containing 2 M urea in 0.1 M phosphate-buffered saline prior to MS analysis. Subsequent modelling of the fluid dynamics in this system has allowed an improved grid-SPE device to be fabricated and tested (Fig. 4) for on-line proteomic sample preparation.32
 | ||
| Fig. 4 A scanning electron micrograph of a grid type micro-extraction device. The channel width is 220 μm and the channel depth is 200 μm. The bead trapping walls are 13 μm wide and spaced 16 μm apart. (Reprinted with permission. Copyright 2002, John Wiley & Sons, Inc.) | ||
Packing columns are often complex and ill-defined. An alternative is to replace conventional stationary phase materials with a continuous, porous bed of support (a porous monolith) formed by in situ polymerisation of organic monomers.33 The process of bed formation is facile, since a low-viscosity monomer solution can be introduced by vacuum or pressure into the microfluidic channel prior to initiation. In addition, the continuous polymer bed is attached to the channels walls, making a retaining frit or weir redundant. To this end, Jean Fréchet and co-workers at the University of California, Berkeley have recently described the preparation of microfluidic devices for SPE using porous monoliths with hydrophobic and ionisable surface chemistries.34,35 High flow rates (up to 10 μL min−1) are achievable due to the facile control of pore sizes, and the authors report the enrichment of a hydrophobic terapeptide and green fluorescent protein with concentration enhancements of up to 103.
Pre-concentration of the analyte of interest
As noted previously, analyte may be present in ‘real’ samples at extremely low concentrations. This combined with the ultra-small detection volumes encountered in microfluidic systems (pL–nL) makes sensitive detection methods a prerequisite for most analyses. Although fluorescence methods provide for extremely low mass and concentration detection limits, their application is restricted to systems containing either intrinsic or extrinsic fluorophores. Other detection methods (including absorption, Raman scattering and electrochemical detection) afford the analysis of a greater range of molecular species, but with significantly inferior detection limits. It is often thus desirable to incorporate sample pre-concentration prior to detection within microfluidic systems. A few of the key developments in this area will now be discussed.Field amplified sample stacking (FASS)
Field amplified sample stacking is a common method for sample pre-concentration in electrophoretic systems.36–43 In FASS, a sample prepared in a low conductivity buffer is injected into the capillary (or channel) filled with a running buffer of higher conductivity. When a voltage is applied the resultant electric field strength is higher in the low-conductivity sample zone than in the running buffer zone, leading to an increased analyte velocity in the sample zone. At the buffer interface, analyte molecules decelerate abruptly and ‘stack’ into a narrow (and concentrated) sample band. This process is described schematically in Fig. 5. Although FASS is well established in conventional capillary electrophoresis, its transferral to a chip-based format is not straightforward. The primary difficulty associated with sample stacking in a microfluidic format is the control of the analyte zone during the stacking and separation procedures. This often necessitates the use of relatively complex channel networks and voltage programs to stack and/or manoeuvre the analyte zone. | ||
| Fig. 5 Principle of sample stacking in capillary electrophoresis: (A) a sample plug of cations is injected in a low conductivity buffer (e.g. de-ionised water). When a voltage is applied, the electric field in the sample solution is higher than in the rest of the capillary, cations migrate rapidly through the sample zone until they reach the low electric field in the separation buffer; (B) the cations then slow down and become stacked at the boundary between the sample region and buffer region. | ||
Stephen Jacobson and Michael Ramsey were the first to address the need for sample stacking techniques in microfluidic devices.44 Employing a field amplified injection method, pre-concentration of the sample is performed at the beginning of a separation channel. Good signal enhancements were reported for dansylated amino acids, with reproducibilities of 2.1% RSD. Other studies by the same group reported the use of field amplified injection techniques to facilitate pre-concentration of metal cations.45 The approach featured a modified ‘gated’ injection scheme, and gave reproducibilities of 1.5% RSD.
It should be noted that the control of sample within fluidic networks develops into more of an issue when performing FASS due to the different velocities the analytes experience in the different zones. To this end Hua Yang and Ring-Ling Chien of Caliper Technologies Corporation have demonstrated that careful control of the electroosmotic forces at the injection intersection within a fluidic network can greatly improve signal enhancements.46 By eliminating electroosmotic flow, the concentration boundary between low and high conductivity zones becomes stationary, except for dispersion induced by diffusion. Signal gains for separations of fluorescein-labelled proteins employing this ‘static sample’ mechanism are in excess of 2 orders of magnitude. In addition, Jan Lichtenberg and associates at the University of Neuchâtel have reported on-chip FASS employing post injection sample stacking (more analogous to FASS in conventional CE).47 The approach provides for the formation of long, volumetrically defined sample plugs, with little electrophoretic bias. Optimisation of channel patterns and voltage control systems yields pre-concentration factors of approximately 65 fold on a time-scale of a few minutes for FITC-labelled amino acids. More recently, researchers at Imperial College, London have proposed a different approach to FASS on-chip.41,48 The authors report the use of a narrow sample channel injector49 to introduce the sample directly into the separation channel. A typical injector is shown in Fig. 6. Narrow sample channel injectors allow sample plugs to be introduced directly into the separation channel, and subsequent stacking and separation can proceed without any need for leakage control. More importantly, stacking and separation occur in a single step negating the requirement for complex channel geometries and voltage switching to control sample plugs during the stacking procedure. Sample plug lengths between 600–1600 μm were studied and signals resulting from FITC-labelled biogenic amines (at concentrations down to 20 pM) were increased by a factor of ∼75.
 | ||
| Fig. 6 SEM micrograph of a typical NSC injector fabricated in PDMS. The widths of the sample and separation channels are 5 μm and 55 μm respectively. | ||
Stacking of neutral analytes
Although popular, FASS methods are only applicable to analysis of charged analytes. Nevertheless, pre-concentration of neutral analytes can be achieved using miceller electrokinetic chromatography (MEKC). One such technique is termed sweeping. Sweeping, initially observed by Gilges50 and further developed by Quirino,51,52 is defined as the picking and accumulating of neutral analyte molecules by pseudostationary phases (e.g. sodium dodecyl sulfate) that enter and fill the sample zone upon application of a voltage. This results in stacked pseudostationary phases carrying neutral analytes at the interface between sample and running buffer zones. In theory, huge improvements in sensitivity can be achieved by the narrowing of neutral analyte zones. However, for significant compression to occur, several requirements must be fulfilled. Firstly, a constant electric field must be maintained along the entire channel. Furthermore, the channel should exhibit negligible electroosmotic flow and no pseudostationary phase should be present in the injected sample.Application of sweeping in conventional CE has resulted in analyte enrichment factors of several thousands. Indeed, Joselito Quirino and Shigeru Terabe from Himeji Institute of Technology have recently reported analyte enrichment factors approaching one million-fold using cation-selective exhaustive injection and sweeping.53 These advancements have led to the demonstration of a number of chip-based systems for sweeping.
James Landers and colleagues at the University of Virginia were first in reporting the online concentration of neutral analytes within microfluidic systems using sweeping techniques.54 The authors exploited electroosmotic flow to inject long sample plugs under continuous and discontinuous co-ion conditions. This allowed for both short injection times, sample plug injections greater than the actual channel length and significant signal enhancements. More recently Shigeru Terabe and co-workers have improved signal enhancement factors to 3 orders of magnitude.55 The authors introduce a charged micelle (anionic micelles generated using sodium dodecyl sulfate) into the running buffer, but not in the sample matrix. On application of an electric field the micelles move through the sample zone ‘sweeping’ the neutral molecules into one discrete band.
Isotachophoresis
Isotachophoresis (ITP) is a technique that can be used for both sample clean-up and sample pre-concentration. Briefly, in ITP a sample is sandwiched between a leading and a terminating electrolyte. The leading electrolyte is chosen such that its ions have a higher mobility than any ions (of the same charge) in the sample. Similarly, the terminating electrolyte is chosen such that its ions have a lower mobility than any ions in the sample. When an electric field is applied, ions in the sample arrange themselves into discrete bands in order of mobility. After solute distribution, an equilibrium is reached, where each ion’s velocity is normalised to the same value and all zones exhibit stable and well-defined boundaries. Importantly, concentration is a direct consequence of velocity normalisation since analyte velocities automatically adjust to the encountered field strength. Consequently, analyte separation and concentration occur concurrently.A number of recent studies have focussed on the application of ITP in microfluidic systems. In an early example, Michael Morris and co-workers at the University of Michigan, Ann Arbor reported isotachophoretic separations of the herbicides in a glass microchip etched channel monitored by normal Raman spectroscopy.56 In addition, Jeff Prest and colleagues at the University of Manchester Institute of Science and Technology reported a miniaturised planar polymer ITP separation system for the analysis of sodium and potassium. The elastomeric chip incorporated a single electrode conductivity detector and afforded sub-nanomolar detection limits.57
Dušan Kaniansky and collaborators at Comenius University and Merck KGaA have published various papers on performing ITP in poly(methyl methacrylate) microchips.58–60 Devices are based on a pair of separation channels in a column coupling arrangement which allow separation to be performed in two stages.61 With the suppression of EOF throughout the fluidic network, rapid separations of anions have been realised at sub-nanomolar concentrations. Using this approach the authors have reported chip-based ITP studies of organic acids and inorganic ions in wine,62 inorganic ions in water samples,59 enantiomeric separations63 and food additives.58 More recently, Ann Wainright and co-workers at ACLARA Biosciences have evaluated microfluidic systems in which large sample volumes can be injected, pre-concentrated by ITP and subsequently separated by zone electrophoresis.64 Using this approach the authors were able demonstrate a 400-fold improvement in detection limits when compared to zone electrophoresis. Furthermore, the method was successfully applied to cell based assays.
In standard formats, ITP is normally incapable of simultaneously analysing anions and cations. Jeff Prest and colleagues have recently utilised the flexibility of micromachining techniques to create planar, plastic chip devices for bi-directional ITP.65 Anions and cations may be concurrently analysed by introducing sample into the centre of a separation microchannel incorporating on-column conductivity detectors at either end. Using this device, the authors were able to concentrate and separate a mixture of three anions and three cations within 1300 seconds. Moreover, the same researchers have highlighted the improved response times that accompany the transferral of ITP from conventional to chip-based formats. Devices cast from silicon rubber and incorporating an integrated conductivity detector were successfully used for the separation of several metal ions in less than half the time taken for the corresponding capillary scale separations.66
Analyte derivatization
In certain circumstances it may be desirable to modify analytes within a sample either before or after a processing operation (such as an electrophoretic or chromatographic separation). Such sample pre-treatment may be required to increase a response at a detector (thus improving analytical sensitivity) or to selectively enhance the detector response by discriminating certain analytes from the sample matrix. As stated, the small volume typical in microfluidic systems dictates that detection methods be both sensitive and selective. Fluorescence has proved a dominant and successful detection method for microscale analysis due to its exquisite sensitivity. However, most molecules are not intrinsically fluorescent and thus must be covalently labelled with a fluorescent tag or reported prior to analysis. Typically, this labelling reaction is performed off-chip, and in many cases can take several hours.67 Consequently, the choice of label and the rapidity of the labelling reaction are of paramount importance when assessing integrated approaches to on-chip sample derivatization.The common approaches to sample derivatization are to ‘tag’ target molecules prior to or subsequent to electrophoretic or chromatographic analysis. An early example of pre-column labelling on-chip was described by Mike Ramsey and co-workers in 1994.68 Using a standard electrophoresis channel network an amino acid sample was separated into component bands, which were then combined with a labelling agent (o-phthaldialdehyde) stream from a side channel. Diffusive mixing of the flow streams afforded efficient labelling of the amino acids, and detection was performed at a given point downstream. The authors also demonstrated the same analysis using a pre-column approach.69 In this case sample was mixed with the fluorescent label in a 1 nL reaction chamber prior to injection and separation along a linear channel. More recently, Jong Hoon Hahn and colleagues at Pohang University of Science and Technology have described integrated pre-column derivatization and MEKC for the analysis of biogenic amines.70 Using a planar poly(dimethylsiloxane) chip, labelling is performed in a 4.5 nL chamber using an amine specific moiety (o-phthaldialdehyde). The proceeding separation is then performed under MEKC conditions resulting in a total analysis time of 1 minute and detection limits of 100 nM.
Many labelling reactions proceed too slowly at ambient conditions and thus must be performed at elevated temperatures. To address this, Andreas Manz and co-workers described the fabrication of a micromachined heated chemical reactor.71 The silicon/glass microreactor contained a 50 μL reactor channel, resistive heaters (providing a heating rate of 2 °C s−1) and integrated resistive heat sensors. To test the efficacy of the device, the authors performed pre-column derivatization of amino acids with 4-fluoro-7-nitrobenzofuran in 2 minutes and at 60 °C, followed by HPLC separation and fluorescence detection. In addition, Michael Ramsey and co-workers have reported the fabrication and operation of a monolithic device for enzymatic reactions, product separation and post-column product labelling.72 Using this device a tryptic digestion of oxidised insulin B-chain was performed within 15 min under stopped flow conditions in a heated channel. This was directly followed by product separation by electrophoresis and subsequently post-column derivatization with naphthalene-2,3-dicarboxaldehyde and detection. The high degree of functional integration (reagent mixing, product separation and post-column labelling) provides an elegant indication of the potential benefits of microfluidic systems when applied to real chemical and biological systems.
For a slightly different application, Valerie Spikmans and collaborators have recently demonstrated the principle of on-chip, post-column derivatization reactions in μ-HPLC hyphenated to electrospray time-of-flight mass spectrometry (ESI-TOFMS).73 Primary and secondary amines were separated using gradient μ-HPLC and labelled on-chip with a positively charged phosphonium complex prior to ESI-TOFMS. Since the optimal flow rates for both μ-HPLC and ESI-TOFMS closely match those encountered within the microfluidic system, the interface between all three components is both facile and flexible.
Biological sample pre-treatment
All of the techniques discussed up to this point can be applied to both chemical and biological analyses. However, the rapid evolution of modern molecular biology has necessitated the concurrent development of specific bioanalytical tools to efficiently and rapidly analyse entities such as proteins, cells, nucleic acids and bacteria. Indeed many of these demands have played an important role in establishing chip-based systems as high-efficiency tools in measurement science. Unfortunately, many biological samples containing species such as cells and spores are difficult to handle and require careful (and often expensive) handling in the analytical laboratory. Consequently, sample pre-treatment of biological samples prior to analysis within microfluidic systems is an area of great interest. Many of these methods have already been discussed in this review, however the following paragraphs list a small selection of sample pre-treatment techniques developed specifically for the analysis of biological fluids. Due to space limitations this discussion is brief and the reader is directed elsewhere for a more complete analysis of the field.9DNA amplification
Since its inception in early 1986, the polymerase chain reaction (PCR) has become an indispensable tool in basic molecular biology, genome sequencing, clinical research and evolutionary studies. At its most basic level PCR serves to amplify or copy DNA in a given sample to levels where it can more easily be detected, analysed or processed. As such its use in sample pre-treatment is undoubted. The mechanistic simplicity of PCR and its dependence on the strict control of experimental parameters mean that PCR is ideally suited to miniaturized formats. The performance gains afforded through transferral to microfluidic platforms lie in improved thermal and mass transfer on a small scale, and have been well-documented elsewhere. Since the focus of a recent mini-review74 in this journal has centred on the development of microsystems for DNA amplification no detailed commentary will be provided here. However, in one study of recent interest Richard Mathies and co-workers at the University of California, Berkeley have reported a microfabricated ‘electrophoretic bioprocessor’ for integrated DNA sequencing sample desalting, template abstraction, pre-concentration and electrophoretic analysis. The authors use a capture matrix to immobilize and pre-concentrate only the extension products of a DNA sequencing reaction. The matrix (containing an acrylamide-copolymerized oligonucleotide) is loaded into a 60 nL capture cell which is connected to the separation channel via a coupling channel. In use, (raw) DNA sequencing reaction products are electrophoretically driven through the chamber, allowing extension products to hydridize to the matrix. Other species, including excess primer, buffering ions and template, are unretained and pass through to the capture outlet. Under optimized conditions, purification can be performed within 2 minutes, with the gel-purified duplex being subsequently released onto the separation column at elevated temperature. This approach yields significant reductions in both cleanup time and reagent volume.75Cell lysis
The release of biological material from clinical samples normally requires lysis of cells prior to analysis. Lysis is generally performed off-chip and achieved through the use of enzymes, detergents, heat or mechanical forces. The integration of cell lysis with downstream processing would clearly extend the applicability of microfluidic devices to analysis of real samples in extra-laboratory environments, and as such is an important target. In 1997, Paul Li and Jed Harrison reported the manipulation and reaction of cells on glass chips using electrokinetic transport.76 As illustrated in Fig. 7, the authors were able to perform erythrocyte cell lysis in less than a second by conjoining a flowing cell stream with a stream of detergent (sodium dodecyl sulfate). Soon after, researchers at Oak Ridge National Laboratory described sequential execution of cell lysis, PCR and electrophoretic analysis on monolithic glass chips. Lysis was effected thermally during the initial stages of PCR and, although not optimised, resulted in sufficient product for CE analysis.77 In addition, more recent studies by Paul Yager have utilised an H-filter arrangement to perform diffusive mixing of a lytic agent stream and a cell suspension stream followed by isolation of the intracellular components.21 | ||
| Fig. 7 Photomicrographs of erythrocyte cell lysis in a microfluidic device. White arrows show direction of flow and the dark bars show the scale (20 µm). Cells enter from the left and SDS from above. A time progression over 0.3 s is illustrated in the three frames. (Adapted with permission. Copyright 1997, The American Chemical Society.) | ||
An area of current concern is the rapid and efficient analysis of bacterial spores such as anthrax. Unfortunately, extraction of endogenous DNA for PCR analysis is generally difficult due to a resilient outer spore cortex. To address this issue Phillip Belgrader and co-workers at Cepheid reported the fabrication and testing of a minisonicator and lysis cartridge.78 Initial results demonstrated the successful disruption of Bacillus spores in 30 seconds with the resulting sample in a PCR compatible form. Further studies by the same group have focused on the development of an integrated cartridge for automation of target concentration, cell disruption and nucleic acid isolation.79,80
Enzymatic digestion
Enzymatic digestion of proteins and nucleic acids is fundamental to the molecular biologist’s toolkit. It is not surprising, therefore, that a number of microdevices for these kinds of reactions have been reported. Stephen Jacobson and Michael Ramsey described an early example of on-chip DNA restriction digestion.81 A planar glass microdevice was used to mix DNA with a restriction enzyme in a 700 pL chamber, and subsequently size the products via gel electrophoresis along a 67 mm microchannel. Using this approach, digestion of a DNA plasmid by the HinfI enzyme and fragment analysis could be achieved within 5 minutes. Protein digestion on chip-based devices has also been reported by a number of groups.72,82–84Conclusion
The example studies outlined in this review clearly indicate that the integration of sample pre-treatment techniques is an active and critical area of research within the field of microfluidics. Indeed, the importance of effective sample pre-treatment will only grow as microfluidic systems are more routinely used to analyse a variety of real (or non-ideal) samples (including bodily fluids and environmental samples). It is evident that highly complex samples may be processed, cleaned, concentrated and prepared for further analysis, however it is worth remembering that sample pre-treatment protocols are highly specific operations, entirely dependent on the nature of the target analyte and downstream processing. This dictates that sample pre-treatment operations should be flexible in both their operation and configuration. Progress to date is encouraging, and more advancements in this area will undoubtedly be key in defining the eventual success and application of microfluidic systems in chemical and biological analysis.References
- S. C. Jakeway, A. J. de Mello and E. Russell, Fresenius’ J. Anal. Chem., 2000, 366, 525 CrossRef CAS.
- S. C. Jacobson, C. T. Culbertson, J. E. Daler and J. M. Ramsey, Anal. Chem., 1998, 70, 3476 CrossRef CAS.
- N. Burggraf, A. Manz, C. S. Effenhauser, E. Verpoorte, N. F. de Rooij and H. M. Widmer, J. High Res. Chromatogr., 1993, 16, 594 Search PubMed.
- K. F. Jensen, Chem. Eng. Sci., 2001, 56, 293 CrossRef CAS.
- A. J. de Mello and R. C. R. Wootton, Lab Chip, 2002, 2, 7N RSC.
- M. A. Burns, B. N. Johnson, S. N. Brahmasandra, K. Hanique, J. R. Webster, M. Krishnan, T. S. Sammarco, P. M. Man, D. Jones, D. Heldsinger, C. H. Mastrangelo and D. T. Burke, Science, 1998, 282, 484 CrossRef CAS.
- E. T. Lagally, I. Medintz and R. A. Mathies, Anal. Chem., 2001, 73, 565 CrossRef CAS.
- M. A. Schwarz and P. C. Hauser, Lab Chip, 2001, 1, 1 RSC.
- J. Lichtenberg, N. F. de Rooij and E. Verpoorte, Talanta, 2002, 56, 233 CrossRef CAS.
- G. Stemme and G. Kittilsland, Appl. Phys. Lett., 1988, 53, 1566 CrossRef CAS.
- G. Kittilsland, G. Stemme and B. Norden, Sens. Actuators A, 1990, 21, 904 CrossRef.
- H. Andersson, W. Wijngaart, P. Enoksson and G. Stemme, Sens. Actuators B, 2000, 67, 203 CrossRef.
- H. Andersson, W. van der Wijngaart and G. Stemme, Electrophoresis, 2001, 22, 249 CrossRef CAS.
- B. He, L. Tan and F. Regnier, Anal. Chem., 1999, 71, 1464 CrossRef CAS.
- W.-H. Chu, R. Chin, T. Huen and M. Ferrari, J. Microelectromech. Syst., 1999, 8, 34 CrossRef CAS.
- J. K. Tu, T. Huen, R. Szema and M. Ferrari, Biomed. Microdev., 1999, 1, 113 Search PubMed.
- J. A. Roberson and C. T. Crowe, Engineering Fluid Mechanics, 5th edn., Wiley, New York, 1993 Search PubMed.
- J. P. Brody, T. D. Osborn, F. K. Forster and P. Yager, Sens. Actuators A, 1996, 54, 704 CrossRef.
- J. P. Brody and P. Yager, Sens. Actuators A, 1997, 58, 13 CrossRef.
- B. H. Weigl and P. Yager, Science, 1999, 283, 346 CrossRef.
- E. A. Schilling, A. E. Kamholz and P. Yager, Anal. Chem., 2002, 74, 1798 CrossRef CAS.
- M. Tokeshi, T. Minagawa and T. Kitamori, Anal. Chem., 2000, 72, 1711 CrossRef CAS.
- K. Sato, M. Tokeshi, T. Sawada and T. Kitamori, Anal. Sci., 2000, 16, 455 CAS.
- H. Hisamoto, T. Horiuchi, M. Tokeshi, A. Hibara and T. Kitamori, Anal. Chem., 2001, 73, 1382 CrossRef CAS.
- H. Hisamoto, T. Horiuchi, K. Uchiyama, M. Tokeshi, A. Hibara and T. Kitamori, Anal. Chem., 2001, 73, 5551 CrossRef CAS.
- J. Shaw, R. Nudd, C. Naik, C. Turner, D. Rudge, M. Benson and A. Garman, Proceedings of Micro-TAS 2000, Kluwer Academic, Dordrecht, 2000, p. 371 Search PubMed.
- J. P. Kutter, S. C. Jacobson and J. M. Ramsey, J. Microcolumn Sep., 2000, 12, 93 CrossRef CAS.
- L. A. Christel, K. Petersen, W. A. McMillan and M. A. Northrup, J. Biomed. Eng., 1998, 121, 22 Search PubMed.
- R. D. Oleschuk, L. L. Shultz-Lockyear, Y. B. Ning and D. J. Harrison, Anal. Chem., 2000, 72, 585–590 CrossRef CAS.
- A. B. Jemere, R. D. Oleschuk, F. Ouchen, F. Fajuyigbe and D. J. Harrison, Electrophoresis, 2002, 23, 3537–3544 CrossRef CAS.
- S. Ekstrom, J. Malmstrom, L. Wallman, M. Lofgren, J. Nilsson, T. Laurell and G. Marko-Varga, Proteomics, 2002, 2, 413 CrossRef CAS.
- J. Bergkvist, S. Ekstrom, L. Wallman, M. Lofgren, G. Marko-Varga, J. Nilsson and T. Laurell, Proteomics, 2002, 2, 422 CrossRef CAS.
- A. J. de Mello, Lab Chip, 2002, 2, 48N RSC.
- C. Yu, M. H. Davey, F. Svec and J. M. J. Frechet, Anal. Chem., 2001, 73, 5088 CrossRef CAS.
- C. Yu, M. C. Xu, F. Svec and J. M. J. Frechet, J. Polym. Sci. Part A—Polym. Chem., 2002, 40, 755 CrossRef CAS.
- F. E. P. Mikkers, F. M. Everaerts and T. P. E. M. Verheggen, J. Chromatogr., 1979, 169, 11 CrossRef CAS.
- R. L. Chien, Anal. Chem., 1991, 63, 2866 CrossRef CAS.
- R. L. Chien and D. S. Burgi, J. Chromatogr., 1991, 559, 141 CrossRef CAS.
- R. L. Chien and J. C. Helmer, Anal. Chem., 1991, 63, 1354 CrossRef CAS.
- D. S. Burgi and R. L. Chien, Anal. Biochem., 1992, 202, 306 CrossRef CAS.
- R. L. Chien and D. S. Burgi, Anal. Chem., 1992, 64, 489A CAS.
- R. L. Chien and J. C. Helmer, Anal. Chem., 1991, 63, 1354 CrossRef CAS.
- D. S. Burgi and R. L. Chien, Anal. Chem., 1991, 63, 2042 CrossRef CAS.
- S. C. Jacobson and J. M. Ramsey, Electrophoresis, 1995, 16, 481 CAS.
- J. P. Kutter, R. S. Ramsey, S. C. Jacobson and J. M. Ramsey, J. Microcolumn Sep., 1998, 10, 313 CrossRef CAS.
- H. Yang and R. L. Chien, J. Chromatogr. A, 2001, 924, 155 CrossRef CAS.
- J. Lichtenberg, E. Verpoorte and N. F. de Rooij, Electrophoresis, 2001, 22, 258 CrossRef CAS.
- N. P. Beard, C. X. Zhang and A. J. de Mello, Electrophoresis, 2003 Search PubMed , in press.
- C. X. Zhang and A. Manz, Anal. Chem., 2001, 73, 2656 CrossRef CAS.
- M. Gilges, Chromatographia, 1997, 44, 191 CAS.
- J. P. Quirino, S. Terabe, K. Otsuka, J. B. Vincent and G. Vigh, J. Chromatogr. A, 1999, 838, 3 CrossRef CAS.
- J. P. Quirino and S. Terabe, Science, 1998, 282, 465 CrossRef CAS.
- J. P. Quirino and S. Terabe, Anal. Chem., 2000, 72, 1023 CrossRef CAS.
- J. Palmer, D. S. Burgi, N. J. Munro and J. P. Landers, Anal. Chem., 2001, 73, 725 CrossRef CAS.
- Y. Sera, N. Matsubara, K. Otsuka and S. Terabe, Electrophoresis, 2001, 22, 3509 CrossRef CAS.
- P. A. Walker, M. D. Morris, M. A. Burns and B. N. Johnson, Anal. Chem., 1998, 70, 3766 CrossRef CAS.
- J. E. Prest, S. J. Baldock, N. Bektas, P. R. Fielden and B. J. Treves Brown, J. Chromatogr. A, 1999, 836, 59 CrossRef CAS.
- R. Bodor, M. Zuborova, E. Olvecka, V. Madajova, M. Masar, D. Kaniansky and B. Stanislawski, J. Sep. Sci., 2001, 24, 802 Search PubMed.
- R. Bodor, V. Madajova, D. Kaniansky, M. Masar, M. Johnck and B. Stanislawski, J. Chromatogr. A, 2001, 916, 155 CrossRef CAS.
- R. Bodor, D. Kaniansky, M. Masar, K. Silleova and B. Stanislawski, Electrophoresis, 2002, 23, 3630 CrossRef CAS.
- B. Grass, A. Neyer, M. Johnck, D. Siepe, F. Eisenbeiss, G. Weber and R. Hergenroder, Sens. Actuators B—Chem., 2001, 72, 249 CrossRef.
- M. Masar, D. Kaniansky, R. Bodor, M. Johnck and B. Stanislawski, J. Chromatogr. A, 2001, 916, 167 CrossRef CAS.
- E. Olvecka, M. Masar, D. Kaniansky, M. Johnck and B. Stanislawski, Electrophoresis, 2001, 22, 3347 CrossRef CAS.
- A. Wainright, S. J. Williams, G. Ciambrone, Q. F. Xue, J. Wei and D. Harris, J. Chromatogr. A, 2002, 979, 69 CrossRef CAS.
- J. E. Prest, S. J. Baldock, P. R. Fielden, N. J. Goddard and B. J. T. Brown, Analyst, 2002, 127, 1413–1419 RSC.
- J. E. Prest, S. J. Baldock, P. R. Fielden and B. J. T. Brown, Analyst, 2001, 126, 433 RSC.
- I. Rodriguez, H. K. Lee and S. F. Y. Li, Electrophoresis, 1999, 20, 118 CrossRef CAS.
- S. C. Jacobson, L. B. Koutny, R. Hergenroder, A. W. Moore and J. M. Ramsey, Anal. Chem., 1994, 66, 3472 CrossRef CAS.
- S. C. Jacobson, R. Hergenroder, A. W. Moore and J. M. Ramsey, Anal. Chem., 1994, 66, 4127 CrossRef CAS.
- K. W. Ro, K. Lim, H. Kim and J. H. Hahn, Electrophoresis, 2002, 23, 1129 CrossRef CAS.
- J. C. T. Eijkel, A. Prak, S. Cowen, D. H. Craston and A. Manz, J. Chromatogr. A, 1998, 815, 265–271 CrossRef CAS.
- N. Gottschlich, C. T. Culbertson, T. E. McKnight, S. C. Jacobson and J. M. Ramsey, J. Chromatogr. B, 2000, 745, 243 CrossRef CAS.
- V. Spikmans, S. J. Lane, B. Leavens, A. Manz and N. W. Smith, Rapid Commun. Mass Spectrom., 2002, 16, 1377 CrossRef CAS.
- A. J. de Mello, Lab Chip, 2001, 1, 24N RSC.
- B. M. Paegel, S. H. I. Yeung and R. A. Mathies, Anal. Chem., 2002, 74, 5092 CrossRef CAS.
- P. C. H. Li and D. J. Harrison, Anal. Chem., 1997, 69, 1564 CrossRef CAS.
- L. C. Waters, S. C. Jacobson, N. Kroutchinina, J. Khandurina, R. S. Foote and J. M. Ramsey, Anal. Chem., 1998, 70, 158 CrossRef CAS.
- P. Belgrader, D. Hensford, G. T. A. Kovacs, K. Venkateswaran, R. Mariella, F. Milanovich, S. Nasarabadi, M. Okuzumi, F. Pourahmadi and M. A. Northrup, Anal. Chem., 1999, 71, 4232 CrossRef CAS.
- P. Belgrader, M. Okuzumi, F. Pourahmadi, D. A. Borkholder and M. A. Northrup, Biosens. Bioelectron., 2000, 14, 849 CrossRef CAS.
- M. T. Taylor, P. Belgrader, B. J. Furman, F. Pourahmadi, G. T. A. Kovacs and M. A. Northrup, Anal. Chem., 2001, 73, 492 CrossRef CAS.
- S. C. Jacobson and J. M. Ramsey, Anal. Chem., 1996, 68, 720 CrossRef CAS.
- C. Wang, R. Oleschuk, F. Ouchen, J. Li, P. Thibault and D. J. Harrison, Rapid Commun. Mass Spectrom., 2000, 14, 1377 CrossRef CAS.
- K. Sakai-Kato, M. Kato and T. Toyo’oka, Anal. Chem., 2003, 74 Search PubMed , in press.
- Q. Xue, Y. M. Dunnayevskiy, F. Foret and B. L. Karger, Rapid Commun. Mass Spectrom., 1997, 11, 1253 CrossRef CAS.
Footnote |
| † For simplicity SPE will be discussed here, rather than in the following section on sample pre-concentration methods. The pre-concentration effect has proven very effective in extending the linear dynamic range of many existing analytical techniques, and has seen particular use in industries concerned with trace analysis. |
| This journal is © The Royal Society of Chemistry 2003 |