From nanocomposite to nanogel polymer electrolytes
M. M. E.
Jacob
,
Emily
Hackett
and
Emmanuel P.
Giannelis
*
Department of Materials Science and Engineering, Cornell University, Ithaca, NY 14853, USA. E-mail: epg2@cornell.edu
First published on 14th November 2002
Abstract
This review discusses recent advances in polymer electrolyte nanocomposites. In particular, we highlight our recent understanding on the structure and dynamics at the polymer/nanoparticle interface and attempt to correlate to macroscopic properties. We also discuss our current effort on nanogel electrolytes synthesized by incorporating layered nanoparticles into a gel. The nanogel electrolytes combine the high ambient conductivity of conventional gel electrolytes with the excellent stability toward the metal electrode and the good mechanical properties of nanocomposite electrolytes.
Introduction
Solid-state, high-density, rechargeable batteries are key to the development of several applications, from portable electronics to electric vehicles to back-up power sources in aircraft. Most present commercial batteries use liquid electrolytes.1 The electrolytes are either aqueous (as in the lead acid and Ni/Cd batteries) or organic (as in the Sony Li-ion cell). Polymer electrolytes are particularly attractive because they can lead to flexible, compact, laminated solid-state structures free from leaks and available in different geometries.2–4 A solid polymer electrolyte serves as both the separator to prevent the electrodes from coming into physical contact, and more importantly, as the ionic conductor.Research in polymer electrolytes has remained strong ever since Armand et al. proposed the use of PEO-Li salts as solid electrolytes.5 In addition to efforts at universities and national labs several large research and development programs have focused on developing polymer electrolyte based Li batteries. These include the Hydro-Quebec/3M program funded by the US Department of Energy for electric vehicles, the Yuasa/NEDO program for distributed energy storage use in Japan, and the JOULE program in Europe.
Despite the volume of R&D work going on at present, practical Li-batteries based on polymer electrolytes have remained elusive. Problems impeding their development include low conductivity of most solid polymer electrolytes at ambient temperature and reactivity with the lithium metal electrode in plasticized systems. While the former problem requires battery operation at temperatures above room temperature, the latter contributes to reliability and safety concerns.
Background
Scrosati and Vincent have divided polymer electrolytes into five categories:6Class 1: polymer/salt complexes. Classical examples are lithium salts like LiClO4 or LiCF3SO3 in a Li+-coordinating polymer like poly(ethylene oxide), PEO.7–9
Class 2: plasticized systems, in which small amounts of liquids are added to Class 1 electrolytes. Plasticized polymer electrolytes represent a compromise between polymer and liquid electrolytes. Liquid plasticizers, however, typically lead to worsening of the mechanical properties of the electrolyte and increasing reactivity towards the metal electrode.
Class 3: gel electrolytes, formed by incorporating an electrolyte solution into a polymer matrix.10,11 Since the electrolyte molecules can preferentially solvate ions, coordinating polymers like PEO are no longer necessary and may be replaced by more robust polymers like poly(vinylidene difluoride), PVDF, polyacrylonitrile, PAN, and poly(methyl methacrylate), PMMA. The small electrolyte molecules form a network through which ion conduction occurs, with the host polymer providing mostly structural support. Gel electrolytes offer high ambient conductivities but suffer the same disadvantages as the plasticized electrolytes — namely release of volatiles and increased reactivity towards the metal electrode.
Class 4: “polymer-in-salt” or “rubbery” electrolytes, discovered by Angell and co-workers.12,13 The salt typically has a low glass transition temperature so that a rubbery material is formed when the salt and the polymer are mixed together. Although high ambient conductivities have already been realized, the salt tends to crystallize at lower temperatures, preventing their practical use.
Class 5: composite electrolytes, in which inorganic(ceramic) particles are introduced into a polymer electrolyte.14–20 In addition to improvements in the conductivity, the mechanical strength and the interfacial stability are also enhanced in the composites. Though the conductivities of composite electrolytes are dramatically improved from those without the nanoparticles, ambient temperature conductivities still remain relatively low for real practical applications.
Another useful way to categorize polymer electrolytes is according to the dominant conduction mechanism.21 In Class 1 or 2, ion transport is coupled to the relaxation of the host polymer. Plasticizing the host polymer by small molecules increases the segmental motion and decreases the glass transition temperature leading to higher conductivity. In a plot of conductivity vs. reciprocal temperature a curve typically indicates a conductivity mechanism in which ionic motion is coupled to the relaxation of the polymer conforming to the Vogel–Tamman–Fulcher (VTF) relationship. In contrast, in Class 3, 4 or 5, ionic transport appears to be decoupled from the polymer relaxation mechanisms, leading to an Arrhenius-type, straight-line plot.
Nanocomposite electrolytes
Despite the activity in the area of (nano)composite electrolytes a complete understanding about the materials or the dominant conduction mechanism has yet to emerge. A major challenge in further developing new electrolytes is the lack of even simple structure–properties models. In the absence of models relating properties to structure, progress in nanocomposite design has remained largely empirical. Similarly, predictions of the ultimate material limits or maximum performance are also difficult at present.A working hypothesis is that the nanoparticles prevent chain reorganization, resulting in a reduction in polymer crystallinity and thus an increase in ionic conductivity. The reduction in crystallinity may be the result of Lewis acid–base interactions between the surface of the nanoparticles and the polymer chains. This allows Li+ ions to move more freely either on the surface of the nanoparticles or through a low-density polymer phase at the interface, which results in enhanced ionic conductivity.17,18
One interesting class of nanocomposite electrolytes is made by intercalating polymers such as PEO between the individual layers of a layered host such as a layered silicate (Fig. 1).22–34 The resulting materials combine relatively high ambient conductivity with single ion conduction characteristics. In these nanocomposite electrolytes, 1 nm thick, negatively-charged silicate layers are separated by interlayers, which contain charge-compensating Li-cations and polymer chains. Because of their massive lateral dimensions, the silicate anions are immobile.
 | ||
| Fig. 1 Schematic of nanocomposite electrolytes containing layered nanoparticles. | ||
An Arrhenius plot of the in-plane ionic conductivity of the PEO/Li+-silicate nanocomposite prepared by melt intercalation containing 40 wt% polymer is shown in Fig. 2.30 For comparison the conductivity of a conventional LiBF4/PEO electrolyte with a comparable O/Li ratio is also shown. As expected the conductivity of the LiBF4/PEO electrolyte decreases several orders of magnitude below the melting temperature. In contrast, the conductivity of the nanocomposite exhibits only a weak temperature dependence over the same temperature range. In addition, the apparent activation energy in the nanocomposite (2.8 kcal mol−1) is similar to that of the molten polymer electrolyte suggesting that the mobility of Li+ in the nanocomposites is comparable to that in the bulk molten electrolyte. Furthermore, the out-of-plane conductivity of the hybrid is only an order of magnitude lower than the in-plane conductivity.
 | ||
| Fig. 2 Arrhenius plots of the ionic conductivity of bulk LiBF4-PEO and PEO nanocomposites.30 | ||
The conductivity of the melt-intercalated hybrid is much higher and more isotropic than that reported previously for PEO/silicate nanocomposites. Note, however, that those samples were prepared by solution intercalation and were thoroughly washed to remove excess polymer.22–28 The enhanced ionic conductivity in our samples is probably due to the presence of excess polymer (40 vs. 30 wt%) that provides an easy conduction path between host particles. Removing excess polymer could lead to chain depletion from the edges of the silicate particles hindering the interparticle mobility of Li+ ions. Furthermore, the tendency of silicate particles to orient parallel to the film surface when processed from solution is minimized in the dry-pressed samples used in our study leading to more isotropic properties.
Even though the conductivity is prohibitively low for practical applications, due to their structural regularity, these nanocomposites are excellent model systems to probe and understand the structure and dynamics at the interface and relate these molecular features to macroscopic properties such as ionic conductivity.35 By combining computer simulations with experiments a detailed picture of the structure and mobility of the polymer and the ions at the interface has started to emerge. Below we summarize our recent results on nanocomposite electrolytes.
Using Monte Carlo, MC, and Molecular Dynamics, MD, computer simulations we have shown that the polymer chains at the interface organize into discrete layers parallel to the crystalline inorganic surface (Fig. 3 and 4).36–38 In the absence of a solvent the cations are basically pinned to the inorganic surface rather than being coordinated with PEO. In the solvated nanocomposites, some cations still reside near the surface but a large number of the Li cations have now moved away from the surface. The cations coordinate primarily to the solvent molecules or the surface but not to the polymer chains. In addition, MD simulations show that the solvated Li ions are much more mobile than those pinned to the surface.
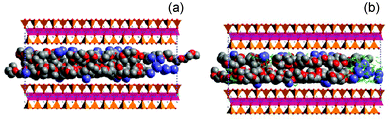 | ||
| Fig. 3 Snapshot of (a) dry and (b) solvated PEO-Li-silicate nanocomposites. The silicate crystals are shown as polyhedra, Li+ as blue spheres, PEO as the grey and red spheres and solvent as the green sticks.37 | ||
 | ||
| Fig. 4 Density profiles for PEO-Li-silicate nanocomposite (a) dry and (b) hydrated. Black line cation, green line PEO oxygen, red line PEO carbon and dashed red line represents the water oxygens. The vertical gray lines represent the outer edges of the silicate oxygen surface.37 | ||
Fig. 3 along with the corresponding density profiles, shown in Fig. 4, illustrate many of the features including the preferred location of ions and the disordered polymer structure. A bilayer structure is quite obvious with the thickness of each layer approximately equal to a PEO chain width. In spite of this ordering, the polymer chains retain a disordered, liquid-like structure consistent with our small angle neutron scattering, SANS, experiments.39 SANS profiles from 15 K to room temperature can only be simulated by assuming a disordered polymer configuration.
In the absence of a solvent the cations are bound to the wall, about 0.15 nm from the center of the surface oxygen atoms, or about 0.45 nm from the central plane of the metal atoms, in very good agreement with the NMR data discussed below. When a solvent like water is present some gallery ions are located <0.2 nm from the surface but there also exists a large number of ions inside the galleries. The location of the cations in the interlayer gallery becomes very important as it affects their mobility and, thus, ionic conductivity.
A complete statistical picture of the gallery species and their coordination is given by combining density profiles with radial pair correlation functions. From pair correlation functions the coordination of gallery ions can be calculated. In a dry system (i.e. in the absence of any solvent) gallery ions are equally coordinated to PEO and surface oxygen atoms. When water is present, however, the gallery ions are primarily coordinated to water molecules and to some extent to the surface oxygens while PEO is outside the coordination sphere of the cations.
Our tools for dynamics include NMR and dielectric relaxation spectroscopy.32,33,40,41 Using surface-sensitive cross polarization with spin-echo experiments in collaboration with D. Zax we have shown that the polymer chains at the interface exhibit a wide range of relaxation times. On one hand, some polymer segments at the interface are more mobile (i.e. more liquid-like) than the bulk polymer at the same temperature. On the other hand, the interface polymer appears to exhibit solid-like character even at temperatures well above the bulk Tg. Similar behavior was observed using dielectric spectroscopy. The polymer at the interface exhibits two relaxation modes. The new mode is much faster than the bulk-polymer α relaxation, with much weaker temperature dependence (Fig. 5).
 | ||
| Fig. 5 Relaxation times as a function of temperature for bulk (green line) and interface polymer in nanocomposites (symbols).41 | ||
Fig. 6 compares the 2H line shapes for bulk PEO and in the nanocomposite. At low temperature, where the local segmental motion of the polymer is quiescent, a typical powder pattern is observed for both the bulk d-PEO and the intercalated d-PEO, indicating the spatial motion of the deuterium is slower than the time scale of the experiment. However, as the temperature increases, a central peak develops at lower temperatures in the intercalated d-PEO than in the bulk polymer. Additionally, the breadth of the central peak from the intercalated d-PEO is substantially narrower than that of the bulk polymer. This central peak results from increased segmental motion, which causes temporal averaging of the signal. The temperature dependence of the line shapes indicates that the intercalated chains have more “freedom” to sample a distribution of local chain configurations, resulting in increased signal averaging. However, at the highest temperatures, the intercalated d-PEO still exhibits a broad base structure reminiscent of the powder pattern, whereas the bulk polymer shows complete motional narrowing of the signal. This indicates that even though the local segmental motion of the intercalated d-PEO appears more dymamic at lower temperatures, the silicate layers still restrict motion such that some local configurations of the chain are not acccessible, and thus complete signal averaging is not possible.
 | ||
| Fig. 6 Comparison of 2H NMR spectra for bulk deuterated, d-PEO and d-PEO nanocomposites as a function of temperature.33 | ||
Intuitively, one might have expected the polymer at the interface to be less mobile compared to the bulk polymer, as “confinement” of the polymer chains within a few nanometers should increase their solid-like character and decrease their mobility. In fact, a decrease in mobility of polymers near the interface has been suggested to account for the presence of two glass transitions (the second at a higher temperature) in polymer composites.42
The results on polymer dynamics in the nanocomposites can be rationalized using the computer simulations discussed above. As the polymer organizes into discrete layers, areas of high and low density coexist in close proximity to each other. The wide range of relaxation times found experimentally reflects the density variation as segments of low and high density would exhibit fast and slow dynamics, respectively. The absence of crystallization and the liquid-like character of the interface polymer definitely support the enhanced ionic conductivity observed in the nanocomposites.
The linewidth of Li NMR spectra unequivocally supports our computer simulation results discussed previously regarding the position of gallery Li ions. At low temperatures where dynamics are absent simulations of the expected 7Li linewidth and shape suggest that the Li+ ions reside along the silicate surface and that the intercalated PEO chains reside primarily towards the middle of the interlayer. Simulations of the NMR spectra provide an average distance between the Li cations and the Fe3+ ions in the octahedral sheet of 0.4 nm, which suggests that the cations are strongly interacting and virtually pinned to the silicate surface in good agreement with the computer simulation results. These results contrast with the generally accepted viewpoint, which suggests that PEO mediates the interactions between the cation and the negatively charged silicate surface and draws the cation away from the surface.
From nanocomposite to nanogel electrolytes
To date, development of new generation ambient Li-batteries has been based almost exclusively on gel electrolytes.43 Several organizations including Bellcore (USA), Mead (USA), Valence (USA) and Yuasa (Japan) have announced lithium batteries based on gel electrolytes. One of the remaining challenges in these systems is to increase the stability with the metal electrode and decrease the interfacial impedance. Gas evolution while cycling and poor mechanical strength makes the use of gel electrolytes in Li-polymer batteries challenging. Nanogel polymer electrolytes, on the other hand, synthesized by incorporating layered nanoparticles in a gel matrix, combine the high ambient conductivity of conventional gel electrolytes with the excellent stability toward the metal electrode and the good mechanical properties of nanocomposite electrolytes.Nanogel polymer electrolytes combine the advantages of both gel and composite electrolytes. The nanogels are synthesized by adding layered silicate nanoparticles like Li-fluorohectorite or Li-fluoromica to gel electrolytes based on poly(vinylidene fluoride) (PVDF) or PVDF-hexafluoropropylene copolymer (PVDF-HFP) containing propylene carbonate (PC) and/or ethylene carbonate (EC) as the solvent and LiCF3SO3 as the salt.44 Both X-ray diffraction and atomic force microscopy suggest that the silicate layers are well dispersed in the gel. The room temperature conductivity (Fig. 7) of 1 M LiCF3SO3 in EC and PC Li-fluoromica containing 5 wt% silicate is 4.4 × 10−4 S cm−1 and becomes 1 × 10−3 S cm−1 at 100 °C. The corresponding conductivities for the neat gel electrolyte (i.e. system containing no nanoparticles) are 1.9 × 10−3 and 5.2 × 10−3 S cm−1, respectively. Interestingly the conductivity of the nanogels remains unchanged after several temperature cycles while that of the gel electrolyte decreases as the solvent evaporates after each thermal cycling.
 | ||
| Fig. 7 Arrhenius plots for gel (1 M LiCF3SO3 in PVDF-co-HFP containing EC/PC) and nanogel electrolytes containing 5 wt% synthetic fluoromica. | ||
Thermogravimetric analysis of the nanogels, held at 100 °C for 10 h, shows a weight loss in the range of 7–14%. The gel electrolyte under similar conditions loses about 40% with 90% of the weight-loss taking place within the first 30 min. It appears that the nanoparticles act as a physical and chemical barrier to the solvent molecules preventing them from evaporating and keeping the solvent in the nanogel. Additionally, in contrast to the gel electrolytes, where the interfacial resistance increases with time, in the nanogels it remains constant for several days, suggesting exceptional stability with the Li metal electrode.
In addition to the above characteristics, the mechanical properties of the nanogel electrolytes are significantly improved. The nanogels exhibit a rubbery behavior with a storage modulus of about 1 GPa (an order of magnitude higher than the corresponding gels). Rubbery behavior and compliance is an important requirement for solid electrolyte materials as charging and discharging during battery operation causes large volumetric changes. Moreover, the nanogels can be processed at much higher temperature as the drop in the modulus due to polymer melting now takes place at 160 °C (the corresponding temperature for the conventional gels is 135 °C).
The nanogels based on layered nanoparticles appear superior to other composite gel systems. For example, gels containing BaTiO3 nanoparticles show a lower conductivity and a much higher weight loss (26% at 100 °C) than the silicate nanogels, suggesting that the dimensionality of the nanoparticles has a large effect on the properties. The combination of high ambient conductivity, mechanical robustness and stability towards Li makes the nanogels very attractive candidates for practical Li batteries.
Acknowledgements
This work was supported by AFOSR. We also acknowledge the use of CCMR facilities supported by NSF-MRSEC. We thank our colloborators S. Anastasiadis and D. Zax.References and notes
- H. Jain, J. O. Thomas and M. S. Whittingham, eds., MRS Bull., 2000, 3, 11 Search PubMed.
- P. G. Bruce, ed., Solid State Electrochemistry, Cambridge University Press, Cambridge, 1995 Search PubMed.
- S. Greenbaum, ed., Electrochim. Acta, 1995, 13–14, 2137.
- B. Scrosati, Chim. Ind. (Milan), 1997, 79, 463 Search PubMed.
- M. B. Armand, J. M. Chabagno and M. Duclot, in Fast Ion Transport in Solids, P. Vashista, J. N. Mundy and G. K. Shenoy, eds., 1979, p. 131 Search PubMed.
- B. Scrosati and C. A. Vincent, MRS Bull., 2000, 25, 28 Search PubMed.
- M. B. Armand, Adv. Mater., 1990, 2, 278 CrossRef CAS.
- Z. Gadjourova, Y. G. Andreev, D. P. Tunstall and P. G. Bruce, Nature, 2001, 412, 520 CrossRef CAS.
- T. H. Epps, T. S. Bailey, H. D. Pham and F. S. Bates, Chem. Mater., 2002, 14, 2002 CrossRef.
- M. Alamgir and K. M. Abraham, in Lithium Batteries: New Materials, Developments, and Perspectives, G. Pistoia, ed., Elsevier, Amsterdam, 1994 Search PubMed.
- J.-M. Tarascon, A. S. Gozdz, C. Schmutz, F. Shokoohi and P. C. Warren, Solid State Ionics, 1996, 86, 49 Search PubMed.
- C. A. Angell, C. Liu and E. Sanchez, Nature, 1993, 362, 137 CrossRef CAS.
- C. A. Angell, K. Xu, S. S. Zhang and M. Videa, Solid State Ionics, 1996, 86-88, 17 Search PubMed.
- W. Wieczorek, K. Such, J. Przyluski and Z. Florianczyk, Synth. Met., 1991, 45, 373 CrossRef CAS.
- B. Kumar and L. G. Scanlon, J. Power Sources, 1994, 52, 261 CrossRef CAS.
- G. B. Appetechi, F. Croce and B. Scrosati, J. Power Sources, 1997, 66, 77 CrossRef.
- F. Croce, G. B. Appetechi, L. Persi and B. Scrosati, Nature, 1998, 394, 456 CrossRef CAS.
- R. Curini, F. Croce, A. Martinelli, L. Persi and B. Scrosati, J. Phys. Chem. B, 1999, 103, 10632 CrossRef.
- M. Riley, P. S. Fedkiw and S. A. Khan, J. Electrochem. Soc., 2002, 149, A667 CrossRef.
- C. J. Leo, G. V. S. Rao and B. V. R. Chowdari, Solid State Ionics, 2002, 148, 159 Search PubMed.
- M. A. Ratner, P. Johansson and D. F. Shriver, MRS Bull., 2000, 25, 31 Search PubMed.
- E. Ruiz-Hitzky and P. Aranda, Adv. Mater., 1990, 2, 545 CrossRef CAS.
- P. Aranda and E. Ruiz-Hitzky, Chem. Mater., 1992, 4, 1395 CrossRef CAS.
- P. Aranda, J. C. Galvan, B. Casal and E. Ruiz-Hitzky, Electrochim. Acta, 1992, 37, 1573 CrossRef CAS.
- E. Ruiz-Hitzky, Adv. Mater., 1993, 5, 334 CrossRef CAS.
- J. Wu and M. M. Lerner, Chem. Mater., 1993, 5, 835 CrossRef CAS.
- E. Ruiz-Hitzky and P. Aranda, Anal. Quím. Int. Ed., 1997, 93, 197 Search PubMed.
- J. C. Hutchinson, R. Bissenur and D. F. Shriver, Chem. Mater., 1996, 8, 1597 CrossRef.
- E. P. Giannelis, Adv. Mater., 1996, 8, 29 CAS.
- R. A. Vaia, S. Vasudevan, W. Krawiec, L. G. Scanlon and E. P. Giannelis, Adv. Mater., 1995, 7, 154 CAS.
- W. Krawiec, L. G. Scanlon, J. P. Fellner, R. A. Vaia, S. Vasudevan and E. P. Giannelis, J. Power Sources, 1995, 54, 310 CrossRef CAS.
- S. Wong, S. Vasudevan, R. A. Vaia, E. P. Giannelis and D. B. Zax, J. Am. Chem. Soc., 1995, 117, 7568 CrossRef CAS.
- S. Wong, R. A. Vaia, E. P. Giannelis and D. B. Zax, Solid State Ionics, 1996, 86–88, 547 Search PubMed.
- R. A. Vaia, B. B. Sauer, O. K. Tse and E. P. Giannelis, J. Polym. Sci. B: Polym. Phys., 1997, 35, 59 Search PubMed.
- E. P. Giannelis, R. Krishnamoorti and E. Manias, Adv. Polym. Sci., 1999, 138, 107 Search PubMed.
- E. Manias, A. Z. Panagiotopoulos, D. B. Zax and E. P. Giannelis, CMS Workshop Lectures, The Clay Minerals Society, Aurora, CO, 2000, vol. 13 Search PubMed.
- E. Hackett, E. Manias and E. P. Giannelis, Chem. Mater., 2000, 12, 2161 CrossRef CAS.
- J. Bujdák, E. Hackett and E. P. Giannelis, Chem. Mater., 2000, 12, 2168 CrossRef CAS.
- R. Krishnamoorti and E. P. Giannelis, unpublished work.
- D. B. Zax, D.-K. Yang, P. A. Santos, H. Hegemann, E. P. Giannelis and E. Manias, J. Chem. Phys., 2000, 112, 2945 CrossRef CAS.
- S. H. Anastasiadis, K. Karatasos, G. Vlachos, E. Manias and E. P. Giannelis, Phys. Rev. Lett., 2000, 84, 915 CrossRef CAS.
- G. Tsagaropoulos and A. Eisenberg, Macromolecules, 1992, 25, 1267 CrossRef.
- K. Murata, S. Izuchi and Y. Yoshihisa, Electrochim. Acta, 2000, 45, 1501 CrossRef CAS.
- In a typical experiment 2 g of PVDF-HFP was first dissolved in 10 ml THF and then added to 5 ml of a 1 M solution of LiCF3SO3 in PC/EC (50∶50) containing the appropriate amount of layered silicate nanoparticles. The mixture was poured into a Petri dish and dried in vacuo at 70 °C for 24 h.
| This journal is © The Royal Society of Chemistry 2003 |