Tube ageing as an important quality factor in the direct determination of Sb in solid samples by ultrasonic slurry sampling-GFAAS
M. J.
Cal-Prieto
,
A.
Carlosena
*,
J. M.
Andrade
,
P.
López-Mahía
,
S.
Muniategui
and
D.
Prada
Department of Analytical Chemistry, University of A Coruña, Campus A Zapateira, E-15071 A Coruña, Spain. E-mail: alatzne@udc.es; Fax: +34-981-167065
First published on 3rd December 2002
Abstract
Although graphite deterioration occurs in all the GFAAS-based techniques, this issue is of most concern whenever solid samples are introduced in the graphite tube as slurries because of the different behaviour they show regarding solutions. In this work several factors were considered to evaluate their influence on the tube ageing, the nature and amount of sample matrix introduced into the atomiser (and so the concomitants) being the most important ones. Scanning electron microscopy and X-ray dispersive analyses were employed to assess the L’vov platform degradation, not only regarding its morphology but the chemical nature of the deposits observed on it. Three modes of sample handling were studied: aqueous solutions, acid extracts and slurries. Changes in the useful lifetime of the tube were studied when analysing Sb in different matrices (soils, sediments, coals, coal fly ashes and slag) and empirical solutions were presented to expand the lifetime of the tube. Coal revealed itself to be the most difficult matrix to deal with and several slurry methodologies were assayed: introduction of an oxidative step, external ashing, enlargement of the temperature program and two extracting media.
Introduction
One of the main factors which can affect the graphite furnace atomic absorption spectrometry (GFAAS) determination is ageing of the graphite tube. Besides, the high cost of pyrolytic graphite tubes greatly affects the cost of the analysis. Therefore, implementing strict control of the atomiser lifetime can attain substantial savings and analytical quality improvements.1 The useful lifetime of the tube is defined as the maximum number of runs at still acceptable signal quality, and it is estimated by repeating the determinations in the same tube.2 While tube endurance is typically determined by the number of firings until the tube actually breaks, the analytical useful lifetime may be substantially shorter. Thus, Welz et al.3 defined the analytical useful lifetime of the graphite tubes as the number of firings which can be made until the analytical signal drops to about 80% of its initial value and/or the relative standard deviation (RSD) begins to deteriorate significantly.The lifetime of the graphite tubes employed for GFAAS determination has been associated to several main factors:3 (a) the temperatures to which a tube is heated during the pyrolysis, atomisation and cleaning steps;4 (b) the time these temperatures are applied; (c) the chemical reactions the sample matrix and the graphite undergo;2,5 (d) the gas phase composition and purge flow-rate; (e) the amount of sample; and (f) sample type, i.e., aqueous, acid extract, slurry or solid. The latter is of great concern as remarkable differences may be found in the lifetime of the tubes when liquid sampling or slurry/solid samples are analysed.
The direct analysis of solid samples by GFAAS (SS-GFAAS) has gained acceptance during the last few years. Of the two well-established procedures, i.e., solid sampling and slurry-sampling, the latter is the preferred one as it combines the significant advantages of the direct solid and liquid sampling methods.6 Notwithstanding, one of its shortcomings is the acceleration of the damage of the graphite tube, which was reported to affect repeatability and sensitivity as well as to interfere with the background correction.7–9
Several approaches have been attempted to lengthen the lifetime of the tube when slurries are analysed by GFAAS. (a) The use of diluted acids10 (stressing the role of the liquid media). (b) The use of suitable temperatures for the mineralisation and/or additional cleaning steps which, when properly selected, can be effective enough to avoid damages in the pyrolytic material.11,12 (c) A previous mild ashing of the sample when analysing materials with high organic contents, although this additional step enlarges the analysis.13,14 (d) The strengthening of the ashing step using an alternative gas flow to achieve a more complete oxidation of the carbonaceous materials.15 Nevertheless, several drawbacks were observed such as the atomic signal depletion16 and the need for a second ashing step.15 Unfortunately, the use of these alternative gases sometimes reduced the atomiser lifetime, mainly for ashing temperatures higher than 600 °C, because graphite becomes steadily more porous when its surface gets oxidised.17
In the present work, the deterioration of the graphite tubes when determining Sb in environmental solid samples as a slurry by GFAAS is investigated by scanning electron microscopy (SEM) combined with X-ray elemental analysis. Different experimental conditions for slurry preparation are assayed and related to the damages observed on the atomiser. A comparison with the effects caused by the analysis of aqueous and acid extract samples is also performed.
Antimony was selected because of its environmental interest. Due to its natural low levels in the earth’s crust, it can be considered as a potential marker of environmental metallic pollution. Besides, few studies have determined this element in complex solid matrices such as soils, sediments, coal, coal combustion residues, etc.
Experimental
Apparatus
A PerkinElmer (Überlingen, Germany) Model 4100 deuterium corrected atomic absorption spectrometer, equipped with a HGA-700 graphite furnace, an AS-70 autosampler and a USS-100 ultrasonic slurry sampler, was employed throughout. Ar was used as the purge gas and pyrolytic graphite tubes with pre-inserted L'vov platforms as atomisers (Z-tek, P/N 4090-0506, Amsterdam, The Netherlands). A hollow cathode lamp (217.6 nm Sb wavelength, 0.2 nm spectral bandwidth) was employed. SEM micrographs were obtained with a Jeol JSM-6400 scanning electron microscope equipped with an X-ray energy dispersion elemental analysis system. A Technics Plasma GmbH 200-G low temperature asher (LTA) was used for ashing coal samples. The LTA conditions were 1.35 mbar, 400 W and 150 °C, N2 as inert gas and O2 as oxidant.Reagents and materials
All the reagents were of analytical grade. High purity water (Milli-Q Water System, Millipore, Madrid, Spain) was employed. Sb standards were prepared on a daily basis by diluting appropriate aliquots of a 1000 mg l−1 stock metal solution (Panreac, Barcelona, Spain). The acids (HNO3, HCl, HF) were of Suprapur grade (J.T. Baker, Deventer, The Netherlands) and the H2O2 grade reagent from Panreac.All glassware, plasticware, pipette tips and storage bottles were soaked in 10% (v/v) HNO3 for 24 h and rinsed with high purity water at least three times prior to use.
Twelve certified reference materials (CRMs) were used throughout: two NCR CRM soils (GBW07401 and GBW07409; National Research Council of Certified Reference Materials, China), two NCR CNRC marine sediments (BCSS-1 and PACS-1; National Research Council of Canada), two BCR CRM geological materials (a calcareous loam soil, CRM 141, and an estuarine sediment, CRM 277; Community Bureau of Reference, Brussels, Belgium), four NIST (National Institute of Standards and Technology, USA) coal related materials (coal fly ashes, SRMs 1633a and 1633b; coals, SRMs 1635 and 1632b) and two NIST atmospheric related materials (urban dust, SRM 1649 and urban particulate matter, SRM 1648). No CRMs were available for slag, so samples from coal combustion power plants were employed.
Sample preparation and analytical procedure
The addition calibration method was always employed for quantification. This method is implemented in the spectrometer software and it holds characteristics from the two traditional calibration modes (aqueous and standard addition calibrations). It implies the development of one standard addition regression for the first sample, and then new samples are quantified by correcting the original regression line by the absorbance of the corresponding new sample.19
Results and discussion
In this work, the ‘maximum number of runs’ which determine the useful lifetime of the tube, following Hauptkorn and Krivan,2 was established by using control charts. Briefly, a control chart is a graphic on which the values of the analytical response (absorbance) are plotted sequentially with respect to time. This representation is defined by a central and two outer lines (also known as action limits). In order to determine them a preliminary step is needed, which consists in measuring a control sample several times (minimum n = 6) while the graphite tube remains new, and computing the average and standard deviation values. Then, two action limits are easily calculated (upper and lower, UCL and LCL; 99% confidence interval).22 Next, a control sample is periodically measured while the tube is being used. The distribution of the control values with respect to the limits provides valuable information. If none of them falls outside the limits, it is concluded that no major assignable causes of variation arose during the measurements. Whenever any point exceeded the action limits, the instrument, sample preparation, weight, etc., were reviewed and the analysis repeated. If the new response lies within the action limits the atomiser is still used. The worst situation is that connected with responses below the LCL which, when confirmed, suggest the end of the lifetime of the tube (which would be verified by evaluating its accuracy). As a consequence, the useful lifetime of the tube can be established as the maximum number of firings that gave an analytical response within the control limits.In this work, the so-called ‘means chart’ was depicted using the average of three consecutive absorption measurements of a control sample. Another type of control chart was simultaneously developed considering the standard deviation for the replicates of each analysis. Thus, the within- and between-run precision of the analyses is also considered.
In the following sections, the effects of several solid matrices on the graphite tube deterioration are assessed when they are analysed as slurries, maintaining one control chart for each tube being used (only one chart is given for each type of sample). A comparison with the effects caused by aqueous and acid extract samples is also performed. Note that these studies are performed for Sb determination and there is no guarantee that exactly the same behaviour will occur when other metals are determined.
Aqueous and acid extract samples
The degradation of the L'vov platforms employed to analyse aqueous Sb standards and soil/sediment acid extracts was monitored using a control chart for each tube. Their performance was highly similar, leading to tube lifetimes of 375–400 cycles, ending with a progressive absorbance shift down (Fig. 1). A positive fact is that severe changes were not observed for the precision (standard deviation charts). | ||
| Fig. 1 Control charts employed to monitor the tube behaviour during the analysis of Sb aqueous standards and soil/sediment acid extracts (UCL = upper control limit, LCL = lower control limit). | ||
Tube ageing was reflected on both the atomic and background peak shapes. Hence, changes on the atomic peak profile (Sb determination in either aqueous and geological acid extract) as well as on the L'vov platform were assessed. Fig. 2 shows the atomic peaks for a 20 ng ml−1 Sb aqueous solution and for a soil/sediment acid extract observed for a new and old tube. The atomic peak shape of the aqueous solutions spreads and broadens after roughly 300 atomisation cycles, and is also noisier. Regarding the soil/sediment acid extracts, the appearance time for both the atomic and background signals gets higher as the tube gets older; this may be due to an increase in the tube porosity as the coating degradation delays the analyte atomisation. High background signals (giving a slight atomic signal over-correction) were obtained either for new and old tubes when acid extracts were determined, maybe by the solubilisation of the concomitants during the digestion of these complex matrices.
 | ||
| Fig. 2 Atomic peaks for (A) a 20 ng ml−1 Sb aqueous solution, (B) a soil/sediment acid extract and (C) a soil slurry (90 mg ml−1). | ||
In order to evaluate the influence of the tube behaviour on the quantification of aqueous standards and soil/sediment samples, calibration curves were compared using new and old tubes. For aqueous standards a significant reduction (95% confidence) on the slope was obtained using peak area (new tube, A s = 0.0031 + 0.0053[Sb] versus old tube, A s = 0.0060 + 0.0039[Sb]). However, peak height values kept the slope almost unchanged. The standard addition method was required and no significant differences were found for the slopes obtained with new nor old tubes. However, the use of peak height led to a better sensitivity (A s = 0.0113 + 0.0040[Sb] versus A = 0.0227 + 0.0079[Sb]). Accordingly, the use of peak height is recommended for quantification purposes.
The degradation of the atomiser as a function of the nature of the aliquot being injected (aqueous or acid extract) was evaluated by observing how the morphology of the pyrolytic platforms changed using scanning electron microscopy (SEM). Fig. 3 (10×- and 100×-magnification) shows a L'vov pyrolytic platform either before and after the analysis of aqueous and soil/sediment acid extract solutions. The new tube presents the typical lamellar structure (Fig. 3(A)) which is very compact and thick, enabling it to reduce the permeability and reactivity of the graphite surface.3
 | ||
| Fig. 3 Inner surface (10×- and 100×-magnification) of a L’vov pyrolytic platform when new (A), and used for the analysis of (B) aqueous Sb standard solutions, (C) soil/sediment acid extracts and (D) soil/sediment slurries. | ||
A homogenous degradation is noticed across the whole of the platform after 150 atomisations when analysing aqueous solutions (about half the lifetime of the tube observed for these samples). The surface seems to be coated by a new layer containing small pores (Fig. 3(B)) which persisted throughout the useful lifetime of the tubes. Fig. 3(C) shows the morphology when acid extracts of geological samples were analysed. A uniform, longitudinal degradation of the lamellar graphite structure can be observed throughout a large area of the platform. Despite these slight differences in the morphology of the degraded tube, the lifetimes observed for acid extracts and aqueous solutions were very similar.
Additionally, X-ray dispersive energy analysis (EDS) of the graphite surface revealed a continuous, similar profile for both types of sample (Fig. 4(A)), which can be attributed to the pyrolytic coating of the graphite tubes. Fig. 4(B) gives an amplified view of this profile in order to reveal that only Cu was detected when acid extracts were analysed; this fact will also be the case for other types of sample, as will be seen below. A sound reason was not found to explain this.
 | ||
| Fig. 4 Elemental analysis by X-ray dispersive energy analysis of the inner surface of the L'vov platform from (A) a new tube and (B) a tube used for aqueous solution/acid digests. | ||
Slurries of soils and sediments
During recent years, geological samples (mostly soils and sediments) have been broadly analysed as slurries.6 In spite of this the analysis of soil/sediment slurries presents several pitfalls,8 among them being the acceleration in tube damage.The control charts developed to monitor the tube behaviour (Fig. 5) revealed a useful lifetime of the tubes of about 90–100 firings (which contrasts with that of the previous section), without significant losses of precision. During the useful lifetime of the tubes no significant variation in the slopes of the standard addition lines was observed, giving satisfactory results for several certified reference materials (see Table 1). Whenever the absorbance signals got outside the action limits, the slopes of the calibration lines diminished as well as the calculated recoveries (<60%).
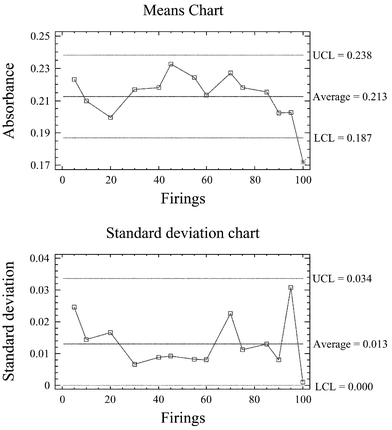 | ||
| Fig. 5 Control charts employed to monitor the tube behaviour during the analysis of soil/sediment slurries (UCL = upper control limit, LCL = lower control limit). | ||
| Sb concentration/µg g−1 (± CI) | |||
|---|---|---|---|
| Material | Content found | Certified value | Recovery (% ) |
| a Non certified value. | |||
| Soil GBW07401 | 0.82 ± 0.08 | 0.87 ± 0.03 | 94 |
| Soil GBW07409 | 0.24 ± 0.02 | 0.21 ± 0.03 | 114 |
| Marine sediment BCSS-1 | 0.58 ± 0.02 | 0.59 ± 0.06 | 98 |
| Marine sediment PACS-1 | 156 ± 16 | 171 ± 14 | 91 |
| Calcareous soil CRM 141 | 0.70 ± 0.06 | (0.70 ± 0.3)a | (100) |
| Estuarine sediment CRM 277 | 3.0 ± 0.2 | (3.9 ± 1.0)a | (77) |
| Coal fly ash SRM 1633a | 7.7 ± 0.1 | 6.8 ± 0.4 | 113 |
| Coal fly ash SRM 1633b | 5.1 ± 0.2 | (6)a | (85) |
| Coal SRM 1635 | 0.12 ± 0.01 | (0.14)a | (86) |
| Coal SRM 1632b | 0.22 ± 0.01 | (0.24)a | (92) |
| Urban dust SRM 1649 | 25.4 ± 1.1 | (29.9)a | (85) |
| Urban particulate matter SRM 1648 | 47.6 ± 1.9 | (45)a | (106) |
Next, the influence of tube ageing on both the Sb peak profile and on the atomiser morphology was investigated for geological slurries. Fig. 2(C) shows the atomic and background signals registered for a new and an old tube employed to analyse soil/sediment slurries (10 µl from a 90 mg ml−1 slurry). Their profiles are noisier and with higher background signals than those of the aqueous and acid extracts. The appearance times of the atomic and background peaks get lower for the aged tube, maybe due to the residue build-up in the atomiser. Hence, the deuterium corrector over-corrected the atomic peak signal, requiring the use of the peak height for quantification. In addition, the analytical and background signals overlapped as the tube became more degraded (Figs. 2(B) and (C)), similar to the case with geological acid extracts.
The SEM analysis revealed a pitting corrosion located at the central area of the platform, just where the sample had been placed (Fig. 3(D)). A 1000×-magnification micrograph showed details about the quite different corrosion patterns for acid extracts, lamellar (Fig. 6(A)), and slurries, forming pits (Fig. 6(B)).
 | ||
| Fig. 6 Different degradation patterns of the L'vov pyrolytic platform (1000×-magnification) observed when analysing (A) soil/sediment acid extracts and (B) soil/sediment slurries. | ||
The tube endurance gets characterised by the appearance of pits, which grow, causing the final tube breakage (Fig. 7(A)). Moreover, many small droplet-like particles can be noticed on the corroded surface areas of the platform pits, which are clearly observed on a detailed view (Fig. 7(B)). They were located just in the middle of the pits and they were examined by EDS. Different profiles were obtained depending on the area of the platform being examined (Fig. 8(A) and (B)), but the same elemental composition was registered. Silicon appears on the profiles and it may be the major factor responsible for tube ageing since graphite deterioration has been widely reported in its presence.12,23 Accordingly, the degradation can be attributed to an important graphite evaporation at high temperatures whenever SiO2 is in the soil/sediment samples, following eqn. 1:
| SiO2 (s) + C(s) → SiO(g) + CO(g) (1400 °C) | (1) |
 | ||
| Fig. 7 Inner surface of a L’vov pyrolytic platform at the end of its life after analysing soil/sediment slurries: (A) 10×- and (B) 1000×-magnification. | ||
 | ||
| Fig. 8 Elemental analysis by X-ray dispersive energy analysis of the inner surface of the L'vov platform from (A) soil/sediment slurries (viewing a ‘clean’ area), (B) soil/sediment slurries (viewing an ‘area with deposits’) and (C) coal slurries. | ||
Further, the silicon signal can be attributed to the build-up of SiC deposits caused by the reaction between the gas phase and the graphite (see eqn. 2). Note that both reactions might well occur at the temperatures used in this work (1700 °C atomisation and 2650 °C cleaning).
| SiO(g) + C(s) → SiC(s) + CO(g) (1600 °C) | (2) |
The atomisers that were used to analyse soils and/or sediments with rather high Si contents (40–66% as SiO2, 10 µl, 90 mg ml−1) had lifetimes of about 90 firings. For soils exhibiting higher Si contents (ca. 73% as SiO2) the tube damage was drastically accelerated, the tubes remaining useful for only 20 atomisations. As a result, it seems clear that high Si contents shortened the lifetimes of the tubes.
It is worth noting the quite high Ti content present in all the samples analysed in this work (from 0.05% for soils to 0.8% for sediments). TiC is formed at high temperatures (by direct reaction of Ti with C) and it has a melting point of 3140 °C. Actually, the TiO2 reaction with C is used for the industrial production of TiC, the reaction starting at 935 °C. It can be presumed that a similar reaction can take place on the graphite platform during the temperature furnace program due to the TiO2 content of the samples. This would explain the Ti signal observed on the platforms. Besides, it was reported that TiC showed a considerable vapour pressure at temperatures above 1700 °C, suggesting that at least part of the TiC residue on the platform may be removed during the atomisation and cleaning steps.5
The effects of Al, Ca, Fe, K, Mg, Na and P on the graphite cannot be disregarded since they can form either superficial or interstitial heteropolar carbides affecting tube ageing.10 As mentioned above, Cu was always detected on the platforms, but we had not found a sensible explanation.
On the other hand, two of the most significant factors reported to influence the lifetime of the tube are the amount of sample mass used for the slurry preparation, in general, the mass/volume relationship (which depends on the analyte content on each sample) and the volume of the aliquot being injected. Table 2 lists several experimental lifetimes (the injected aliquots were always representative, >50 particles)24 observed when either different slurry aliquots or mass/volume relationships were employed. When soil/sediment slurries with low Sb levels are analysed, a high mass/volume ratio is needed (90 mg ml−1). The injection of 10 µl aliquots led to a tube lifetime of 90–100 firings, which increased by about 50% using only 5 µl of slurries. For samples with very high Sb contents much lower mass/volume ratios (5 mg/2 ml) had to be used, following which the lifetime of the tubes increased up to four-fold.
| Material | [Sb]/µg g−1 | m/v (mg ml−1) | Injected volume/µl | Useful lifetime of the tubes (firings) |
|---|---|---|---|---|
| Soil GBW07401 | 0.87 | 90/1 | 10 | 90–100 |
| Soil GBW07401 | 0.87 | 90/1 | 5 | 140 |
| Marine sediment PACS-1 | 171 | 5/2 | 5–10 | 400 |
| Coal fly ash samples | 1–30 | 5–35/1 | 2–5 | 230–250 |
| Slag samples | 0.15–8 | 25–150/2 | 5–10 | 90–100 |
Finally, it would be wise to use as little sample mass as possible for the slurry preparation and to inject low slurry volumes, looking for a compromise among the atomiser lifetime, sample representativeness, sensitivity and precision.
Slurries of coal combustion residues
Coal residues from coal-combustion power plants, such as coal fly ashes and slag, are of great concern for the environment.25 Their effects can be either negative (leaching of metals and other chemicals, changes in soil composition and vegetation, accumulation through the food chain, etc.) or positive (enhancing soil fertility and the water-holding capacity).26 Although there are not many works dealing with such matrices, SS-GFAAS has been proposed as a suitable technique to determine trace metals on them.27,28 In our laboratory, a previous analytical method19 was successfully extended to measure Sb in coal fly ash and slag and their effects on the graphite tube are investigated here.The lifetime of the tubes used for slag samples was stated at 90–100 firings, similar to that of soil/sediment slurries (Fig. 9). The precision was good during this time and the accuracy was checked by calculating the analytical recovery; good figures were found (ca. 114%). In some cases, after 100 atomisations, large holes were viewed on the L'vov platform (Fig. 10(A)). This degradation is similar to that for soil/sediment slurries and it can be attributed to the analogous contents of Sb and Si (45% as SiO2). The EDS analysis showed analogous profiles to those presented on Figs. 8(A) and 8(B) for soil/sediments slurries.
 | ||
| Fig. 9 Control charts employed to monitor the tube behaviour during the analysis of slag slurries (UCL = upper control limit, LCL = lower control limit). | ||
 | ||
| Fig. 10 Inner surface of L'vov pyrolytic platforms used to analyse (A) slag and (B) coal fly ashes as slurries. | ||
Regarding the tubes used for coal fly ash samples, they exhibited a longer lifetime, 230–250 firings, showing good precision (Fig. 11) and accurate results employing two certified coal fly ashes (Table 1). As for the samples studied above, no significant changes were observed on the slopes during the lifetime of the tubes.
 | ||
| Fig. 11 Control charts employed to monitor the tube behaviour during the analysis of coal fly ash slurries (UCL = upper control limit, LCL = lower control limit). | ||
A detailed view (100× magnification) of the aged platforms used for fly ash slurries (Fig. 10(B)) showed a pitting corrosion along the surface without any hole. This different degradation pattern can be attributed to several reasons, mainly the use of more diluted slurries and small aliquots (because these samples have high Sb contents), as well as the lower particle size of these samples (0.2–90 µm), most obvious when compared with soils/sediments (5–140 µm). Therefore, as can be deduced from Table 2, the more concentrated the slurries, the more concomitants and the shorter the lifetime of the tubes.
Slurries of coals
Despite coals showing naturally low levels of many toxic metals, they have to be strictly controlled as trace metals accumulate in the coal combustion residues. The main hindrance in the direct analysis of coals by SS-GFAAS is their huge carbonaceous content and, accordingly, the very low concentration of the mineral fraction.The direct introduction of coal slurries (100 mg ml−1) into the atomiser produced a large amount of residue after only 20 injections (6 µl each). Fig. 12(A) shows a general view (10× magnification) of the platforms where many deposits can be observed across all of the surface. This residue caused the Sb signal to drop from 0.133 to 0.052 A for a control sample, largely because it obstructed the light beam. When the platforms were analysed by EDS (Fig. 8(C)), a curved baseline very similar to that observed for a new tube (Fig. 4) was obtained while more pronounced. This shape appears to be caused by the carbonaceous residue on the platform. Therefore, the highly concentrated slurries (required by the low Sb content) and the nature of the coal itself quickly deteriorated the tube, hindering the direct Sb determination by USS-GFAAS in this type of sample.
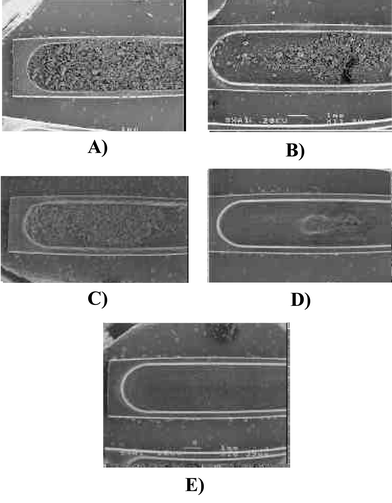 | ||
| Fig. 12 Inner surface (10×- magnification) of a L'vov pyrolytic platform used for the analysis of coal slurries prepared in (A) 0.5% v/v HNO3, (B) in 40% v/v H2O2, (C) previous low temperature ashing for 4 h, (D) previous low temperature ashing for 15 h, (E) previous high temperature ashing (450 °C, constant weight, 6–18 h). | ||
In order to avoid (or reduce) the carbonaceous residue build-up several assays were made. The furnace temperature program was modified by increasing either the cleaning hold time (from 3 to 7 s) or the pyrolysis hold time (from 30 to 60 s). Although this approach was successful for other applications,29 here the strategy failed because no significant residue reduction was attained.
A second alternative consisted in performing a sample pre-oxidation step. Two modes were tried: (i) using an oxidant reagent, and (ii) performing a previous ashing-step.
The first one employed H2O2 (40% v/v) as diluent due to its oxidant properties. Its effectiveness was slight since the tube became useless after 60 heating cycles and no satisfactory accuracy was achieved (ca. 120%). As presented in Fig. 12(B), a residue build-up similar to that of Fig. 12(A) was observed, though to a lesser extent, therefore this way was not successful (despite Viñas et al.7 reporting good results when combining H2O2 with HNO3 for As determination in baby foods).
Regarding the second mode, the samples underwent a previous ashing step in a low temperature ashing system (LTA). This system avoids losses of volatile elements because the temperature is fixed at 150 °C. Two ashing times were tried, 4 and 15 h. The SEM micrographs obtained for the tubes employed (Figs. 12(C) and (D)) revealed that the higher the ashing time, the lesser the residue. Nevertheless, the absorbance value for the control sample shifted down from 0.110 to 0.062 A after only 35 atomisation cycles, which represents an important reduction in sensitivity and makes this approach unsuitable.
Another ashing method was tested. Coal samples were ashed in a crucible furnace until constant weight (450 °C for 6–18 h). This sample pretreatment concentrated the Sb in the coal ashes, allowing us to prepare more diluted slurries than those when coals were directly analysed (7–15 mg ml−1 for coal ashes versus 100 mg ml−1 for coals). The analysis of these slurries did not give carbonaceous residues on the platform (Fig. 12(E)) during the useful lifetime of the tubes, which was established at 250 heating cycles (Fig. 13). The standard deviation chart shown satisfactory precision values; accuracy was good (Table 1).
 | ||
| Fig. 13 Control charts employed to monitor the tube behaviour during the analysis of ashed coal slurries (UCL = upper control limit, LCL = lower control limit). | ||
Ultrasound-assisted extraction of slurried samples
Finally, the ultrasound-assisted extraction of slurries coupled to GFAAS was investigated as an alternative to direct slurry sampling-GFAAS analysis. One of its main benefits was an improvement in the graphite lifetime of the tubes,30,31 either because the organic matrices did not produce carbonaceous residues and the effects of the inorganic concomitants are largely reduced. This approach was tested for soils, sediments and coals (representing samples with different organic contents).Two acid media were investigated to achieve a quantitative Sb extraction into the liquid phase. The use of a 65% (v/v) aqua regia solution as extractant damaged the external surface of the injection hole (Fig. 14) after only 40–50 atomisation cycles, although the inner platform did not appear to be affected and, in fact, the Sb atomic signal did not diminish after more than 200 firings. Nevertheless, this mixture did not give a quantitative Sb extraction from soil, sediment or coal samples, leading to recoveries lower than 30% in all cases.
 | ||
| Fig. 14 External deterioration of a pyrolytic graphite tube when 65% (v/v) aqua regia soil slurry extracts were analysed. | ||
When a 20% (v/v) HF liquid medium was used the usefulness of the atomisers was significantly improved as regards that attained when soil/sediment slurries were directly analysed (ca. 300 versus ca. 100 firings). In both cases, the sample mass used to prepare the slurries was the same (20–90 mg ml−1) but the amount of matrix introduced into the atomiser diminished drastically for the slurry extracts. Fig. 15 shows a smooth and uniform deterioration of the L’vov platform surface when HF extracts were atomised. Regarding accuracy, this acid provided a good Sb solubilisation, giving recoveries between 105 and 115% for certified reference soils and sediments, although the results were unsatisfactory for coals.
 | ||
| Fig. 15 Inner surface of a L'vov pyrolytic platform used for the analysis of soil/sediment slurry extracts using HF 20% v/v: (A) 10×- and (B) 100×-magnification. | ||
Therefore, the ultrasound-assisted extraction of slurried samples appears to be a suitable procedure for samples with low organic contents, and it extends the analytical lifetime of the tubes. Nevertheless, the search for the appropriate extraction conditions requires much time and effort and prolonged contact between the ultrasonic probe and the HF should be avoided (due to the incompatibility of this acid with Ti-based materials).
Acknowledgements
This work was partially supported by the ‘Vicerrectorado de Investigación’ of the University of A Coruña. The authors are indebted to the SXAIN (Servicios Xerais de Apoio á Investigación) for its technical advice in the electronic microscopy and X-ray analyses. M.J.C.P. acknowledges the Xunta de Galicia for a PhD grant.References
- M. J. Cal-Prieto, J. M. Andrade, A. Carlosena, S. Muniategui, P. López-Mahía and D. Prada, Quím. Anal., 1999, 18, 137 Search PubMed.
- S. Hauptkorn and V. Krivan, Spectrochim. Acta, Part B, 1994, 49, 221 CrossRef.
- B. Welz, G. Schlemmer, H. M. Ortner and W. Wegsheider, Analyst, 1994, 121, 111 Search PubMed.
- N. S. Thomaidis and E. A. Piperaki, Analyst, 1996, 121, 111 RSC.
- H. M. Dong, V. Krivan, B. Welz and G. Schlemmer, Spectrochim. Acta, Part B, 1997, 52, 1747 CrossRef.
- M. J. Cal-Prieto, M. Felipe-Sotelo, A. Carlosena, J. M. Andrade, P. López-Mahía, S. Muniategui and D. Prada, Talanta, 2002, 56, 1 CrossRef CAS.
- P. Viñas, M. Pardo-Martínez and M. Hernández-Córdoba, J. Anal. At. Spectrom., 1999, 14, 1215 RSC.
- E. C. Lima, F. Barbosa and F. J. Krug, J. Anal. At. Spectrom., 1999, 14, 1913 RSC.
- A. A. Almeida and J. L. F. C. Lima, J. Anal. At. Spectrom., 2000, 15, 1019 RSC.
- U. Schäffer and V. Krivan, Spectrochim. Acta, Part B, 1996, 52, 1211 CrossRef.
- P. Bermejo-Barrera, A. Moreda-Piñeiro, J. Moreda-Piñeiro and A. Bermejo-Barrera, Anal. Chim. Acta, 1997, 349, 319 CrossRef CAS.
- S. Hauptkorn and V. Krivan, Spectrochim. Acta, Part B, 1996, 51, 1197 CrossRef.
- M. A. Z. Arruda, M. J. Quintela, M. Gallego and M. Valcárcel, Analyst, 1994, 119, 1695 RSC.
- P. Viñas, N. Campillo, I. López-García and M. Hernández-Córdoba, At. Spectrosc., 1995, 16, 86 Search PubMed.
- P. Bermejo-Barrera, J. Moreda-Piñeiro, A. Moreda-Piñeiro and A. Bermejo-Barrera, Anal. Chim. Acta, 1994, 296, 181 CrossRef CAS.
- P. Bermejo-Barrera, A. Moreda-Piñeiro, J. Moreda-Piñeiro and A. Bermejo-Barrera, J. Anal. At. Spectrom., 1997, 12, 301 RSC.
- L. Ebdon, A. S. Fisher, H. G. M. Parry and A. A. Brown, J. Anal. At. Spectrom., 1990, 5, 321 RSC.
- M. J. Cal-Prieto, A. Carlosena, J. M. Andrade, S. Muniategui, P. López-Mahía and D. Prada, Afinidad, 1999, LVI, 105 Search PubMed.
- M. J. Cal-Prieto, A. Carlosena, J. M. Andrade, S. Muniategui, P. López-Mahía, E. Fernández and D. Prada, J. Anal. At. Spectrom., 1999, 14, 703 RSC.
- M. J. Cal-Prieto, A. Carlosena, J. M. Andrade, S. Muniategui, P. López-Mahía and D. Prada, At. Spectrosc., 2000, 21(3), 93 Search PubMed.
- M. J. Cal-Prieto, PhD thesis, Department of Analytical Chemistry, University of A Coruña, Spain, 2002.
- ASTM, Manual on Presentation of Data and Control Chart Analysis, 1986.
- A. B. Volynsky, Spectrochim. Acta, Part B, 1998, 53, 509 CrossRef.
- N. J. Miller-Ihli, Fresenius' J. Anal. Chem., 1993, 345, 482 CrossRef CAS.
- I. M. Smith, Trace Elements from Coal Combustion: Emissions, IEA Coal Research, London, 1987 Search PubMed.
- C. L. Carlson and D. Adriano, J. Environ. Qual., 1993, 22, 227 Search PubMed.
- M. Felipe-Sotelo, A. Carlosena, E. Fernández, P. López-Mahía, S. Muniategui and D. Prada, Fresenius' J. Anal. Chem., 2001, 371, 1139 Search PubMed.
- N. J. Miller-Ihli, J. Anal. At. Spectrom., 1997, 12, 205 RSC.
- E. C. Lima, F. J. Krug and K. W. Jackson, Spectrochim. Acta, Part B, 1998, 53, 1791 CrossRef.
- J. L. Capelo, I. Lavilla and C. Bendicho, J. Anal. At. Spectrom., 1998, 13, 1285 RSC.
- J. L. Capelo, A. V. Filgueiras, I. Lavilla and C. Bendicho, Talanta, 1999, 50, 905 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2003 |