
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHypervalent iodine-promoted twofold oxidative coupling of amines with amides and thioamides: chemoselective pathway to oxazoles and thiazoles†
Jiang
Nan
 *a,
Xin
Ren
a,
Qiang
Yan
a,
Shilei
Liu
a,
Jing
Wang
a,
Yangmin
Ma
a and
Michal
Szostak
*b
*a,
Xin
Ren
a,
Qiang
Yan
a,
Shilei
Liu
a,
Jing
Wang
a,
Yangmin
Ma
a and
Michal
Szostak
*b
aThe Youth Innovation Team of Shaanxi Universities, Shaanxi Key Laboratory of Chemical Additives for Industry, College of Chemistry and Chemical Engineering, Shaanxi University of Science and Technology, Xi'an 710021, China. E-mail: nanjiang@sust.edu.cn
bDepartment of Chemistry, Rutgers University, 73 Warren Street, Newark, New Jersey 07102, USA
First published on 27th February 2023
Abstract
Direct functionalization of the C(O)–N amide bond is one of the most high-profile research directions in the last few decades; however oxidative couplings involving amide bonds and functionalization of thioamide C(S)–N analogues remain an unsolved challenge. Herein, a novel hypervalent iodine-induced twofold oxidative coupling of amines with amides and thioamides has been established. The protocol accomplishes divergent C(O)–N and C(S)–N disconnection by the previously unknown Ar–O and Ar–S oxidative coupling and highly chemoselectively assembles the versatile yet synthetically challenging oxazoles and thiazoles. Employing amides instead of thioamides affords an alternative bond cleavage pattern, which is a result of the higher 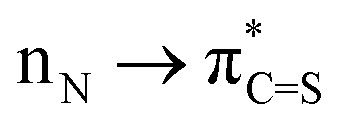 conjugation in thioamides. Mechanistic investigations indicate ureas and thioureas generated in the first oxidation as pivotal intermediates to realize the oxidative coupling. These findings open up new avenues for exploring oxidative amide and thioamide bond chemistry in various synthetic contexts.
conjugation in thioamides. Mechanistic investigations indicate ureas and thioureas generated in the first oxidation as pivotal intermediates to realize the oxidative coupling. These findings open up new avenues for exploring oxidative amide and thioamide bond chemistry in various synthetic contexts.
Introduction
The amide bond undoubtedly belongs to one of the most inert chemical bonds owing to the intramolecular conjugation and high resonance stabilization (Scheme 1a, left).1a–h Notwithstanding that the amide bond is well-recognized as the most stable carboxylic acid derivative, the highly selective C(O)–N activation of amides that enables us to sequentially perform chemical operations has attracted great attention in organic synthesis over the past few decades (Scheme 1a, right).2a–g This is mostly due to the ubiquitous presence of amide bonds in biochemistry, pharmaceuticals, and diverse bulk and fine chemicals as well as their versatility in synthetic settings.3a–f Indeed, amide bond activation has been experiencing major progress by means of carefully controlled steric repulsion, conformational restriction and electronic effects to successfully activate the N–C(O) bond by ground-state-destabilization of amidic resonance (Scheme 1b, left).4,5a–d,6,7 This mode of activation broadly relies upon disrupting
conjugation and high resonance stabilization (Scheme 1a, left).1a–h Notwithstanding that the amide bond is well-recognized as the most stable carboxylic acid derivative, the highly selective C(O)–N activation of amides that enables us to sequentially perform chemical operations has attracted great attention in organic synthesis over the past few decades (Scheme 1a, right).2a–g This is mostly due to the ubiquitous presence of amide bonds in biochemistry, pharmaceuticals, and diverse bulk and fine chemicals as well as their versatility in synthetic settings.3a–f Indeed, amide bond activation has been experiencing major progress by means of carefully controlled steric repulsion, conformational restriction and electronic effects to successfully activate the N–C(O) bond by ground-state-destabilization of amidic resonance (Scheme 1b, left).4,5a–d,6,7 This mode of activation broadly relies upon disrupting 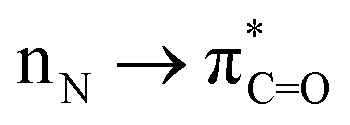 conjugation to increase negative charge at the nitrogen atom, while this mode should be contrasted with the tremendously important field of electrophilic activation of amides.4 The current well-developed reaction types are predominantly restricted to sterically twisted amides8 or electronically activated amides9 featuring electron-withdrawing groups (EWGs) that facilitate acyl addition or oxidative addition of the C(O)–N bond to a low-valent metal. Comparatively, formamides are much more broadly available than such activated amides; however, there are few examples of the site-specific C(O)–N activation of non-activated formamides (Scheme 1b, right).10
conjugation to increase negative charge at the nitrogen atom, while this mode should be contrasted with the tremendously important field of electrophilic activation of amides.4 The current well-developed reaction types are predominantly restricted to sterically twisted amides8 or electronically activated amides9 featuring electron-withdrawing groups (EWGs) that facilitate acyl addition or oxidative addition of the C(O)–N bond to a low-valent metal. Comparatively, formamides are much more broadly available than such activated amides; however, there are few examples of the site-specific C(O)–N activation of non-activated formamides (Scheme 1b, right).10
 | ||
| Scheme 1 Amide functionalization and context of this work. | ||
Formamides characterized by planarity and featuring electron-donating groups (EDGs) are not amenable to the ground-state-destabilization pathway. The present amide bond activation has been most successful using metal-catalyzed coupling and only implemented to generate architecturally linear molecules by single bond formation thus far.11
Thioamides, the closest O to S isosteres of amides in the strictest sense, have served as highly valuable motifs to accomplish synthetic processes.12 With respect to the structural and electronic characteristics, thioamides feature a shorter C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S bond than the analogous CO bond in amides. The weaker electronegativity of sulfur and the higher
S bond than the analogous CO bond in amides. The weaker electronegativity of sulfur and the higher  conjugation provide the higher contribution of the polar resonance form. This results in a stronger C(S)–N bond in thioamides, which together with potential side-reactions, such as desulfurization, electrophilic addition, and cyclization, poses a major challenge to chemoselectively cleave the C(S)–N bond (Scheme 1c).13 Indeed, studies on the activation of twisted thioamide bonds are exceedingly scarce, and chemoselective C(S)–N bond activation of non-twisted and non-electronically activated thioamides has not been documented to date.
conjugation provide the higher contribution of the polar resonance form. This results in a stronger C(S)–N bond in thioamides, which together with potential side-reactions, such as desulfurization, electrophilic addition, and cyclization, poses a major challenge to chemoselectively cleave the C(S)–N bond (Scheme 1c).13 Indeed, studies on the activation of twisted thioamide bonds are exceedingly scarce, and chemoselective C(S)–N bond activation of non-twisted and non-electronically activated thioamides has not been documented to date.
In continuation of our long-standing interest to discover new modes of activation of amide bonds,7 herein, we disclose our findings on novel hypervalent iodine-induced twofold oxidative coupling of amines with amides and thioamides (Scheme 1d and e). Of note, the protocol accomplishes divergent C(O)–N and C(S)–N disconnection by the previously unknown Ar–O and Ar–S oxidative coupling. The method represents the first example of combining the inert C(O)–N/C(S)–N coupling with C–H functionalization in a single operation, providing a powerful platform to assemble the versatile yet synthetically challenging oxazoles and thiazoles that are common in pharmaceuticals and agrochemicals14a,b but currently lack a practical synthetic route.15 Employing amides instead of thioamides affords an alternative bond cleavage pattern, which is a result of the higher  conjugation in thioamides, furnishing C2–NR2-substituted benzoxazoles (Scheme 1f).16 This oxidative pathway outperforms the current dominant tactic of decorating the benzoxazole scaffold (Scheme 1g).17 Overall, these findings open up new avenues for exploring oxidative amide and thioamide bond chemistry in various synthetic contexts.
conjugation in thioamides, furnishing C2–NR2-substituted benzoxazoles (Scheme 1f).16 This oxidative pathway outperforms the current dominant tactic of decorating the benzoxazole scaffold (Scheme 1g).17 Overall, these findings open up new avenues for exploring oxidative amide and thioamide bond chemistry in various synthetic contexts.
Results and discussion
Initially, we explored the reactivity of commercially available N,N-dimethylthioamide 2 with β-naphthylamine 1a (Table 1). After comprehensive screening of reaction parameters, we were delighted to find that upon treatment with 2-iodoxybenzoic acid (IBX) as an oxidant, these two substrates underwent a formal [3 + 2] cyclization to produce thiazole derivative 3a in 70% yield under metal-free and solvent-free conditions (entry 1). The reaction accomplished a series of C(S)–N/C–H cleavages and C–N/C–S bond forming events. Control experiments showed that IBX played an indispensable role in simultaneously realizing this novel C(S)–N thioamide conversion with thiazole ring formation in a single operation (entry 2). Other broadly utilized high-valent I3+ oxidants, such as PIDA, PIFA or PhIO, were all feasible but afforded lower yields (entries 3–5). Changes in the reaction temperature (110 °C) or reaction atmosphere were detrimental (entries 6 and 7). Decreasing the amount of IBX or 2 afforded inferior results (entries 8 and 9). Interestingly, employing solvents adversely affected the reaction (entries 10–12). It is noteworthy that solvent-free reactions have recently attracted increasing attention due to low-cost and green chemistry considerations.18 Most notably, during the screening of all reaction parameters, the side-product 3a′ was not observed, indicating a highly efficient oxidative C(S)–N bond cleavage through breaking resonance.
resonance.
|
|
||
|---|---|---|
| Entry | Variations from the standard conditions | Yield of 3ab (%) |
| a Performed on a 0.20 mmol scale. b Isolated yields. PIDA = phenyliodine(III) diacetate, PIFA = phenyliodine(III) bis(trifluoroacetate), and PhIO = iodosylbenzene. | ||
| 1 | None | 70 |
| 2 | Without IBX | 0 |
| 3 | PIDA instead of IBX | 23 |
| 4 | PIFA instead of IBX | 12 |
| 5 | PhIO instead of IBX | 10 |
| 6 | At 110 °C | 34 |
| 7 | O2 atmosphere | 40 |
| 8 | Reducing loading of IBX to 1.0 equiv. | 52 |
| 9 | Reducing loading of 2 to 2.0 equiv. | 45 |
| 10 | In DCE (0.25 M) | 17 |
| 11 | In 1,4-dioxane (0.25 M) | nd |
| 12 | In DMSO (0.25 M) | 26 |

Having identified optimal reaction parameters, we next sought to evaluate the substrate scope of this novel transformation by coupling various naphthylamines with N,N-dimethylthioformamide 2 (Table 2). As depicted in Table 2, a range of 2-naphthylamines reacted smoothly with full regioselectivity at the α-site of amine substrates, delivering thiazoles 3a–s in good yields. The electronically distinct substituents at 6- and 7-positions, such as methyl (3b), methoxy (3c and 3h), methoxycarbonyl (3d), cyclopropyl (3e), bromo (3f and 3g), furyl (3i), thienyl (3j), thianthrenyl (3k), and a series of diverse-substituted arene system (3l–3p) moieties, all proved to be competent substrates. The structure of 3b was unambiguously assigned by X-ray single crystallography. Notably, the high-value but usually challenging in oxidative coupling alkenyl (3q) group was retained intact under these conditions, thus providing a versatile handle for downstream modification. Likewise, π-electron-rich phenanthryl- and anthryl-derived thiazoles (3r–3s) were equally compatible and prepared in 70% and 78% yields, respectively. Furthermore, the use of 1-naphthylamine to engage in this protocol also furnished the target molecule 3aa in 56% yield. The naphthyl ring could be diversely decorated with cyclopropyl (3ab) or aromatics substituted with alkyl (3ad), sulfuryl (3ae), or nitro (3af) groups. It should be noted that 4-bromo-1-naphthylamine was not tolerated, but delivered the debrominated product 3aa. However, the readily oxidized dihydro-acenaphthylene unit was well-compatible and generated product 3ah in 56% yield. At the present stage, the reaction is not compatible with simple anilines (3ai) probably because of the more stable aromaticity of than that of naphthylamines.

We next employed N,N-dimethylformamide (DMF) as the structurally closest analog of N,N-dimethylthioformamide to participate in this novel methodology (Table 2). To our surprise, reacting 2-naphthylamine 1a with DMF resulted in the formation of NMe2-substituted naphthyl-fused oxazole 4a, wherein the alternative bond cleavage pattern occurred chemoselectively. Considering the unique selectivity with DMF to accomplish the synthesis of 2-amino-substituted benzoxazoles by oxidative coupling, we optimized the reaction conditions (details in the ESI†). By slightly tuning the conditions by replacing IBX with PIDA, the target 4a was isolated in 81% yield, and its structure was unambiguously confirmed by X-ray crystallography.
With the optimized conditions in hand, the scope of this benzoxazole formation was investigated (Table 2). As shown, various naphthylamine counterparts were amenable to this divergent reaction. Specifically, a collection of functionalized naphthylamines, encompassing methyl, methoxy, ester, cyclopropyl, and bromo groups, were subjected to this oxidative process to afford the expected products 4b–4h in 59–81% yields. Importantly, in this variant by-products resulting from C–N dissociation were not observed, indicating the key difference between  conjugation resonance under oxidative conditions (vide infra). Strikingly, the sensitive vinyl fragment (4i–4j) was well-compatible. An array of electronically distinct aryl systems (4k–4p) and even heterocycles (4q–4r) were also feasible while the simple amine behaved unsatisfactorily (4s). In addition, the substrate scope of amides was subsequently assessed. The results showed that linear (4a′–4d′), sterically bulky (4e′), and cyclic (4f′–4h′) tertiary amides all underwent smooth cyclization. Notably, this reaction could also be extended to secondary amides, thus resulting in the corresponding NH-secondary amine-tethered heterocycles (4i′–4k′) with high value in organic synthesis.
conjugation resonance under oxidative conditions (vide infra). Strikingly, the sensitive vinyl fragment (4i–4j) was well-compatible. An array of electronically distinct aryl systems (4k–4p) and even heterocycles (4q–4r) were also feasible while the simple amine behaved unsatisfactorily (4s). In addition, the substrate scope of amides was subsequently assessed. The results showed that linear (4a′–4d′), sterically bulky (4e′), and cyclic (4f′–4h′) tertiary amides all underwent smooth cyclization. Notably, this reaction could also be extended to secondary amides, thus resulting in the corresponding NH-secondary amine-tethered heterocycles (4i′–4k′) with high value in organic synthesis.
Next, we evaluated further functionalization of several pharmaceutical synthons by these two protocols (Scheme 2). Thus, DL-menthol and (−)-borneol derivatives could be easily converted into the corresponding thiazoles 5a and 5b in high yields. Likewise, the industrially important diacetone-β-fructose (DAF) and (−)-borneol synthons also performed well in this transformation to synthesize value-added oxazoles 5c and 5d in 77% and 75% yields.
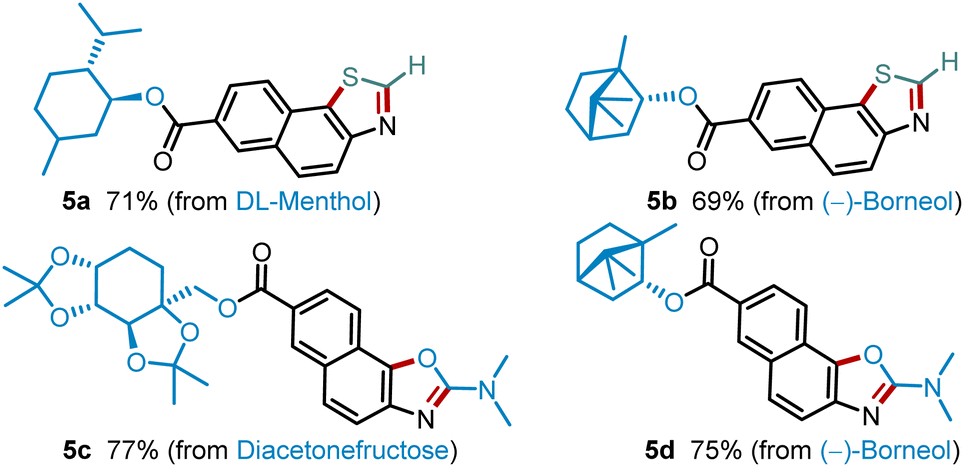 | ||
| Scheme 2 Further modification of bioactive synthons. | ||
To showcase the versatility and synthetical value of this new methodology, additional experiments were next conducted. First, scaling-up these two reactions to 5.0 mmol proved to be readily feasible, and both thiazole 3a and oxazole 4a were obtained in high yields, attesting to the scalability of the protocols (Scheme 3a). Second, shortening the reaction time was feasible to deliver oxazole 4a in 53% yield after one minute reaction time, which demonstrated exceedingly fast oxidative coupling (Scheme 3b). Next, although the direct synthesis of C2-NR2 substituted thiazoles was not feasible by this method, the product could be readily obtained through functionalizing the C–H bond of 3a with DMF as the aminating reagent (Scheme 3c).19a Furthermore, using copper catalysis,19b C–H arylation of 3a with iodobenzene was feasible to generate C2-arylated 7 in 64% yield (Scheme 3d). Likewise, despite the presence of several coordinating heteroatoms, Suzuki–Miyaura cross-coupling of 4f proceeded smoothly to deliver 8 in 89% yield (Scheme 3e). Next, the secondary naphthylamine 1′ could be cyclized to generate oxazole product 4a in 53% yield via C–N bond cleavage, ascribed to the in situ formed free naphthylamine (Scheme 3f). Finally, when formamide was used, the oxazole 9 was not formed, but the reaction produced 78% yield of formylation product 10 (Scheme 3g). This outcome proved the stability of 10 in the presence of hypervalent iodine and provided insights into the reaction mechanism.
 | ||
| Scheme 3 Applicability of the oxidative methodology. | ||
To shed light on this highly chemoselective reaction pathway, several sets of control experiments as well as studies with key intermediates were performed (Scheme 4). As depicted in Scheme 4a and b, C2-free oxazole 11 failed to deliver product 4a, and conversely, C2–NMe2 thiazole 6 did not undergo the C–N bond cleavage to afford product 12. These two results clearly illustrate the divergence of oxidative mechanisms to build oxazole and thiazole rings. To further probe the key reaction intermediates, several possible derivatives were prepared and subjected to the reaction conditions. As such, amidine 13 with high-activity20 did not afford the cyclization product, while urea 12 was transformed into the target molecule 4a with a comparatively high efficiency cf. the reaction between 1a and DMF (Scheme 4c). On the other hand, employing amine-derived thioformamide 14 resulted in full recovery, which ruled out the possible intermediacy of thioformamide. However, the use of thiourea 15 resulted in the formation of the target product 3a with efficiency comparable to the reaction of 1a and N,N-dimethylthioamide (Scheme 4d). Finally, a deuterium labelling study with d7-N,N-dimethylthioamide showed in 22% D incorporation in the C2 position (Scheme 4e).
 | ||
| Scheme 4 Studies of the reaction mechanism. | ||
Overall, these mechanistic studies indicate that urea and thiourea species act as pivotal intermediates to enable the oxidative assembly of oxazoles and thiazoles. The key mechanistic difference results from the divergent  conjugation, where the stronger conjugation of the thioamide bond induces N–C cleavage under oxidative conditions. A tentative mechanism is proposed in Scheme 5. The reaction commences with electrophilic activation of the CS and CO bonds by the hypervalent iodine reagent,21 which is followed by the nucleophilic addition of amine. Subsequently, the first oxidation takes place to give thiourea 15 or urea 12. At this point, with respect to the generation of thiazoles, the electron-rich intermediate 15 reacts with the hypervalent iodine(VI) to induce a hydride shift to afford A, which undergoes the second oxidative coupling by C–H/C–S cyclization. For the generation of oxazoles, the product is obtained by electrophilic activation, the second oxidative coupling by C–H/C–O cyclization and isomerization in the presence of hypervalent iodine(III). In the case of thioamides, hypervalent iodine promotes amine dehydrogenation to form iminium.22a The key difference in reactivity between amides and thioamides stems from higher reactivity of thioamides to electrophiles22b–d and high affinity of sulfur towards hypervalent iodine reagents23a with a weaker thiocarbonyl bond that is more susceptible to reduction.23b
conjugation, where the stronger conjugation of the thioamide bond induces N–C cleavage under oxidative conditions. A tentative mechanism is proposed in Scheme 5. The reaction commences with electrophilic activation of the CS and CO bonds by the hypervalent iodine reagent,21 which is followed by the nucleophilic addition of amine. Subsequently, the first oxidation takes place to give thiourea 15 or urea 12. At this point, with respect to the generation of thiazoles, the electron-rich intermediate 15 reacts with the hypervalent iodine(VI) to induce a hydride shift to afford A, which undergoes the second oxidative coupling by C–H/C–S cyclization. For the generation of oxazoles, the product is obtained by electrophilic activation, the second oxidative coupling by C–H/C–O cyclization and isomerization in the presence of hypervalent iodine(III). In the case of thioamides, hypervalent iodine promotes amine dehydrogenation to form iminium.22a The key difference in reactivity between amides and thioamides stems from higher reactivity of thioamides to electrophiles22b–d and high affinity of sulfur towards hypervalent iodine reagents23a with a weaker thiocarbonyl bond that is more susceptible to reduction.23b
 | ||
| Scheme 5 Possible mechanism. | ||
Conclusions
In summary, we have developed an unprecedented hypervalent iodine-induced twofold oxidative coupling of amines with amides. This coupling proceeds via a series of C–N/C–O and C–N/C–S forming events enabled by hypervalent iodine oxidants. The previously unknown Ar–O and Ar–S oxidative coupling assembles a collection of versatile yet synthetically challenging oxazoles and thiazoles with high chemoselectivity. Mechanistic studies demonstrated the importance of thiourea and urea species as the key reaction intermediates. This methodology represents the first example of chemoselective divergent oxidative bond functionalization in amides and thioamides to construct valuable heteroarenes. These findings open up new avenues for exploring oxidative amide and thioamide bond chemistry in various synthetic contexts.Data availability
All experimental and characterization data, as well as NMR spectra are available in the ESI.† Crystallographic data for compound 3b and 4a have been deposited in the Cambridge Crystallographic Data Centre under accession number CCDC 2224291 and 2223988.Author contributions
J. N. and Y. M. M. conceived and directed the project. X. R. designed and performed the experiments. Q. Y., S. L. L. and J. W. helped with the collection of some new compounds and data analysis. J. N., and M. S. wrote the paper. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the NSFC (21801159 and 22171171), the Innovative Talent Promotion Plan-Young Science and Technology Star Project (2022KJXX-15), and the Young Talent Fund of the University Association for Science and Technology in Shaanxi, China (20200605). M.S. thanks Rutgers University and the NSF (CAREER CHE-1650766) for support.Notes and references
- (a) M. Szostak, Amide bond activation: concepts and reactions, Wiley-VCH., New York, 2022 CrossRef; (b) S. Davey, Nat. Chem., 2015, 7, 682 CrossRef CAS; (c) D. Kaiser, A. Bauer, M. Lemmerer and N. Maulide, Chem. Soc. Rev., 2018, 47, 7899 RSC; (d) G. C. Li and M. Szostak, Chem. Rec., 2019, 20, 649 CrossRef PubMed; (e) G. C. Li, S. Y. Ma and M. Szostak, Trends Chem., 2020, 2, 914 CrossRef CAS; (f) T. B. Boit, A. S. Bulger, J. E. Dander and N. K. Garg, ACS Catal., 2020, 10, 12109 CrossRef CAS PubMed; (g) G. R. Meng, J. Zhang and M. Szostak, Chem. Rev., 2021, 121, 12746 CrossRef CAS PubMed; (h) M. H. Feng, H. Q. Zhang and N. Maulide, Angew. Chem., Int. Ed., 2022, 61, e202212213 CAS.
- (a) G. Pelletier, W. S. Bechara and A. B. Charette, J. Am. Chem. Soc., 2010, 132, 12817 CrossRef CAS PubMed; (b) J. F. Hu, Y. Zhao, J. J. Liu, Y. M. Zhang and Z. Z. Shi, Angew. Chem., Int. Ed., 2016, 55, 8718 CrossRef CAS; (c) N. A. Weires, E. L. Baker and N. K. Garg, Nat. Chem., 2016, 8, 75 CrossRef CAS; (d) J. A. Walker Jr., K. L. Vickerman, J. N. Humke and L. M. Stanley, J. Am. Chem. Soc., 2017, 139, 10228 CrossRef PubMed; (e) C.-L. Ji and X. Hong, J. Am. Chem. Soc., 2017, 139, 15522 CrossRef CAS PubMed; (f) A. A. Kadam, T. L. Metz, Y. Q. Qian and L. M. Stanley, ACS Catal., 2019, 9, 5651 CrossRef CAS; (g) G. J. Wang, Q. Q. Shi, W. Y. Hu, T. Chen, Y. Y. Guo, Z. L. Hu, M. H. Gong, J. C. Guo, D. H. Wei, Z. Q. Fu and W. Huang, Nat. Commun., 2020, 11, 946 CrossRef CAS PubMed.
- (a) A. Greenberg, C. M. Breneman and J. F. Liebman, The amide linkage: Structural significance in chemistry, biochemistry and materials science, Wiley-VCH., New York, 2000 Search PubMed; (b) A. B. Hughes, Amino acids, peptides and proteins in organic chemistry, Wiley, Weinheim, 2011 CrossRef; (c) K. Brix and W. Stocker, Proteases: structure and function, Springer, Verlag, Wien, 2013 Search PubMed; (d) B. J. Burkhart, C. J. Schwalen, G. Mann, J. H. Naismith and D. A. Mitchell, Chem. Rev., 2017, 117, 5389 CrossRef CAS PubMed; (e) A. Karmakar and A. J. L. Pombeiro, Coord. Chem. Rev., 2019, 39, 86 CrossRef; (f) V. Pattabiraman and J. Bode, Nature, 2011, 480, 471 CrossRef CAS PubMed.
- (a) S. A. Ruider and N. Maulide, Angew. Chem., Int. Ed., 2015, 54, 13856 CrossRef CAS; (b) D. Kaiser, C. J. Teskey, P. Adler and N. Maulide, J. Am. Chem. Soc., 2017, 139, 16040 CrossRef CAS; (c) H. Takezawa, K. Shitozawa and M. Fujita, Nat. Chem., 2020, 12, 574 CrossRef CAS.
- (a) C. P. Chen, P. Liu, M. M. Luo and X. M. Zeng, ACS Catal., 2018, 8, 5864 CrossRef CAS; (b) C. C. D. Wybon, C. Mensch, C. Hollanders, C. Gadais, W. A. Herrebout, S. Ballet and B. U. W. Maes, ACS Catal., 2018, 8, 203 CrossRef CAS; (c) C. Hollanders, E. Renders, C. Gadais, D. Masullo, L. V. Raemdonck, C. C. D. Wybon, C. Martin, W. A. Herrebout, B. U. W. Maes and S. Ballet, ACS Catal., 2020, 10, 4280 CrossRef CAS; (d) Y. Long, Y. L. Zheng, Y. Xia, L. Qu, Y. H. Yang, H. F Xiang and X. G. Zhou, ACS Catal., 2022, 12, 4688 CrossRef CAS.
- (a) K. Tani and B. M. Stoltz, Nature, 2006, 441, 731 CrossRef CAS PubMed; (b) B. Sliter, J. Morgan and A. Greenberg, J. Org. Chem., 2011, 76, 2770 CrossRef CAS PubMed; (c) S. A. Glover and A. A. Rosser, J. Org. Chem., 2012, 77, 5492 CrossRef CAS PubMed; (d) T. Shiba, T. Kurahashi and S. Matsubara, J. Am. Chem. Soc., 2013, 135, 13636 CrossRef CAS PubMed; (e) H. Nagae, T. Hirai, D. Kato, S. Soma, S. Akebi and K. Mashima, Chem. Sci., 2019, 10, 2860 RSC.
- (a) S. C. Shi, G. R. Meng and M. Szostak, Angew. Chem., Int. Ed., 2016, 55, 6959 CrossRef CAS PubMed; (b) F. Hu, R. Lalancette and M. Szostak, Angew. Chem., Int. Ed., 2016, 55, 5062 CrossRef CAS; (c) C. W. Liu and M. Szostak, Angew. Chem., Int. Ed., 2017, 56, 12718 CrossRef CAS; (d) G. R. Meng, S. C. Shi, R. Lalancette, R. Szostak and M. Szostak, J. Am. Chem. Soc., 2018, 140, 727 CrossRef CAS PubMed; (e) T. L. Zhou, C.-L. Ji, X. Hong and M. Szostak, Chem. Sci., 2019, 10, 9865 RSC; (f) G. C. Li, C.-L. Ji, X. Hong and M. Szostak, J. Am. Chem. Soc., 2019, 141, 11161 CrossRef CAS PubMed; (g) C.-A. Wang, M. M. Rahman, E. Bisz, B. Dziuk, R. Szostak and M. Szostak, ACS Catal., 2022, 12, 2426 CrossRef CAS; (h) D. X. Zuo, Q. Wang, L. Liu, T. Z. Huang, M. Szostak and T. Q. Chen, Angew. Chem., Int. Ed., 2022, 61, e202202794 CAS.
- (a) J. Aubé, Angew. Chem., Int. Ed., 2012, 51, 3063 CrossRef; (b) P. Lei, G. R. Meng and M. Szostak, ACS Catal., 2017, 7, 1960 CrossRef CAS; (c) L. B. Fu, M. Z. Xu, J. Y. Yu and W. R. Gutekunst, J. Am. Chem. Soc., 2019, 141, 2906 CrossRef CAS PubMed; (d) H. Takezawa, K. Shitozawa and M. Fujita, Nat. Chem., 2020, 12, 574–578 CrossRef CAS PubMed.
- (a) G. R. Meng, P. Lei and M. Szostak, Org. Lett., 2017, 19, 2158 CrossRef CAS PubMed; (b) Y. M. Liu, S. C. Shi, M. Achtenhagen, R. Z. Liu and M. Szostak, Org. Lett., 2017, 19, 1614 CrossRef CAS PubMed; (c) S. Yu, T. Shin, M. S. Zhang, Y. Z. Xia, H. Kim and S. Lee, Org. Lett., 2018, 20, 7563 CrossRef CAS PubMed; (d) J. J. Chen, Y. Z. Xia and S. Lee, Org. Lett., 2020, 22, 3504 CrossRef CAS PubMed; (e) Y. Long, Y. L. Zheng, Y. Xia, L. Qu, Y. H. Yang, H. F. Xiang and X. G. Zhou, ACS Catal., 2022, 12, 4688 CrossRef CAS.
- (a) C. Liana Allen, B. N. Atkinson and J. M. J. Williams, Angew. Chem., Int. Ed., 2011, 51, 1383 CrossRef PubMed; (b) T. M. E. Dine, D. Evans, J. Rouden and J. Blanchet, Chem. - Eur. J., 2016, 22, 5894 CrossRef PubMed; (c) R. B. Sonawane, N. K. Rasal and S. V. Jagtap, Org. Lett., 2017, 19, 2078 CrossRef CAS; (d) T. Ghosh, S. Jana and J. Dash, Org. Lett., 2019, 21, 6690 CrossRef CAS PubMed; (e) J. W. Yin, J. Y. Zhang, C. Q. Cai, G.-J. Deng and H. Gong, Org. Lett., 2019, 21, 387 CrossRef CAS PubMed; (f) M.-H. Zhu, Z. R. Cheng, J. L. Wei, H. Tan and N. Jiao, CCS Chem., 2022 DOI:10.31635/ccschem.022.202202585.
- (a) W. S. Bechara, G. Pelletier and A. B. Charette, Nat. Chem., 2012, 4, 228 CrossRef CAS PubMed; (b) H. Yue, L. Guo, S.-C. Lee, X. Liu and M. Rueping, Angew. Chem., Int. Ed., 2017, 56, 3972 CrossRef CAS PubMed; (c) J. Zhuo, Y. Zhang, Z. J. Li and C. Li, ACS Catal., 2020, 10, 3895 CrossRef CAS; (d) G. S. Lee, J. Won, S. Choi, M.-H. Baik and S. H. Hong, Angew. Chem., Int. Ed., 2020, 59, 16933 CrossRef CAS PubMed.
- (a) T. Murai, The Chemistry of Thioamides, Springer, Singapore, 2019 CrossRef; (b) T. S. Jagodziński, Chem. Rev., 2003, 103, 197 CrossRef; (c) S. Fukagawa, Y. Kato, R. Tanaka, M. Kojima, T. Yoshino and S. Matsunaga, Angew. Chem., Int. Ed., 2018, 58, 1153 CrossRef PubMed; (d) J. Zhang, Z. L. Liu, Z. Yin, X. F. Yang, Y. M. Ma, R. Szostak and M. Szostak, Org. Lett., 2020, 22, 9500 CrossRef CAS PubMed; (e) M. Saito, S. Murakami, T. Nanjo, Y. Kobayashi and Y. Takemoto, J. Am. Chem. Soc., 2020, 142, 8130 CrossRef CAS.
- (a) A. N. Desnoyer and J. A. Love, Chem. Soc. Rev., 2017, 46, 197 RSC; (b) J. Lou, Q. Wang, P. Wu, H. Wang, Y.-G. Zhou and Z. Yu, Chem. Soc. Rev., 2020, 49, 4307 RSC; (c) R. W. Newberry, B. VanVeller, I. A. Guzei and R. T. Raines, J. Am. Chem. Soc., 2013, 135, 7843 CrossRef CAS PubMed; (d) J. Luo, M. Rauch, L. Avram, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2020, 142, 21628 CrossRef CAS PubMed.
- (a) E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257 CrossRef CAS PubMed; (b) C. A. Grice, K. L. Tays, B. M. Savall, J. M. Wei, C. R. Butler, F. U. Axe, S. D. Bembenek, A. M. Fourie, P. J. Dunford, K. Lundeen, F. Coles, X. H. Xue, J. P. Riley, K. N. Williams, L. Karlsson and J. P. Edwards, J. Med. Chem., 2008, 51, 4150 CrossRef CAS PubMed.
- (a) D. Ma, X. Lu, L. Shi, H. Zhang, Y. Jiang and X. Liu, Angew. Chem., Int. Ed., 2010, 50, 1118 CrossRef PubMed; (b) Y. Sun, H. Jiang, W. Wu, W. Zeng and X. Wu, Org. Lett., 2013, 15, 1598 CrossRef CAS PubMed; (c) P. Dang, W. L. Zeng and Y. Liang, Org. Lett., 2015, 17, 34 CrossRef CAS PubMed.
- (a) K. G. Liu, J. R. Lo, T. A. Comery, G. M. Zhang, J. Y. Zhang, D. M. Kowal, D. L. Smith, L. Di, E. H. Kerns, L. E. Schechter and A. J. Robichaud, Bioorg. Med. Chem. Lett., 2009, 19, 1115 CrossRef CAS PubMed; (b) G. Pochetti, N. Mitro, A. Lavecchia, F. Gilardi, N. Besker, E. Scotti, M. Aschi, N. Re, G. Fracchiolla, A. Laghezza, P. Tortorella, R. Montanari, E. Novellino, F. Mazza, M. Crestani and F. Loiodice, J. Med. Chem., 2010, 53, 4354 CrossRef CAS PubMed.
- (a) J. Y. Kim, S. H. Cho, J. Joseph and S. Chang, Angew. Chem., Int. Ed., 2010, 49, 9899 CrossRef CAS PubMed; (b) S. Wertz, S. Kodama and A. Studer, Angew. Chem., Int. Ed., 2011, 50, 11511 CrossRef CAS PubMed; (c) J. Liu and J. M. Hoover, Org. Lett., 2019, 21, 4510 CrossRef CAS PubMed.
- (a) C. Villa, E. Mariani, A. Loupy, C. Grippo, G. C. Grossia and A. Bargagna, Green Chem., 2003, 5, 623 RSC; (b) V. Polshettiwar and R. S. Varma, Acc. Chem. Res., 2008, 41, 629 CrossRef CAS PubMed; (c) A. Sarkar, S. Santra, S. K. Kundu, A. Hajra, G. V. Zyryanov, O. N. Chupakhin, V. N. Charushinb and A. Majee, Green Chem., 2016, 18, 4475 RSC.
- (a) Q. Sun and X. G. Bao, Chem. Commun., 2022, 58, 2216 RSC; (b) L. M. Bouchet, A. A. Heredia, J. E. Argüello and L. C. Schmidt, Org. Lett., 2020, 22, 610 CrossRef CAS PubMed.
- (a) P. Debnath, M. Baeten, N. Lefèvre, S. V. Daele and B. U. W. Maes, Adv. Synth. Catal., 2015, 357, 197 CrossRef CAS; (b) M. Baeten and B. U. W. Maes, Adv. Synth. Catal., 2016, 358, 826 CrossRef CAS.
- (a) C. Zhu, Y. Liang, X. Hong, H. Sun, W.-Y. Sun, K. N. Houk and Z. Shi, J. Am. Chem. Soc., 2015, 137, 7564 CrossRef CAS PubMed; (b) T. Otani, A. Tsuyuki, T. Iwachi, S. Someya, K. Tateno, H. Kawai, T. Saito, K. S. Kanyiva and T. Shibata, Angew. Chem., Int. Ed., 2017, 56, 3906 CrossRef CAS PubMed; (c) L. Habert and K. Cariou, Angew. Chem., Int. Ed., 2020, 60, 171 CrossRef PubMed; (d) Y. Wang, M. Yang, Y.-Y. Sun, Z.-G. Wu, H. Dai and S. H. Li, Org. Lett., 2021, 23, 8750 CrossRef CAS PubMed.
- (a) K. C. Nicolaou, C. J. N. Mathison and T. Montagnon, J. Am. Chem. Soc., 2004, 126, 5192 CrossRef CAS PubMed; (b) N. Mahanta, D. M. Szantai-Kis, E. J. Petersson and D. A. Mitchell, ACS Chem. Biol., 2019, 14, 142 CrossRef CAS; (c) H. J. Kim, D. W. Graham, A. A. DiSpirito, M. A. Alterman, N. Galeva, C. K. Larive, D. Asunskis and P. M. A. Sherwood, Science, 2004, 305, 1612 CrossRef CAS PubMed; (d) A. Okano, R. C. James, J. G. Pierce, J. Xie and D. L. Boger, J. Am. Chem. Soc., 2012, 134, 8790 CrossRef CAS PubMed.
- (a) D. Kaiser, A. Bauer, M. Lemmerer and N. Maulide, Chem. Soc. Rev., 2018, 47, 7899 RSC; (b) K. B. Wiberg and D. J. Rush, J. Am. Chem. Soc., 2001, 123, 2038 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2223988 and 2224291. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc00301a |
| This journal is © The Royal Society of Chemistry 2023 |