Speciation of arsenic in sulfidic waters
Richard T. Wilkina, Dirk Wallschlägerb and Robert G. Forda
aU.S. Environmental Protection Agency, Office of Research and Development, National Risk Management Research Laboratory, 919 Kerr Research Drive, Ada, OK 74820, USA
bEnvironmental and Resource Studies Program, Trent University, 1600 West Bank Drive, Peterborough, ON K9J 7B8, Canada
First published on 7718th March 2003
Abstract
Formation constants for thioarsenite species have been determined in dilute solutions at 25 °C, ΣH2S from 10−7.5 to 10−3.0 M, ΣAs from 10−5.6 to 10−4.8 M, and pH 7 and 10. The principal inorganic arsenic species in anoxic aquatic systems are arsenite, As(OH)30, and a mononuclear thioarsenite with an S/As ratio of 3 ∶ 1. Thioarsenic species with S/As ratios of 1 ∶ 1, 2 ∶ 1, and 4 ∶ 1 are lesser components in sulfidic solutions that might be encountered in natural aquatic environments. Thioarsenites dominate arsenic speciation at sulfide concentrations > 10−4.3 M at neutral pH. Conversion from neutral As(OH)30 to anionic thioarsenite species may regulate the transport and fate of arsenic in sulfate-reducing environments by governing sorption and mineral precipitation reactions.
1. Introduction
The chemical speciation of arsenic in natural waters determines its reactivity, toxicity, and transport in the environment.1–4 Historically, the formation of soluble thioarsenic species has been recognized as an important factor governing arsenic chemistry in reducing environments.5–17 This observation has been paralleled by efforts to identify and quantify the chemistry controlling the formation of thioarsenic species in nature, yet reliable analytical strategies for these species are not fully developed.18 Speciation models derived from thermodynamic analysis of arsenic sulfide solubility in aqueous systems support the existence of thioarsenite species,9,12–15 as do more recent molecular orbital theory calculations and Raman spectroscopic data.16,17,19 Despite these extensive efforts, available data are limited for many practical applications because they provide an indirect quantification of thioarsenite stability and stoichiometry at conditions that are often unrepresentative of aquatic environments, i.e., at saturation with respect to an arsenic sulfide. Excluding some low-pH environments,20 natural systems are usually found to be highly undersaturated with respect to arsenic sulfides such as orpiment.21,22 Development of thermodynamic data for mineral and aqueous species is critical towards assessment of arsenic chemistry in sulfate-reducing environments. This geochemical setting is commonly encountered in organic-rich surface and groundwater systems, e.g., landfill leachate plumes,23 hydrocarbon contaminant plumes,24 and lacustrine to marine systems.25,26In this paper, we provide new experimental data that explore arsenic speciation in sulfidic waters and examine the results of previous solubility studies in light of the new experimental evidence. Reported herein is a direct analytical quantification of thioarsenite species formed under environmentally relevant conditions.
2. Experimental
In order to make accurate predictions of arsenic mobility in reducing natural systems, an understanding is needed of the stoichiometry and stability of dissolved arsenic species in sulfide-deficient and sulfide-bearing solutions. We examined the change in As(III) speciation from arsenite to a distribution of thioarsenic species with increasing sulfide concentrations in aqueous solution. The switch in arsenic speciation from oxyanionic to sulfoxyanionic forms was reversible and triggered by adding aliquots of a 1 mM sodium bisulfide buffer to solutions containing about 2 to 15 µM arsenite from NaAsO2. The concentrations of arsenite and individual thioarsenic species were determined by ion chromatography–inductively coupled plasma–mass spectrometry (IC-ICP-MS). Solutions were prepared at nominal pH values of 7 and 10 with ΣH2S/ΣAs ranging from 0.002 to 350 on a molar basis. Actual pH values varied by as much as 0.8 units from the nominal values. Experimental conditions were selected to be undersaturated with respect to amorphous As2S3. Based upon Eary's solubility model for amorphous As2S3,15 arsenic concentrations were between 0.02 and 50% of saturation at pH 7 (Fig. 1a). At pH 10, solutions were highly undersaturated with respect to amorphous As2S3; total arsenic concentrations were always less than 0.1% of those expected at saturation (Fig. 1b).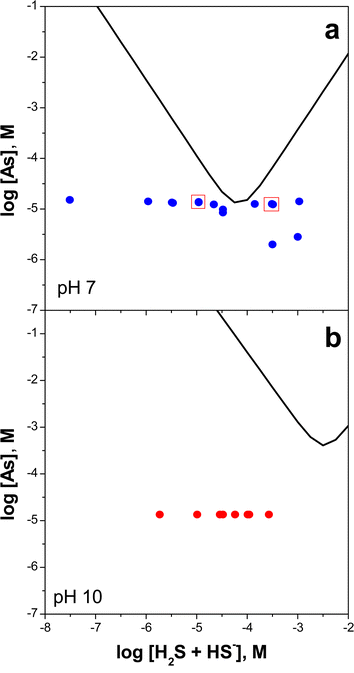 | ||
| Fig. 1 Saturation state of experiments conducted at (a) pH 7 and (b) pH 10 relative to the predicted solubility of amorphous As2S3 (bold V-shaped curves, ref. 15). Boxed points in (a) correspond to the chromatographic traces shown in Fig. 2. | ||
All solutions were prepared using deoxygenated and distilled water (Milli-Q 18 MΩ). Removal of dissolved oxygen to concentrations < 0.2 mg L−1 was verified by using rhodazine D colorimetric test kits (Chemetrics, K-7501). Sodium bisulfide solutions were prepared by purging dilute oxygen-free sodium hydroxide solution with high-purity 1% (v/v) hydrogen sulfide gas. The resulting buffer solution had a pH of 7.0 and a ΣH2S concentration of 1.1 mM. In all cases, bisulfide solutions were used within 12 h of preparation. Arsenite solutions were prepared by dissolving quantities of NaAsO2 (Fisher Reagent Grade) into nitrogen-purged water.
Arsenic and bisulfide solutions were mixed and sealed in 45 mL glass reaction vessels within an anaerobic glove box and allowed to equilibrate for 1 to 4 h. After this time period, solutions were flash-frozen in liquid nitrogen and kept frozen until analysis to insure species preservation. Experiments were conducted at pH 7 (±0.3) and pH 10 (±0.8). In the high-pH experiments, pH was adjusted by adding aliquots of 0.01 M NaOH. All experiments were conducted at room temperature (23 ± 1.5 °C). Reversibility was tested by spiking solutions with arsenite immediately prior to analysis and tracking the re-equilibration of arsenic speciation. The results of these tests indicated that rates of equilibration are rapid among the oxyanion and thiooxyanions of arsenic in sulfidic solutions.
Arsenic speciation was determined by ion chromatography coupled on-line to inductively-coupled plasma mass spectrometry (IC-ICP-MS). Chromatographic separation was achieved for 1 mL samples using gradient elution with dilute NaOH on a high-capacity anion exchange column, similar to a method previously used for selenium speciation in natural waters.27 A DX-500 ion chromatograph (Dionex) and an Elan 6000 ICP-MS (Perkin Elmer) were used for these experiments. Arsenic and sulfur were simultaneously detected by monitoring the m/z = 75 (As) and 48 (SO) signals, and quantified based on peak area. From the obtained signals, the As/S ratios for the separated thioarsenic species were calculated in each sample, and then averaged over all samples in which no apparent interferences on either signal were observed. At constant ΣH2S/ΣAs, variations in sample pH yielded differing speciation trends, evidence that equilibration of the sample during chromatographic elution did not direct the outcome of the speciation results. The analytical method is described in detail in a forthcoming publication (Wallschläger et al., in prep.).
3. Results and discussion
Thioarsenite species
Experimental conditions and results are listed in Table 1. Results indicate that, in addition to arsenite, sulfidic solutions may contain up to four distinct thioarsenic species with average S/As ratios of 1 ∶ 1, 2 ∶ 1, 3 ∶ 1, and 4 ∶ 1 (Fig. 2, Table 2). The same species appear in solutions highly undersaturated with respect to amorphous As2S3 and in near-saturated solutions. Recovery of arsenic during analysis was in all cases between 86 and 102%, which indicates that there was no major loss of arsenic during chromatographic separation and that the principal arsenic species are accounted for in our analysis. At pH 7 and ΣAs = 10−4.9 M, the crossover from arsenite- to thioarsenic-dominated speciation occurred at ΣH2S of about 10−4.3 M. The crossover in arsenic speciation is in excellent correspondence with the ΣH2S concentration where the slope on a log [As] versus log [ΣH2S] solubility diagram transitions from a negative to a positive value (Fig. 1a). Eary15 proposed that the change in slope at higher total sulfide concentrations indicates a change in arsenic speciation from arsenite to thioarsenite, either the mononuclear 3 ∶ 1 thioarsenite species (As(SH)30), the trinuclear 2 ∶ 1 species (As3(SH)3S30), or their respective deprotonated forms. The results of this study suggest that in fact multiple species with S/As ratios ranging from 1 ∶ 1 to 4 ∶ 1 account for the solubility of orpiment in sulfidic solutions.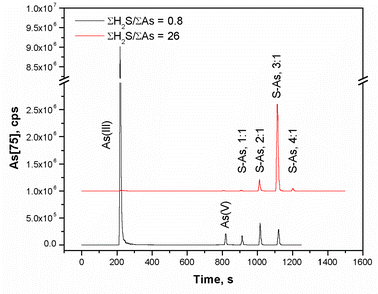 | ||
| Fig. 2 Mass spectral chromatograms of mass 75(As) showing the change in arsenic speciation at pH 7 from arsenite- to thioarsenite-dominated with increasing ΣH2S/ΣAs. | ||
| Exp. | pH | log ΣH2S | log ΣAs | Fractional abundances | |||||
|---|---|---|---|---|---|---|---|---|---|
| As(III) | As(V) | S ∶ As 1 ∶ 1 | S ∶ As 2 ∶ 1 | S ∶ As 3 ∶ 1 | S ∶ As 4 ∶ 1 | ||||
| a Concentrations in M. Concentrations of ΣH2S were calculated based on gravimetric mixing ratios of the stock NaHS and arsenite solutions, and the measured thioarsenite speciation; ΣH2S concentration in the stock NaHS solution was determined from the average of quadruplicate analyses using the methylene blue colorimetric method. Concentrations of ΣAs were determined by inductively coupled plasma–optical emission spectroscopy on unacidified samples. Precision of measurements: pH ∼ 0.03; ΣH2S ∼ 5%; ΣAs ∼ 5%; species fractional abundance (F) ∼5% for F > 0.1, ∼10% for 0.1 > F > 0.01, ∼20% for F < 0.01. | |||||||||
| 1 | 7.24 | −7.51 | −4.82 | 0.998 | 0.002 | 0.000 | 0.000 | 0.000 | 0.000 |
| 2 | 7.30 | −5.49 | −4.87 | 0.988 | 0.005 | 0.001 | 0.005 | 0.002 | 0.000 |
| 3 | 7.20 | −4.96 | −4.87 | 0.917 | 0.015 | 0.012 | 0.032 | 0.024 | 0.000 |
| 4 | 7.01 | −4.28 | −4.93 | 0.102 | 0.011 | 0.005 | 0.072 | 0.811 | 0.000 |
| 5 | 7.20 | −3.85 | −4.90 | 0.007 | 0.001 | 0.001 | 0.027 | 0.962 | 0.003 |
| 6 | 7.12 | −3.51 | −4.91 | 0.009 | 0.002 | 0.007 | 0.139 | 0.838 | 0.005 |
| 7 | 7.04 | −3.49 | −4.91 | 0.011 | 0.003 | 0.004 | 0.094 | 0.865 | 0.023 |
| 8 | 7.17 | −2.97 | −4.85 | 0.000 | 0.004 | 0.000 | 0.063 | 0.847 | 0.086 |
| 9 | 7.16 | −3.50 | −5.63 | 0.000 | 0.004 | 0.004 | 0.078 | 0.902 | 0.012 |
| 10 | 7.00 | −3.00 | −5.55 | 0.000 | 0.012 | 0.006 | 0.125 | 0.805 | 0.053 |
| 11 | 6.79 | −5.96 | −4.85 | 0.994 | 0.010 | 0.000 | 0.000 | 0.000 | 0.000 |
| 12 | 7.24 | −5.47 | −4.88 | 0.973 | 0.016 | 0.002 | 0.004 | 0.000 | 0.000 |
| 13 | 7.05 | −4.96 | −4.87 | 0.960 | 0.005 | 0.014 | 0.021 | 0.000 | 0.000 |
| 14 | 7.20 | −4.66 | −4.91 | 0.896 | 0.011 | 0.038 | 0.054 | 0.001 | 0.000 |
| 15 | 7.30 | −4.48 | −4.94 | 0.748 | 0.030 | 0.026 | 0.089 | 0.095 | 0.000 |
| 16 | 9.13 | −5.73 | −4.88 | 0.942 | 0.033 | 0.012 | 0.005 | 0.009 | 0.000 |
| 17 | 10.02 | −4.99 | −4.94 | 0.840 | 0.000 | 0.114 | 0.045 | 0.003 | 0.000 |
| 18 | 9.83 | −4.48 | −4.89 | 0.650 | 0.132 | 0.127 | 0.185 | 0.009 | 0.000 |
| 19 | 9.80 | −3.80 | −4.89 | 0.290 | 0.035 | 0.024 | 0.198 | 0.454 | 0.000 |
| 20 | 10.90 | −3.57 | −4.86 | 0.054 | 0.034 | 0.041 | 0.251 | 0.620 | 0.000 |
| 21 | 9.51 | −4.54 | −4.96 | 0.800 | 0.000 | 0.124 | 0.063 | 0.005 | 0.000 |
| 22 | 9.67 | −4.24 | −4.87 | 0.710 | 0.000 | 0.200 | 0.090 | 0.000 | 0.000 |
| 23 | 9.20 | −3.96 | −4.94 | 0.470 | 0.050 | 0.071 | 0.221 | 0.184 | 0.000 |
| Exp. | Species | Elution time/s | Arsenic/µmol L−1 | Sulfur/µmol L−1 | Molar ratio (S/As)a |
|---|---|---|---|---|---|
| a Among all samples variability of molar S/As ratios for the 2 ∶ 1, 3 ∶ 1, and the 4 ∶ 1 complexes was 2.1 ± 0.3, 3.1 ± 0.7, and 4.2 ± 0.3, respectively.b The first S–As complex is likely to be a 1 ∶ 1 complex but in most cases the sulfur from this thioarsenite species co-eluted with sulfur from other sulfur species. This was evidenced by noncoincident retention times for As and S peaks. n.d., not detected. | |||||
| 3 | Arsenite | 217 | 12.3 | n.d. | – |
| Arsenate | 827 | 0.20 | n.d. | – | |
| S–As (1 ∶ 1) | 922 | 0.17 | b | – | |
| S–As (2 ∶ 1) | 1024 | 0.43 | 0.97 | 2.25 | |
| S–As (3 ∶ 1) | 1129 | 0.32 | 1.06 | 3.31 | |
| S–As (4 ∶ 1) | – | n.d. | n.d. | – | |
| 7 | Arsenite | 231 | 0.14 | n.d. | – |
| Arsenate | 815 | 0.04 | n.d. | – | |
| S–As (1 ∶ 1) | 914 | 0.05 | b | – | |
| S–As (2 ∶ 1) | 1016 | 1.16 | 2.78 | 2.40 | |
| S–As (3 ∶ 1) | 1119 | 10.6 | 30.5 | 2.88 | |
| S–As (4 ∶ 1) | 1205 | 0.28 | 1.09 | 3.91 | |
At pH 10, the observed crossover in arsenic speciation from oxyanionic to sulfoxyanionic forms shifted to a slightly higher ΣH2S concentration of about 10−3.5 M. Based on the solubility model for amorphous As2S3,15 the crossover point at pH 10 is predicted to be ΣH2S = 10−2.5. However, it should be noted that the 25 °C As2S3 solubility determinations of Eary,15 Webster,12 and Mironova et al.14 were in all cases determined below pH 9, hence the solubility curve shown on Fig. 1a has been extrapolated outside of the measured range.
Analysis of the isothermal pH- and ΣH2S-dependent solubility of crystalline and amorphous As2S3 represents the method most often used to determine the formation constants of thioarsenic species.11–16 Proposed arsenic species in sulfidic waters include mononuclear (e.g., AsS2−), dinuclear (e.g., As2S42−), and trinuclear thioarsenites (e.g., H2As3S6−). Trimeric thioarsenite species have frequently been selected as the solubility-controlling species of disordered and crystalline orpiment in sulfidic solutions.11,12,15 However, as noted above solubility data for orpiment are consistent with the formation of either mononuclear 3 ∶ 1 species, trinuclear 2 ∶ 1 thioarsenite species, or both. Underlying thermodynamic analysis of mineral-buffered experiments in the system As–S–O–H is the fact that the chemical potential of As2S3 is fixed by equilibration with a solid (orpiment); consequently, it is not possible to accurately distinguish between monomeric and polymeric species.15,16 In related studies, molecular orbital theory has been used to calculate bond distances, vibrational frequencies, gas-phase energetics, and proton affinities for various thioarsenite molecules to aid interpretation of EXAFS and Raman spectra of arsenic in concentrated 1 M NaHS solutions.16,28 Based on modeling results, the existence of dimeric thioarsenic species has been rejected in solutions saturated with As2S3 in favor of monomeric or trimeric species.16 In the context of the present study, the trends in chromatographic elution times (Fig. 2), the fact that the same series of thioarsenic species were observed in solutions ranging from highly undersaturated to near-saturated with respect to amorphous As2S3, and the dominance of the 3 ∶ 1 complex are all consistent with the presence of mononuclear thioarsenite species at the conditions of our experiments (ΣAs from 10−5.6 to 10−4.9 M); conditions typical of groundwater and surface water environments. With increasing pH and ΣH2S, arsenic concentrations at saturation with orpiment easily exceed 10−2 M; conditions where polynuclear species might be anticipated.
The 4 ∶ 1 thioarsenite compound observed at pH 7 and ΣH2S/ΣAs > 20 is somewhat unexpected although this complex stoichiometry has been previously proposed.7 Arsenic(III) compounds are expected to involve 3 bonded atoms plus a sterically active but non-bonding electron pair. The 4 ∶ 1 species, if it is an As(III) complex, would apparently not be able to accommodate the non-bonding electron pair. An alternative explanation would be a thioarsenate, AsS4Hx(x−3), with no non-bonding electron pair. The formation of a 4 ∶ 1 thioarsenate would imply that oxidation of As(III) to As(V) occurred in relatively reducing solutions at near-neutral pH but not in weakly sulfidic and moderately alkaline conditions where faster oxidation kinetics might be expected and arsenate was sometimes detected. The 4 ∶ 1 species was not detected in experiments at pH 10 and ΣH2S < 10−3.3. Reaction of arsenate with bisulfide and tetrasulfide solutions (from Na2S4) under similar conditions yielded only minor quantities of the S ∶ As 1 ∶ 1, 2 ∶ 1, and 3 ∶ 1 species and none of the S ∶ As 4 ∶ 1 species. This observation suggests that the S ∶ As 4 ∶ 1 species is a true thioarsenite and that the observed 1 ∶ 1, 2 ∶ 1 and 3 ∶ 1 thioarsenites were generated in experiments with sulfides and polysulfides through reduction of arsenate and complexation with sulfide.
In order to evaluate formation constants for thioarsenite species, we measured concentrations of arsenite, arsenate, and the four principal thioarsenite species in the experimental solutions using IC-ICP-MS spectra. Although arsenate was detected in most solutions analyzed, the fractional abundance of arsenate was typically negligible (av. = 0.018, n = 23) and arsenate was not considered as a component of arsenic speciation in sulfidic systems, either as an oxyanion or as a thioarsenate.17 Greater arsenate fractional abundance was found in the pH 10 experiments (0.032 ± 0.045, n = 8) as compared to the pH 7 experiments (0.009 ± 0.008, n = 15). This suggests that arsenate was likely an oxidation product of arsenite and we suspect that most arsenate production occurred during chromatographic separation.
At each sulfide concentration investigated, the species distribution of arsenic may be defined as the sum of all arsenic(III) species present in solution:
| ΣAs = ΣAs(OH)30 + ΣAs(OH)2(SH)0 + ΣAs(OH)(SH)20 +ΣAs(SH)30 + ΣAs(SH)4H0 | (1) |
 | ||
| Fig. 3 (a) Distribution of arsenite and thioarsenite species at pH 7 as a function of the concentration of bisulfide based on the formation constants determined in this study (Table 4). Data points correspond to the measured fractional abundance of arsenite at pH 7 (open circles). (b)–(e) Modeled distribution (pH 7, red curves; pH 10, blue curves) and measured fractional abundances of S ∶ As 1 ∶ 1, 2 ∶ 1, 3 ∶ 1, and 4 ∶ 1 species at pH 7 (open circles) and pH 10 (filled circles). Standard errors show uncertainty in the fractional abundance of arsenic species. | ||
| S/As | Charge (0) | Charge (1−) | Charge (2−) | Charge (3−) |
|---|---|---|---|---|
| 0 | As(OH)30 | As(OH)2O− | As(OH)O22− | AsO33− |
| 1 | As(OH)2(SH)0 | As(OH)2S− | AsO2HS2− | AsO2S3− |
| 2 | As(OH)(SH)20 | As(OH)S2H− | As(OH)S22− | AsOS23− |
| 3 | As(SH)30 | AsS3H2− | AsS3H2− | AsS33− |
| 4 | As(SH)4H0 | As(SH)4− | As(SH)3S2− | As(SH)2S23− |
| Reaction | log K 25°, I = 0 |
|---|---|
| a Results are mean values ± standard error (2σ) determined from the fractional abundance of arsenite and thioarsenites in model solutions. Activity coefficients for charged species were estimated using the Davies equation. | |
| As(OH)30 + HS− + H+ ⇌ As(OH)2(SH)0 + H2O | 8.69 ± 0.29 (n = 10) |
| As(OH)30 + HS− ⇌ As(OH)2S− + H2O | 3.54 ± 0.36 (n = 8) |
| As(OH)2S− + HS− ⇌ As(OH)S22− + H2O | 5.06 ± 0.49 (n = 17) |
| As(OH)S22− + HS− + H+ ⇌ AsS3H2− + H2O | 11.78 ± 0.89 (n = 11) |
| As(OH)S22− + HS− ⇌ AsS33− + H2O | 3.89 ± 0.41 (n = 6) |
| AsS3H2− + HS− + 2H+ ⇌ As(SH)4− | 16.16 ± 0.29 (n = 6) |
The 4 ∶ 1 complex was only detected in experiments at pH 7. Although the fractional abundance of the 4 ∶ 1 species was never > 0.086, it was present at quantifiable concentrations at ΣH2S concentrations above 0.14 mM (n = 6). At pH 7, the S ∶ As 4 ∶ 1 species is predicted to become the dominant thioarsenite at ΣH2S > 10−2 M. Unfortunately, this concentration is outside the range that can be currently investigated with the IC-ICP-MS method without sample dilution. Natural sulfidic systems rarely exceed ΣH2S > 10−3 M so that the 4 ∶ 1 species is unlikely to be a major component of arsenic speciation in aquatic environments.
Orpiment solubility
The results of this study suggest that the solubility of amorphous or crystalline As2S3 in sulfidic waters is controlled by at least four thioarsenite species, each with multiple protonation states. In addition to the polynuclear complexes documented in previous studies, additional solubility expressions are possible for amorphous As2S3:| 0.5As2S3 (am) + 2H2O ⇌ As(OH)2S− + 0.5HS− + 1.5H+ | (2) |
| 0.5As2S3 (am) + 0.5HS− + H2O ⇌ As(OH)S22− + 1.5H+ | (3) |
| 0.5As2S3 (am) + 1.5HS− ⇌ AsS3H2− + 0.5H+ | (4) |
| 0.5As2S3 (am) + 2.5HS− + 1.5H+ ⇌ As(SH)4− | (5) |
| 0.5As2S3 (am) + 3H2O ⇌ As(OH)30 + 1.5HS− + 1.5H+ | (6) |
Estimated log K values for reactions (2)–(5) are −18.9 ± 0.4, −13.8 ± 0.5, −2.0 ± 0.9, and 14.1 ± 0.3, respectively. Arsenic solubility predicted by reactions (2)–(5) and (6) at pH 5 is plotted and compared in Fig. 4a to 25 °C solubility measurements reported in Eary.15 Visual inspection of Fig. 4a indicates that the 5-species model is in reasonable agreement with the solubility measurements as compared to the 2-species model shown in Fig. 4b. Note that no attempt was made to improve the fit to the orpiment solubility data and that data in Table 4 derived from experiments at pH 7 and 10 were extrapolated to pH 5. Fig. 4a was constructed using only reaction (6) and data that were derived from an independent set of measurements made in the absence of a solid phase. The calculated slope, ∂log[As]/∂log[H2S0], based on linear regression analysis of the solubility data with positive trend ranges from 1.75 to 1.91 depending on how the transition point is selected. In addition, regression analysis of amorphous As2S3 solubility data15 collected at 40 to 90 °C yields slopes that range from 1.64 to 2.15. These non-integer values are perhaps unlikely to be the result of a single thioarsenite species and may be the result of the presence of multiple thioarsenic species with varying S ∶ As ratios in solutions saturated with As2S3. We suggest that because of the complexity of arsenic speciation in sulfidic solutions as indicated here and in previous studies,17 speciation analysis using direct methods is necessary to yield accurate speciation models from solubility measurements. Data sets provided in previous reports still provide detailed insight into total arsenic concentrations at saturation with crystalline and amorphous As2S3 and as such will continue to provide the basis for speciation studies.
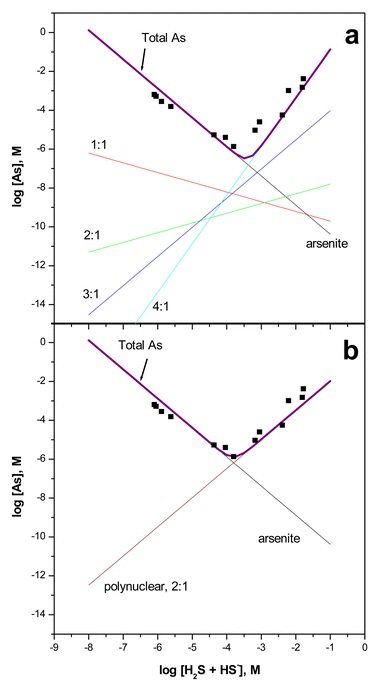 | ||
| Fig. 4 Experimental amorphous As2S3 solubilities at 25 °C from ref. 15 (squares) compared to predicted solubilities based upon (a) thioarsenite formation constants in Table 4 and selected thermodynamic constants (see text) and (b) the 2-species model, for example, as proposed in ref. 15. | ||
Thioarsenite species in the environment
At near-neutral to mildly alkaline conditions the fractional abundance of arsenite relative to the sum of all thioarsenic species increases with increasing pH at a given ΣH2S concentration (Fig. 5a). This trend correlates with greater solubility of orpiment at near neutral to alkaline pH compared to low pH.12,14,15 Experimental results indicate that thioarsenite forms dominate over arsenite when sulfide concentrations are greater than about 0.1 to 1 mM. In general, concentrations of dissolved sulfide in aquatic systems are controlled by the rate of bacterial sulfate reduction and by the nature and abundance of iron in the solid matrix. Concentrations of dissolved sulfide are typically low when reactive iron is abundant, i.e., iron present in iron oxyhydroxides, iron oxides, or Fe-bearing silicates. Dissolved sulfide concentrations are able to increase only after the supply of reactive iron is exhausted via reductive dissolution processes and subsequent iron monosulfide precipitation.30 Dissolved iron and sulfide are usually near saturation with respect to mackinawite (Fe1 + xS) or greigite (Fe3S4)31,32 and this relationship can be used to understand the conditions in which thioarsenites are expected to occur in natural environments (Fig. 5b). In solutions saturated with mackinawite over the pH range from 6 to 10, thioarsenites will dominate arsenic speciation only when dissolved ferrous iron concentrations are low (10−10 M to 10−6 M). In other words, the presence of reactive iron will generally preclude the formation of thioarsenic species in sulfate-reducing systems by maintaining low dissolved sulfide concentrations. Environments in which the abundance of reactive iron is limited will favor sulfide accumulation and thioarsenite formation. | ||
| Fig. 5 (a) Modeled fractional abundance of arsenite relative to thioarsenite species as a function of pH and sulfide concentration based on formation constants determined in this study (Table 4). (b) Predominance area diagram for aqueous arsenic species. The contours represent the activity of ferrous iron in equilibrium with mackinawite (FeS) at 25 °C. | ||
The mobility of arsenic in aquatic systems is governed mainly by redox conditions.1–4,33,34 Arsenic sulfides, iron–arsenic sulfides, sulfosalts, and iron oxides are believed to be the principal sources of arsenic to anaerobic ground waters through microbial and non-microbial processes.4,35,36 Based on the results of this study, dissolution reactions of arsenic-bearing materials should involve thioarsenite species in weakly to strongly sulfidic systems (Table 4; Fig. 5a). Although the eventual fate and distribution of thioarsenic species requires more investigation, these species are likely to be involved in adsorption, precipitation, and/or oxidation reactions. For example, precipitation of authigenic pyrite (FeS2) is known to scavenge arsenic from solution.37,38 However, it is not clear if arsenic partitioning to pyrite is controlled by arsenite or by thioarsenite species. Precipitation of orpiment is another possible sink for thioarsenite in low pH environments. Orpiment has a molecular structure of linked pyramidal AsS3 groups.39,40 The covalently bonded AsS3 units in orpiment are bound together by weaker chemical forces that give rise to the lack of hardness and low melting point of this mineral. Condensation of ΣAs(SH)30 thioarsenite species provides a reasonable pathway for nucleation and growth of orpiment.
Thioarsenite species may act as multi-dentate chelating agents that enhance the transport of highly-polarized (B-type) cations, such as Cu(II), Au(I,III), Ag(I), Tl(I), and Hg(II).41 Copper concentrations in sulfidic wetland systems, in addition to concentrations of zinc and cadmium, are too high to be accounted for by the solubility of simple metal sulfides and investigations suggest that existing thermodynamic data are too incomplete to accurately describe the factors that govern metal mobility in sulfidic systems.22 Yet when solubility behavior in ternary systems has been studied, increases of several orders of magnitude in copper solubility can occur due to the formation of mixed Cu–thioarsenite species in the sulfide concentration range of 0.001 to 0.1 mM.41 Our limited understanding of ternary metal–As–S systems is significant considering the potential importance of such species for regulating contaminant transport and fate processes in natural systems and at hazardous waste sites. Furthermore, the association between the distributions of arsenic and gold in shallow hydrothermal deposits has long been recognized. Gold is often found in low-temperature (<200 °C) sulfide deposits associated with arsenian pyrite,42 silver–arsenic sulfosalts,43 and as discrete arsenic sulfides.44 Solubility studies are needed in the ternary system Au–As–S to evaluate the possible role of gold transport by thioarsenic species. Results documented herein provide the basis for reliable and accurate determination of ternary metal–thioarsenite complexes.
4. Conclusions
Our study provides direct analytical confirmation of the complexity of the As–S–O–H system as indicated in previous studies. Monomeric thioarsenic species with S/As ratios of 1 ∶ 1, 2 ∶ 1, 3 ∶ 1, and 4 ∶ 1 were detected in model sulfidic solutions. Thioarsenites dominate arsenic speciation at sulfide concentrations > 10−4.3 M at neutral pH and results of this study suggest that thioarsenite species will only persist in iron-limited environments. The formation and fate of thioarsenite species in the environment are not fully understood. New experimental data are needed to explore arsenic sulfide solubility in the context of the multiple thioarsenite species documented here. Because natural aquatic systems are often highly undersaturated with respect to pure arsenic sulfide phases, adsorption and co-precipitation processes involving thioarsenites require investigation.Acknowledgements
The U. S. Environmental Protection Agency through its Office of Research and Development funded the research described here. It has not been subjected to Agency review and therefore does not necessarily reflect the views of the Agency, and no official endorsement should be inferred. Mention of trade names or commercial products does not constitute endorsement or recommendation for use. The manuscript was improved by suggestions from an anonymous reviewer.References
- J. F. Ferguson and J. Gavis, Water Res., 1972, 6, 1259 CrossRef CAS.
- W. R. Cullen and K. J. Reimer, Chem. Rev., 1989, 89, 713 CrossRef.
- N. Korte and Q. Fernando, Crit. Rev. Environ. Control, 1991, 21, 1 Search PubMed.
- P. L. Smedley and D. G. Kinniburgh, Appl. Geochem., 2002, 17, 517 CrossRef CAS.
- R. Höltje, Z. Anorg. Chem., 1929, 181, 395 Search PubMed.
- A. K. Babko and G. S. Lisetskaya, Russ. J. Inorg. Chem., 1956, 1, 95.
- H. N. Srivastava and S. Ghosh, J. Indian Chem. Soc., 1958, 35, 165 Search PubMed.
- J. Angeli and P. Souchay, C. R. Acad. Sci. Paris, 1960, 250, 713 Search PubMed.
- B. W. Weissberg, F. W. Dickson and G. Tunell, Geochim. Cosmochim. Acta, 1966, 30, 815 CrossRef CAS.
- V. E. Thilo, K. Hertzog and A. Winkler, Z. Anorg. Chem., 1970, 373, 111 Search PubMed.
- N. F. Spycher and M. H. Reed, Geochim. Cosmochim. Acta, 1989, 53, 2185 CrossRef CAS.
- J. G. Webster, Geochim. Cosmochim. Acta, 1990, 54, 1009 CrossRef CAS.
- R. E. Krupp, Geochim. Cosmochim. Acta, 1990, 54, 3239 CrossRef CAS.
- G. D. Mironova, A. V. Zotov and N. I. Gul'ko, Geochem. Int., 1991, 27, 61 Search PubMed.
- L. E. Eary, Geochim. Cosmochim. Acta, 1992, 56, 2267 CrossRef CAS.
- G. R. Helz, J. A. Tossell, J. M. Charnock, R. Pattrick, D. J. Vaughan and C. D. Garner, Geochim. Cosmochim. Acta, 1995, 59, 4591 CrossRef CAS.
- S. A. Wood, C. D. Tait and D. R. Janecky, Geochem. Trans., 2002, 2002, 4 RSC.
- G. Schwedt and M. Rieckhoff, J. Chromatogr., A, 1996, 736, 341 CrossRef CAS.
- J. A. Tossell, Inorg. Chem., 2001, 40, 6487 CrossRef CAS.
- H. W. Langer, C. R. Jackson, T. R. McDermott and W. P. Inskeep, Environ. Sci. Technol., 2001, 35, 3302 CrossRef.
- L. S. Balistrieri, J. W. Murray and B. Paul, Geochim. Cosmochim. Acta, 1994, 58, 3993 CrossRef CAS.
- C. H. Gammons and A. K. Frandsen, Geochem. Trans., 2001, 2001, 1 RSC.
- T. H. Christensen, P. L. Bjerg, S. A. Banwart, R. Jakobsen, G. Heron and H. Albrechtsen, Appl. Geochem., 2001, 16, 659 CrossRef CAS.
- D. A. Vroblesky and F. H. Chapelle, Water Resour. Res., 1994, 30, 1561 CrossRef CAS.
- C. M. Wicks, J. S. Herman and A. L. Mills, Appl. Geochem., 1991, 6, 213 CrossRef CAS.
- M. B. Goldhaber and I. R. Kaplan, in Marine Chemistry, ed. E. D. Goldberg, Wiley-Interscience, New York, 1974, pp. 569–655 Search PubMed.
- D. Wallschläger and R. Roehl, J. Anal. At. Spectrom., 2001, 16, 922 RSC.
- J. A. Tossell, Environ. Sci. Technol., 2000, 34, 1483 CrossRef CAS.
- D. D. Wagman, W. H. Evans, V. B. Parker, R. H. Schumm, I. Halow, S. M. Bailey, K. L. Churney and R. L. Nutall, J. Phys. Chem. Ref. Data, 1982, 11, 1 CAS.
- D. E. Canfield, Geochim. Cosmochim. Acta, 1989, 53, 619 CrossRef CAS.
- R. T. Wilkin and H. L. Barnes, Am. J. Sci., 1997, 297, 620 Search PubMed.
- L. E. Bågander and R. Carmen, Appl. Geochem., 1994, 9, 379 CrossRef.
- A. W. Hounslow, Ground Water, 1980, 18, 331 Search PubMed.
- A. H. Welch, M. S. Lico and J. Hughes, Ground Water, 1988, 26, 333 Search PubMed.
- M. Kim, J. Nriagu and S. Haack, Environ. Sci. Technol., 2000, 34, 3094 CrossRef.
- D. Ahmann, L. R. Krumholz, H. F. Hemond, D. R. Lovely and F. M. M. Morel, Environ. Sci. Technol., 1997, 31, 2923 CrossRef CAS.
- R. Raiswell and J. Plant, Econ. Geol., 1980, 75, 684 Search PubMed.
- N. Belzile and J. Lebel, Chem. Geol., 1986, 54, 279 CrossRef CAS.
- N. Morimoto, Mineral. J., 1954, 1, 160 Search PubMed.
- C. J. Brabec, Phys. Rev. B, 1991, 44, 13332 CrossRef CAS.
- M. B. Clarke and G. R. Helz, Environ. Sci. Technol., 2000, 34, 1477 CrossRef CAS.
- J. S. Cline, Econ. Geol., 2001, 96, 75 Search PubMed.
- P. Heald, N. K. Foley and D. O. Hayba, Econ. Geol., 1987, 82, 1 Search PubMed.
- R. E. Krupp and T. M. Seward, Econ. Geol., 1987, 82, 1109 Search PubMed.
| This journal is © The Royal Society of Chemistry 2003 |