Two thousand years of atmospheric rare earth element (REE) deposition as revealed by an ombrotrophic peat bog profile, Jura Mountains, Switzerland†
Michael
Krachler
*a,
Carola
Mohl
b,
Hendrik
Emons
b and
William
Shotyk
a
aInstitute of Environmental Geochemistry, University of Heidelberg, Im Neuenheimer Feld 236, 69120 Heidelberg, Germany. E-mail: krachler@ugc.uni-heidelberg.de; Fax: +49 62 21 54 49 56; Tel: +49 62 21 54 48 48
bInstitute of Phytospheric Research, Research Centre Juelich, Juelich, Germany
First published on 11th December 2002
Abstract
A peat core from a Swiss bog represents 2110 14C years of peat accumulation and provides a continuous record of atmospheric rare earth element (REE) deposition. This is the first study providing a time-series of all REE originating from the atmosphere. Concentrations of the 14 REE (La, Ce, Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, Yb and Lu) were determined using inductively coupled plasma-mass spectrometry (ICP-MS) after dissolution of 200 mg aliquots of age-dated peat samples with 3 ml HNO3 and 0.1 ml HBF4 at 240 °C in a microwave autoclave. Strict quality control schemes were applied to ensure the accuracy of the applied analytical methodology. Previous analyses of selected REE by instrumental neutron activation analysis (INAA) in the same set of peat samples revealed that INAA frequently under- or overestimated REE concentrations in a systematic manner. Concentration profiles obtained for all REE were almost identical, except for Ce and Eu. Calculation of enrichment factors (EF) revealed a distinct depletion of heavy REE relative to light REE in peat samples since the beginning of the 19th century which marks the onset of the Industrial Revolution in Europe, suggesting a pronounced influence by anthropogenic activities. Enrichments of REE calculated using Sc as a reference element exceeded unity, relative to the Upper Continental Crust. Overall, EF in all peat samples ranged from 1.96 for Sm to 2.34 for Gd, with considerably lower EF for Ce (1.82) and Eu (1.44), respectively. A significant enrichment of all REE which may have been caused by military activities, was observed in the peat sample dating from World War II (1944); this exceptional sample, however, is not enriched in Ce. The concentration profiles of REE were similar but not identical to those of other lithogenic, conservative reference elements such as Sc, Y, Al, Zr and Ti. While it has been suggested that individual REE concentrations or the sum of REE can be used as a reference parameter to calculate crustal EF in environmental samples the data presented here indicates that anthropogenic emissions of REE cannot simply be ignored.
Aim of the investigation
Today, human activity is the dominant source affecting the global exogenic cycling of many elements. Anthropogenic fluxes of several elements to the atmosphere clearly exceed the natural fluxes. In this respect, knowledge of past metal deposition allows present-day deposition to be quantified, and seen in a temporal perspective.Peat cores from ombrotrophic bogs have repeatedly proved to be meaningful archives of ancient atmospheric metal deposition.1–6 In this context, Pb has received considerable attention, but also elements such as Cu, Zn, Cd, Hg, Au, As, Sb, etc. have been investigated in more detail.1–7 An ombrotrophic bog is a domed peatland in which the surface peat layers are hydrologically isolated from the influence of local ground and surface waters, and are fed exclusively by atmospheric deposition. Previous geochemical studies revealed that the peat core investigated in the present study is truly ombrotrophic and maintains its status with increasing depth beneath the surface of the bog.1,5
Rare earth elements (REE) are widely used as robust and powerful tracers for a variety of processes in cosmochemistry, igneous petrology and sedimentology because of their low solubility, transport occurring largely in the particulate phase in the atmosphere and their origin from natural sources.8–14 Recently, REE have witnessed growing interest in their use as tracers of environmental processes. For example, REE have been used as a tracer of petroleum refining9 and Gd as a tracer for medical waste.15
Apart from substitution in primary rock forming minerals, the REE occur in a wide variety of REE-rich accessory minerals, such as apatite, titanite, monazite and allanite.10 The concentrations of the REE in monazite and bastnäsite ores, respectively, are summarised in Table 1. The mobility and fractionation of the REE during weathering is highly dependent on their distribution among these minerals, since minerals weather at different rates. However, monazite (a REE phosphate), for example, is comparatively dense. Therefore, one might expect the REE to strongly fractionate, both during chemical weathering in soils and in the air during transport as dust particles.
| Element | World production/t year−1 | Estimated world reserves/t × 106 | Concentration in UCCa (ppm) | Monazite ore (%) | Bastnäsite ore (%) |
|---|---|---|---|---|---|
| a UCC Upper Continental Crust, data from ref. 17. | |||||
| Sc | 0.050 | ? | 16 | ||
| La | 12000 | 6 | 30 | 20 | 33 |
| Ce | 23000 | ? | 60 | 43 | 49 |
| Pr | 2500 | 2 | 6.7 | 4.5 | 4.3 |
| Nd | 7000 | 8 | 27 | 16 | 12 |
| Sm | 700 | 2 | 5.3 | 2.5 | 0.8 |
| Eu | 100 | 0.15 | 1.3 | 0.1 | 0.1 |
| Gd | 400 | ? | 4.0 | 1.5 | 0.2 |
| Tb | 10 | 0.30 | 0.65 | 0.05 | 0.016 |
| Dy | 100 | ? | 3.8 | 0.6 | 0.03 |
| Ho | 10 | 0.40 | 0.8 | 0.005 | 0.005 |
| Er | 500 | ? | 2.1 | 0.2 | 0.0035 |
| Tm | 50 | 0.10 | 0.35 | 0.02 | 0.001 |
| Yb | 50 | 1 | 2.0 | 0.1 | 0.0005 |
| Lu | 10 | 0.20 | 0.35 | 0.02 | 0.0001 |
The increasing industrial applications of REE such as their use in the production of high technology alloys could reasonably be expected to cause increased releases of REE to the environment. To some extent, the amount of each individual REE produced annually reflects the abundance of each REE in the Upper Continental Crust (UCC) (Table 1). Losses of REE through the use of zeolite catalysts in oil refineries might also have contributed to enrichments of REE in atmospheric aerosols during the last century. These losses account for 7600 t of REE per year in the USA, which amounts to 43% of the total US use.9 Rare-earth zeolite catalysts are used for “cracking” crude oil to produce shorter-chain molecules in petrol refining. The determination of REE in the peat core may reveal whether or not the growing industrial use of REE is reflected in the peat bog profile which extends from today back to the Roman Period.
Reliable analytical data are one important prerequisite for a meaningful reconstruction of ancient REE deposition rates. A major analytical problem is frequently caused by the dissolution of siliceous matter present in largely varying amounts in peats, by hydrofluoric acid (HF).18 “Excess HF” will cause the precipitation of REE fluorides, which, in turn, will consequently lead to low REE yields.18 Addition of a saturated solution of boric acid (H3BO3) to the digestion solution can partly overcome this problem, however, this causes a cascade of several other problems.19,20 Therefore, we recently developed a new digestion procedure based on the use of nitric acid and tetrafluoroboric acid (HBF4) combining the positive effects of both HF and H3BO3, by simultaneously attacking the silicates while preventing the precipitation of REE fluorides.18
The main goal of this study is to determine the REE concentration profiles in age-dated peat samples from an ombrotrophic bog extending back to Roman times. The results obtained in this study for REE using the newly developed microwave digestion-ICP-MS procedure will be compared with previous results for selected REE from instrumental neutron activation analysis (INAA) to further assess the accuracy and precision of both analytical methodologies. The calculation of enrichments or depletions of REE during more than 2000 years of atmospheric dust deposition will shed more light on the influence of natural and anthropogenic activities on the geochemical cycle of REE.
Experimental
Instrumentation
Aliquots (∼200 mg) of powdered peat and plant samples were dissolved in a microwave autoclave (ultraCLAVE II, MLS, Leutkirch, Germany) at elevated pressure.ICP-MS measurements were carried out with an ELAN 5000 (PerkinElmer-Sciex) equipped with an autosampler (AS 90, PerkinElmer) and an ultrasonic nebulizer (USN) with membrane desolvation (U-6000 AT+, Cetac Technologies, Omaha, NE, USA). Gas flows and the position of the torch were adjusted to obtain lowest possible oxide formation rates for Ce and Ba and maximum count rates for 103Rh. Typical ratios obtained for CeO/Ce and BaO/Ba were around 0.07% and 0.02%, respectively. Under the optimised operating conditions, summarised in Table 2, the formation of doubly charged ions—assessed via the ratio of Ba2+/Ba—amounted to about 0.3%.
| Instrument: | ELAN 5000 (PerkinElmer, Norwalk, CT, USA) |
| Forward power | 1200 W |
| Cones | Nickel |
| Plasma gas | 15.0 l min−1 |
| Nebulizer gas | ∼1.0 l min−1, daily optimised to obtain maximum 103Rh signal intensity and minimum oxide formation rates |
| Auxiliary gas | ∼0.8 l min−1 daily optimised to obtain maximum 103Rh signal intensity and minimum oxide formation rates |
| Data acquisition | Peak hoping mode, 50 sweeps per reading, 1 reading per replicate, 3 replicates, dwell time 20 ms for all isotopes except 40 ms for Eu, Tb, Ho, Yb and 60 ms for Tm |
| Sample uptake | ∼2 ml min−1 |
| Nebulizer | U-6000AT+ (Cetac Technologies, Omaha, NE, USA) |
| Sweep gas (argon) | ∼2.85 l min−1 (depending on the condition of the membrane) |
| Heating temperature | 140 °C |
| Desolvation temperature | 140 °C |
| Cooling temperature | 2 °C |
Scandium and selected REE in the peat profile were determined previously, using instrumental neutron activation analysis (INAA) at a commercial laboratory (ACTLABS, Activation Laboratories Ltd., Ancaster, ON, Canada). An energy-dispersive miniprobe multielement analyzer (EMMA) was used to determine Ti, Y and Zr and a wavelength dispersive X-ray fluorescence spectrometer to measure Al in the peat samples.21,22
Reagents and standards
For the preparation of all solutions, high-purity water (18.2 MΩ cm) from a MilliQ-system (Millipore, Milford, MA, USA) was used. Nitric acid (65%, analytical-reagent grade, Merck, Darmstadt, Germany) was further purified by sub-boiling distillation (MLS). Additionally, tetrafluoroboric acid solution (HBF4, ∼50%, purum, Fluka, Buchs, Switzerland) and hydrogen peroxide (30%, Baker analysed, J.T. Baker, Deventer, Holland) were employed for sample digestion.Calibration solutions (0.2; 0.5; 1; 5; 10 µg l−1) were prepared daily by adequate dilution of a multi-element stock standard solution (SPEX, Metuchen, NJ, USA, 5% nitric acid) containing 10 mg l−1 of each element under investigation with 0.42 mol l−1 high-purity nitric acid. To correct for instrumental drifts and plasma fluctuations all analyte solutions were spiked with a rhodium and rhenium standard solution (SPEX) to a final concentration of 5 µg l−1 of each element. Final dilution factors amounted to approximately 400.
Collection and preparation of peat samples
On 26 August 1991, a complete vertical peat core was collected from Etang de la Gruère (EGR), a raised bog in the Jura Mountains of Switzerland. Details of the sample collection procedure, the preparation of the peat samples and previous geochemical studies have been published elsewhere.5 Briefly, the samples were taken in the central part of the bog where it is strongly domed and peat accumulation is more than 6 m. The top 100 cm of peat was sampled using a Wardenaar peat profile sampler to remove a monolith 10 × 10 × 100 cm (core 2F) and this was sectioned by hand into 3 cm slices using a bread knife. Selected samples from deeper peat layers of the 2F core were dated using 14C, and the uppermost peat layers of the 2F core using 210Pb. Starting from sample number 2F14, all 14C age dates have been calibrated to calendar years.5Acid digestion in the microwave autoclave
Sample aliquots (approx. 200 mg) were weighed to 0.1 mg into 20 ml PTFE vessels. The acid mixtures contained 3 ml HNO3 and 0.1 ml HBF4. Up to 40 samples can simultaneously be digested under identical experimental conditions in the microwave autoclave. A starting pressure of 50 bar (with Ar) was applied to the reaction chamber. Vessels were then heated in the microwave autoclave for 76 min, increasing the temperature up to 240 °C; during this phase the pressure in the reaction chamber reached about 120 bar, as described in detail earlier.18 After cooling for about 35 min well below the boiling point of the acid mixture at atmospheric pressure, the reaction chamber was opened. Colourless, homogenous digestion solutions were obtained which indicates efficient destruction of the organic matter. The contents of the digestion vessels were quantitatively transferred into graduated 15 ml polypropylene tubes (Falcon®, Becton Dickinson, Meylan Cedex, France) and filled to the mark with high-purity water.Quality control
A peat reference material with certified elemental concentrations is currently not available. Therefore, a plant reference material with certified or recommended REE concentrations (GBW 07602 Bush Branches and Leaves, Institute of Geophysical and Geochemical Exploration, Langfang, China) was analysed with every batch of samples to ensure the accuracy of the applied analytical procedures, which was also evaluated against the reference material CRM 670 Aquatic Plant (BCR, Brussels, Belgium) for which certified values for all REE are available. A detailed description of the newly developed analytical methodology for the determination of REE in peat is given elsewhere, along with all relevant quality control data.18Each peat sample of the 2F core was digested twice. Each diluted digestion solution was analysed for REE by ICP-MS twice on different days, yielding a total of four analytical values per sample; these normally agreed within an uncertainty of <5%.
Correction for the moisture content
All reference materials and peat samples were used as bottled. Results were corrected for the moisture content in the reference materials as determined on 200 mg aliquots of each material by an electronic moisture analyser (MA 100 H, Sartorius, Göttingen, Germany).23 As only a limited amount of each peat sample from the 2F peat core was available, no correction of the moisture content of the dry powders (previously dried at 105 °C, then stored in air-tight low density polyethylene bags) was performed.Results and discussion
Abundance and distribution of REE
Concentration profiles of the 14 REE determined by the microwave digestion-ICP-MS approach in the 2F core are summarised as electronic supplementary information (ESI)†. The shapes of all REE concentration profiles in the peat core are very similar and therefore, as an example, the depth profile of the concentration of La is highlighted in Fig. 1. Because of their similar behaviour in the peat column, it might be possible to select a single REE for analyses, and to estimate the corresponding concentrations of all REE using as conversion factor data of the composition of the Upper Continental Crust (UCC) or chondritic meteorites. To test this hypothesis, such a calculation has been performed on three randomly selected peat samples using the experimental results of Lu. The calculated values, summarised in Table 3, demonstrate that in most cases an agreement of ±10% between the found and the calculated concentration can be obtained using this approach. Europium, however, is an exception and cannot be reasonably estimated by this approximation. Calculated concentrations of Eu exceeded the found value by 30% to 50%. For all other REE, an analyst might choose the REE which can be determined best with a particular method and might use this analytical result to calculate the concentration of the other REE in the sample. | ||
| Fig. 1 Concentration profile of La, Sc and the La/Sc ratio in the 2F core at Etang de la Gruère. | ||
| REE | Peat #2F14 | Peat #2F24 | Peat #2F31 | |||
|---|---|---|---|---|---|---|
| Found | Calculated | Found | Calculated | Found | Calculated | |
| Lu | 35.7 | — | 13.2 | — | 18.8 | — |
| La | 3190 | 3060 | 1060 | 1130 | 1720 | 1610 |
| Ce | 5750 | 6120 | 1770 | 2260 | 2990 | 3220 |
| Pr | 720 | 683 | 215 | 253 | 367 | 360 |
| Nd | 3010 | 2750 | 909 | 1020 | 1550 | 1450 |
| Sm | 608 | 541 | 175 | 200 | 293 | 285 |
| Eu | 96.2 | 133 | 31.1 | 49.0 | 49.9 | 69.8 |
| Gd | 470 | 408 | 167 | 151 | 273 | 215 |
| Tb | 67.4 | 66.3 | 24.7 | 24.5 | 40.5 | 34.9 |
| Dy | 431 | 388 | 153 | 143 | 252 | 204 |
| Ho | 82.8 | 81.6 | 30.6 | 30.2 | 49.9 | 43.0 |
| Er | 234 | 214 | 86.0 | 79.2 | 142 | 113 |
| Tm | 37.3 | 30.6 | 12.6 | 11.3 | 20.3 | 16.1 |
| Yb | 253 | 204 | 78.9 | 75.4 | 127 | 107 |
In general, lowest concentrations were found in samples from the entire last century (depth 0–30 cm) with the youngest sample from 1991 having the lowest concentrations of REE (Fig. 1). This uppermost sample, collected at the surface of the bog, consists mainly of plant material (Sphagnum moss) that has not yet converted to peat. Concentrations of REE in this plant-derived sample are in good agreement with recent results of REE in various plant matrices from Switzerland and Bulgaria.24,25 The corresponding age dates of the peat samples are summarised in Fig. 1 and Table 4, respectively. Roughly, the REE concentration profiles consist of four distinct maxima: during the 1950s, at AD 1661, at AD 1020 and at a depth of −88.5 cm, which corresponds to approximately AD 250. By far the highest concentrations were found in samples originating from AD 1661. As the concentration profile of REE alone does not allow a conclusive interpretation of the results, the concentration profiles have to be normalised to an immobile reference element as described in the following.
| Sample | Depth/cm | Age datea | Ash (%) | Alb (%) | Scb/µg g−1 | Yc/µg g−1 | Zrc/µg g−1 | Tic/µg g−1 |
|---|---|---|---|---|---|---|---|---|
| a 2F1–2F13 are 210Pb age dates, the others are calibrated 14C age dates (in calendar years). b Determined by instrumental neutron activation analysis (INAA).5 c Determined by energy-dispersive miniprobe multielement analyzer (EMMA).21,22 d Below detection limit. e Not determined. | ||||||||
| 2F1 | −1.5 | 1991 ± 0 | 2.4 | 0.083 | 0.12 | 1.2 | 1.0 | 67.3 |
| 2F2 | −1.5 | 1989 ± 2 | 1.6 | 0.080 | 0.11 | 0.7 | —d | 51.2 |
| 2F3 | −4.5 | 1985 ± 2 | 1.9 | 0.094 | 0.14 | 1.4 | 1.9 | 58.7 |
| 2F4 | −7.5 | 1979 ± 2 | 2.0 | 0.080 | 0.13 | 1.0 | 2.1 | 56.1 |
| 2F5 | −10.5 | 1967 ± 2 | 2.5 | 0.126 | 0.23 | 1.9 | 3.1 | 98.3 |
| 2F6 | −13.5 | 1953 ± 2 | 2.0 | 0.111 | 0.18 | 0.8 | 1.5 | 90.3 |
| 2F7 | −16.5 | 1944 ± 2 | 1.6 | 0.084 | 0.14 | 1.2 | 1.1 | 57.2 |
| 2F8 | −19.5 | 1936 ± 3 | 1.5 | 0.078 | 0.13 | 0.7 | 1.4 | 81.6 |
| 2F9 | −22.5 | 1929 ± 3 | 1.5 | 0.081 | 0.14 | 0.6 | 2.3 | 56.7 |
| 2F10 | −25.5 | 1920 ± 4 | 1.7 | 0.110 | 0.16 | 1.0 | —d | 63.3 |
| 2F11 | −28.5 | 1905 ± 6 | 2.6 | 0.177 | 0.26 | 1.7 | 2.1 | 102 |
| 2F12 | −31.5 | 1879 ± 11 | 4.6 | 0.308 | 0.47 | 2.2 | 7.5 | 204 |
| 2F13 | −34.5 | 1843 ± 25 | 8.6 | 0.552 | 0.89 | 3.4 | 20.9 | 455 |
| 2F14 | −37.5 | AD 1661 | 7.2 | 0.529 | 0.91 | 2.9 | 21.2 | 438 |
| 2F15 | −40.5 | AD 1439 | 4.1 | 0.384 | 0.56 | 1.8 | 11.9 | 276 |
| 2F16 | −43.5 | AD 1290 | 2.3 | 0.309 | 0.36 | 1.4 | 4.6 | 164 |
| 2F17 | −46.5 | AD 1192–1208 | 2.8 | 0.293 | 0.42 | 1.3 | 6.8 | 192 |
| 2F18 | −49.5 | AD 1020 | 4.2 | 0.390 | 0.69 | 1.9 | 12.4 | 378 |
| 2F19 | −52.5 | 3.9 | 0.378 | 0.64 | 1.5 | 14.2 | 294 | |
| 2F20 | −55.5 | AD 891 | 3.4 | 0.324 | 0.52 | —e | —e | —e |
| 2F21 | −58.5 | 1.6 | 0.240 | 0.29 | 1.0 | 4.2 | 141 | |
| 2F22 | −61.5 | 1.9 | 0.235 | 0.30 | 1.0 | 3.8 | 150 | |
| 2F23 | −64.5 | AD 669 | 1.4 | 0.208 | 0.22 | 0.7 | 1.6 | 80.9 |
| 2F24 | −67.5 | 1.7 | 0.205 | 0.27 | 0.8 | 3.1 | 112 | |
| 2F25 | −70.5 | 1.6 | 0.194 | 0.26 | 0.5 | 4.2 | 122 | |
| 2F26 | −73.5 | AD 653 | 1.4 | 0.184 | 0.20 | 0.7 | 2.4 | 86.2 |
| 2F27 | −76.5 | 1.2 | 0.185 | 0.19 | 0.7 | 2.1 | 90.9 | |
| 2F28 | −79.5 | 1.4 | 0.182 | 0.19 | 0.6 | 3.7 | 98.3 | |
| 2F29 | −82.5 | AD 433 | 1.3 | 0.178 | 0.18 | 0.9 | 3.5 | 90.9 |
| 2F30 | −85.5 | 2.4 | 0.251 | 0.37 | 1.1 | 8.4 | 205 | |
| 2F31 | −88.5 | 2.7 | 0.293 | 0.40 | 1.3 | 6.9 | 222 | |
| 2F32 | −91.5 | AD 128 | 2.0 | 0.277 | 0.34 | 1.2 | 4.3 | 153 |
| 2F33 | −94.5 | 1.8 | 0.268 | 0.34 | 1.1 | 4.8 | 155 | |
| 2F34 | −97.5 | AD 151–117 BC | 1.8 | 0.264 | 0.37 | 0.9 | 3.9 | 153 |
| 2F35 | −100.5 | AD 151–117 BC | 1.6 | 0.215 | 0.28 | 0.8 | 3.8 | 120 |
Enrichments of REE relative to scandium
Natural variations in the abundance of mineral matter generally have an important effect on elements in peats, especially in low-ash Sphagnum peats from ombrotrophic bogs. Therefore, substantially more relevant information can be derived from the concentration profiles of any element when these are compared with some reference element. Consequently, concentration values of a specific element in a peat profile are often normalised to a lithogenic, conservative element.2–7 Scandium can be successfully employed for this purposes because it acts as a surrogate for atmospheric soil dust.4–6 Normalisation to Sc helps to assess whether a peak in the concentration profile is caused by an enrichment of a particular element or whether it may simply be due to increased soil dust input to the peat column. Additionally, the ash content of the peat samples (Table 4) also reflects the varying concentration of mineral matter in peat samples.4–6Scandium (Fig. 1) may serve as an example: The calculated La/Sc ratios reach their lowest values between 1843 and 1905, respectively. This period exactly matches with the highest peak in the concentration profile of La. As the Sc concentration also increases during this period, the La/Sc ratio remains almost unchanged. Therefore, at this time the elevated La concentrations indicate that simply more aerosol particles were deposited to the surface of the peat bog, however, no enrichment of La relative to Sc occurred. Interestingly, the La/Sc ratios (range: 2.67–5.91) in all samples of the peat profile were higher than their corresponding value in the UCC (1.88).
Calculation of enrichment factors
To emphasise that the REE/Sc ratios in all peat samples from the 2F core at EGR exceed the crustal ratio, the enrichment factor (EF) was calculated for all REE as| EF REE = ([REE]/[Sc])Sample/([REE]/[Sc])UCC | (1) |
 | ||
| Fig. 2 Enrichment factors (EF, see text for calculation) of 14 rare earth elements during the last 2000 years, as revealed by the 2F core at Etang de la Gruère. | ||
Several reasons might be responsible for the observed Ce anomaly in the peat sample from 1944: First, aerosols may already have been depleted in Ce before they were deposited to the bog, e.g. Ce was fractionated during weathering. This remains relatively unlikely because this effect was only observed in the sample from World War II and not for prolonged periods. Second, Ce is perhaps mobile in the peat profile. However, this possibility also can be most probably excluded because for the remaining peat samples the behaviour of Ce was comparable to all other REE. Therefore, an anthropogenic influence might explain the observed Ce anomaly in the peat sample and because it dates from 1944, emission sources related to military activities are suspected.
The two negative peaks in Fig. 3 (−61.5 cm and −82.5 cm) indicating a depletion of REE can presently not be explained. However, it is important to note that the presence or absence (in the case of Ce) of all maxima/minima in Fig. 2 were supported by INAA data (which will be discussed later in the manuscript) and cannot be regarded as analytical artefacts.
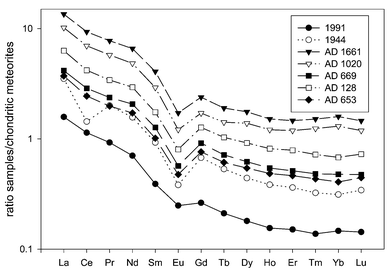 | ||
| Fig. 3 Chondrite-normalised REE plot of selected peat samples from Etang de la Gruère. | ||
Besides the aforementioned changes, a pronounced shift in EF can be observed centred upon the sample dating from 1843 (depth: −34.5 cm) approaching crustal values (Fig. 2). Following this time, there is a sudden shift of the EF to distinctly lower values which are 2 to 3 times lower than the EF relative to Sc for the previous 1900 years. This significant shift is most pronounced for the heavier REE (e.g., Lu EF in Fig. 2) and dates from the beginning of the Industrial Revolution, suggesting that the origin of the shift is anthropogenic. Sample 2F14 (36–39 cm) was dated using decay counting at 240 ± 20 14C year BP which has a calibrated age of AD 1661,5 and sample 2F13 (33–36 cm) was dated using 210Pb at 1843 ± 25. As the greatest shift toward lower REE EF takes place between these two samples, the change must have occurred at least two, if not three centuries ago which roughly corresponds to the first phase of the Industrial Revolution which began ca. 1760 in Central Europe. However, this period could be characterised by urbanisation and as the beginning of intensive consumption of coal for the production of steel; if this process contributed significantly to the REE flux to the bog surface, then there should be a trend in the REE inventory (or EF) which reflects the general increase in industrialisation with time. While such trends have been found for As and Pb concentrations,1 for example, the REE EF does not show a clear trend, but rather a shift from one fairly constant EF value, to another (Fig. 2).
It is important to note here that the Franches Montagnes region of the Jura Mountains was settled in the 17th century, and this resulted in a significant change to the landscape, with much of the original forest cover being cleared for agriculture. Forest clearing would have accelerated the erosion of local soils; these are derived from sedimentary rocks (mainly limestones) which, in general, are poor in REE compared with crystalline rocks or siliceous sedimentary rocks.14 The elevated flux of atmospheric soil dust, relatively poor in REE, may help to explain the shift in REE EF from one fairly constant value to a second, lower value. This hypothesis, although speculative, suggests that there may be a link between the atmospheric flux of REE to the bog, and local land use history.
Interestingly, the shift towards lower EF after 1843 is more pronounced for heavier REE (∼2.5) than for lighter REE (∼2.1). As replicate digestion/quantification of all REE in the peat samples agreed within <5%, the difference in EF between lighter and heavier REE becomes significant. In other words, starting from 1843 (onset of the Industrial Revolution), heavier REE have been depleted relative to lighter REE. This finding is in good agreement with data of Rahn26 who found that light REE are often more abundant in the atmosphere relative to heavy REE than is predicted by crustal abundance patterns, especially among fine airborne particles. Rahn proposed that these REE anomalies might arise from fractionation during high temperature processes, such as coal burning.26 As coal combustion increased dramatically during the Industrial Revolution, aerosol particles deposited to the surface of the peat bog at EGR might reflect the environmental changes occurring during this period. With the introduction of heating oil in Switzerland in 1930 and the gradual replacement of coal, the EF steadily increased again, reaching present day values. However, if coal burning was a significant source for REE, then the REE enrichment should be increasing steadily with time as global coal consumption has grown since the Industrial Revolution to 4 billion t year−1 today. As the EF only shows a weak trend of a slight increase during the last 100 years, coal and heating oil combustion are most probably not the main source of REE in the aerosols of the 20th century. In fact, it almost looks as though there has been a strong shift in the predominant source of the REE.
Besides the calculation of the EF, normalisation of REE concentrations to those of chondritic meteorites is a convenient way to compare different data sets. The even atomic-numbered elements such as Ce, Nd and Sm are about one order of magnitude more abundant than the adjacent odd-numbered elements (“Oddo-Harkins effect”). Therefore, REE concentrations in a sample are often normalised to chondritic meteorites which represent the primordial earth. These chondrite-normalised plots should normally result in a smooth curve as demonstrated in Fig. 3 for selected peat samples from EGR. The so called “negative Eu anomaly” which is commonly observed in the chondrite-normalised plots is also present in all peat samples, except in the uppermost sample, representing Sphagnum moss, which only shows a slight depletion compared to all other samples. Selective uptake of REE by Sphagnum moss might be the reason for this finding. Of particular importance is the negative peak of Ce in the peat sample from 1944 which indicates a depletion of Ce. However, as the calculation of the EF for all REE revealed (Fig. 2), the EF for Ce remains fairly unchanged over time, but all other REE have been enriched in that particular peat sample. Therefore, chondrite-normalised plots display only the relative behaviour of REE to each other and absolute statements about enrichments or depletions of single REE should only be made with great caution to avoid misinterpretation of the data.
The chondrite-normalised plots of all other peat samples, some of which are displayed in Fig. 3 showed a very unique behaviour. Roughly, the slope of the curves decreased reaching a minimum at Eu, followed by almost horizontal curves for the REE Gd to Lu.
Potential use of REE as reference elements
As mentioned above, normalisation of elemental concentration profiles obtained from a peat core to an immobile, conservative element provides deeper insights about atmospheric metal deposition than the consideration of the concentration profile itself alone. Moreover, normalisation of elemental concentrations is the basis for the calculation of EF which allows the quantification of the changing enrichment or depletion of an element with time. However, frequently the question arises which element should be chosen to normalise concentration profiles in a peat bog. A detailed discussion of the geochemistry and reasons to prefer Sc over other lithogenic elements for this purpose was reported earlier.5 Briefly, Sc is neither enriched or depleted comparing the Continental Crust with the primitive mantle and thus behaves more conservative during weathering than Al, for example. Besides Sc, Y, Al, Zr and Ti are commonly used as reference elements.6Fig. 4 displays the concentration profiles of these elements in the 2F core of EGR together with that of Cu. Copper was chosen as an example for an “anthropogenic” element to demonstrate the different behaviour of other elements in the peat core. As can be seen in Fig. 4, the concentration profiles of these potential reference elements (except for Cu) are very similar and perhaps more important, almost identical with the La profile presented in Fig. 1 (to compare with other REE refer to ESI†). This finding is particularly important in the light of the analytical difficulties posed by the reliable determination of Sc and Y, which are frequently used to normalise elemental concentrations in a peat core. Before the introduction of inductively coupled plasma-sector field-mass spectrometry (high resolution ICP-MS), concentrations of Sc and Y could be only reliably determined in environmental samples at low concentrations by INAA and XRF, respectively. To date, the occurrence of severe spectral interferences on the signals of Sc and Y excludes the use of ICP-MS instruments equipped with quadrupole mass filters for this purpose.19 Having said this, and taking into account the similarity of the REE and Sc plots, owners of ICP-QMS instruments who cannot measure Sc may wish to consider using the concentrations of REE instead, as a surrogate for atmospheric soil dust. | ||
| Fig. 4 Comparison of the concentration profiles of the lithogenic elements Sc, Al, Y, Zr and Ti as well as Cu in the 2F core at Etang de la Gruère. | ||
The use of the sum of all REE as a reference parameter in geochemical studies was emphasised by Chiarenzelli et al.8 because of their exclusive transport in the particulate phase, lack of significant anthropogenic sources, coherent group chemistry and upper crustal signatures. These authors suggested that the sum of the concentrations of all REE provides a more robust normalising factor than any single elemental concentration that can be used as a reference to gauge the relative enrichment or depletion of other elements.8 However, as frequently reported for REE patterns and as also observed in the present study (Fig. 2), REE anomalies are often observed for Ce and Eu. These anomalies are caused by the fact that these two elements can form different ions in nature (Ce4+, Eu2+), besides the trivalent ions occurring for all REE. Moreover, as depicted in Table 1, almost 50 000 t of REE are produced each year, and therefore, the possible importance of the anthropogenic influence cannot be ignored. Finally, REE may become relatively enriched during cold climate phases, as reduced soil cover and greater wind strength promote soil erosion and atmospheric dust transport.5
To evaluate the potential of various parameters to normalise enrichments of particular elements, the EF of Cu was calculated relative to each single REE, the sum of REE as well as Sc, Al, Zr, Y and Ti. The results of these calculations are summarised in Fig. 5A and B. As can be seen in Fig. 5A, the REE behave uniformly, with the exception of Ce. Cerium, in turn, does not show the negative peak at −16.5 cm corresponding to 1944 and revealed a very similar pattern as Sc and Al as depicted in Fig. 5B. Using the sum of REE to calculate Cu EF as indicated by Chiarenzelli et al.8 also reveals this negative peak which suggests that Ce alone may be a better reference element than the sum of all REE. Zirconium and Ti also do not fit very well. Additionally, Zr is depleted in aerosols during weathering, relative to the rocks from which they are derived, leading to EF that are too high, whereas Ti is enriched and thus yields too low EF. Concentrations of Y in the peat samples were often at the detection limit of XRF. Therefore, Y values show high fluctuations and have to be interpreted with caution (Fig. 5B).
 | ||
| Fig. 5 Enrichment factors calculated for Cu normalised by (A) single REE, and (B) Sc, sum of REE, Al, Y, Zr and Ti. | ||
Clearly, neither the sum of REE nor any single REE is an appropriate reference parameter to calculate elemental EF. While Ce might eventually be worth considering as a reference element, this requires much more study. Scandium, followed by Al, is certainly the conservative element of choice to calculate elemental EF because it is uniformly distributed in the UCC and the primitive mantle and shows no fractionation in any compartment.5 Titanium and Zr, in contrast, are unsuitable because they are found in specific minerals such as biotite, ilmenite, rutile, titanite and zircon which may fractionate strongly.
Comparison of REE results obtained by INAA and ICP-MS
For a meaningful reconstruction of atmospheric REE deposition rates accurate and precise analytical results of peat analyses are an essential prerequisite. Therefore, all ICP-MS results from the 2F core were compared with data for selected REE (La, Ce, Nd, Sm, Yb, Lu) which had been previously determined using INAA, i.e. a direct analytical method requiring no digestion of peat.5As observed previously for arsenic and antimony in peat, INAA often provides precise results, but is frequently lacking accuracy.27,28 For example, depth profiles obtained from analysis of As and Sb in samples of selected peat cores by INAA, ICP-MS and HG-AAS displayed very similar trends, but the absolute values differed sometimes by up to 50%.28 Similar observations were made for REE analyses in peat in the present study. Correlation coefficients (R) for La, Ce, Nd, Sm, Yb and Lu for INAA and ICP-MS results in all peat samples always exceeded 0.85, as depicted in Fig. 6.
 | ||
| Fig. 6 Correlation between the results of La, Ce, Nd, Sm, Yb and Lu in the 2F core determined by instrumental neutron activation analysis (INAA) and inductively coupled plasma mass spectrometry (ICP-MS). | ||
However, the slopes of the regression lines in the graphs of Fig. 6 are highly variable. If ICP-MS and INAA analysis would give comparable results, one would expect a slope of the regression curves in Fig. 6 close to unity. As can be clearly seen in Fig. 6, slopes of the regression curves vary from 1.175 to 0.721 indicating that INAA results for La are almost 18% higher, whereas Sm values are about 28% lower than their corresponding ICP-MS results. Concentrations of Ce, Nd and Yb fall between these extremes. Ironically, while interference from U is claimed to hamper the reliable determination of Lu by INAA,8 Lu values compared very well (slope = 0.97). In the present study all REE have been determined in the diluted digests simultaneously by ICP-MS using a multi-element standard solution containing all REE for calibrating the ICP-MS. Therefore, any potential error occurring during the dilution of the digests, the preparation of calibration solutions, or problems associated with the ICP-MS measurements, would affect the results of all REE more or less to the same extent. Having said this, it is obvious from the variable slopes of the INAA/ICP-MS plots that calibration of INAA seems to be problematic for peat samples and effects each REE differently. Additionally, INAA produced several outliers further deteriorating the correlation between INAA and ICP-MS as is depicted in Fig. 6. Detection limits (LOD) for INAA using 8 g of each sample (La: 0.01 µg g−1, Ce: 0.3 µg g−1, Nd: 0.5 µg g−1, Sm: 0.05 µg g−1, Eu: 0.005 µg g−1, Yb: 0.005 µg g−1, Lu: 0.001 µg g−1) were always higher by at least one order of magnitude than corresponding method detection limits of the microwave digestion-ICP-MS procedure used in this study. The main reason for the especially poor correlation between INAA and ICP-MS results in Fig. 6 can be related to several Nd concentrations that were at or below the high LOD for Nd by INAA (0.5 µg g−1). Neodymium values, which were below the LOD of INAA, but never below the LOD of ICP-MS, were excluded from Fig. 6.
Similar discrepancies between the REE results in several plant samples determined by using ICP-MS and INAA (employing the same commercial laboratory as in this study) were reported recently.8 However, the authors of that study did not include Lu in the list of REE determined by ICP-MS and indicated that an agreement between INAA and ICP-MS on the level of ±25% was “excellent”. While this might be acceptable to the mineral exploration industry or for some environmental surveys, it is unacceptable for most other studies. For high resolution time series reconstructions of atmospheric REE depositions, for example, quantitative analyses of the elements of interest must be of greater accuracy and precision.
During the development of the microwave digestion-ICP-MS approach the quality of the obtained analytical data was extensively checked with adequate reference materials.18 The excellent agreement between certified and experimentally determined REE concentrations proves the high quality of the results from this study. However, only recently a few plant derived reference materials with certified or recommended concentration values for all REE became commercially available. At the time of the INAA measurements, only soil and rock reference materials with much higher REE concentrations were obtainable. As these reference materials did not match the peat matrix very well and because the REE concentrations were much higher than actual values in the peat samples, the quality of the INAA procedures could not be adequately controlled leading to the discrepancies described before.
In conclusion, although a digestion step and consequently dilution of the digests is necessary for ICP-MS analyses of REE, results provided by this analytical approach are far more sensitive and more accurate than INAA values from a commercial laboratory. Moreover, the INAA analyses yielded usable data for only five of the REE which is of limited use, whereas ICP-MS measurements yield all 14 elements simultaneously.
Conclusions
The determination of REE in the 2F core at EGR revealed that natural and anthropogenic activities only marginally influenced the geochemical cycle of REE during the last 2000 years. Although the industrial applications of REE have tremendously increased over the last decades, this elevated emission of REE is not strongly reflected in an increase of REE EF in the peat core. The EF ranged from 1.44 to 2.34 relative to crustal rocks, but these are very small values compared to EF of other elements with strong anthropogenic influence such as Pb (EF up to 100 and more).1–6 However, the EF data reveal a pronounced shift in the predominant source of REE corresponding to the start of the Industrial Revolution. This, as well as some other changes in the profile, require further study.As regards the importance of the analytical quality of the results, this is demonstrated by comparative analyses of REE by ICP-MS and INAA, respectively. Although ICP-MS requires dissolution of the powdered peat samples by acids, results for REE determined by ICP-MS are far more reliable than those from INAA.
The determination of REE in a new peat core collected at Etang de la Gruère in 2002, which will yield a much better temporal resolution, will provide both deeper insights of atmospheric REE deposition and information about the REE enrichments of last ten years which are not covered by the present peat core.
Acknowledgements
Financial support to M. K. from the Forschungspool of the University of Heidelberg (Project: “Trace element analyses of geological archives: sediments, peat, and ice”) and for C. M. from the Deutsche Forschungsgemeinschaft (DFG project EM 51/3-1) are gratefully acknowledged.References
- W. Shotyk, Water Air Soil Pollut., 1996, 90, 375 CrossRef CAS.
- W. Shotyk, A. K. Cheburkin, P. G. Appleby, A. Frankhauser and J. D. Kramers, Earth Planet. Sci. Lett., 1996, 145, E1 CrossRef.
- D. Weiss, W. Shotyk, P. Appleby, J. D. Kramers and A. K. Cheburkin, Environ. Sci. Technol., 1999, 33, 1340 CrossRef CAS.
- W. Shotyk, D. Weiss, P. G. Appleby, A. K. Cheburkin, R. Frei, M. Gloor, J. D. Kramers, S. Reese and W. O. van der Knapp, Science, 1998, 281, 1635 CrossRef CAS.
- W. Shotyk, D. Weiss, J. D. Kramers, R. Frei, A. K. Cheburkin, M. Gloor and S. Reese, Geochim. Cosmochim. Acta, 2001, 65, 2337 CrossRef CAS.
- W. Shotyk, M. Krachler, A. Martinez-Cortizas, A. K. Cheburkin and H. Emons, Earth Planet. Sci. Lett., 2002, 199, 21 CrossRef CAS.
- A. Martinez-Cortizas, X. Pontevedra-Pombal, E. Garcia-Rodeja, J. C. Novoa-Munoz and W. Shotyk, Science, 1999, 284, 939 CrossRef CAS.
- J. Chiarenzelli, L. Aspler, C. Dunn, B. Cousens, D. Ozarko and K. Powis, Appl. Geochem., 2001, 16, 245 CrossRef CAS.
- I. Olmez and G. E. Gordon, Science, 1985, 229, 966 CAS.
- M. Land, B. Öhlander, J. Ingri and J. Thunberg, Chem. Geol., 1999, 160, 121 CrossRef CAS.
- M. Aström, Chem. Geol., 2001, 175, 249 CrossRef CAS.
- A. Bechtel, A. M. Ghazi, W. C. Elliott and S. Oszczepalski, Appl. Geochem., 2001, 16, 375 CrossRef CAS.
- M. Steinmann, P. Stille, K. Mengel and B. Kiefel, Appl. Geochem., 2001, 16, 351 CrossRef CAS.
- S. R. Taylor, in The Encyclopedia of Geochemistry and Environmental Sciences, ed. R. W. Fairbridge, Dowden, Hutchinson, and Ross, Stroudsburg, Pennsylvania, 1972 Search PubMed.
- K. Kummerer and E. Helmers, Environ. Sci. Technol., 2000, 34, 573 CrossRef.
- J. Emsley, Nature’s Building Blocks: An A–Z Guide to the Elements, Oxford University Press Inc., New York, 2001 Search PubMed.
- K. H. Wedepohl, Geochim. Cosmochim. Acta., 1995, 59, 1217 CrossRef CAS.
- M. Krachler, C. Mohl, H. Emons and W. Shotyk, J. Anal. At. Spectrom., 2002, 17, 844 RSC.
- T. Prohaska, S. Hann, C. Latkoczy and G. Stingender, J. Anal. At. Spectrom., 1999, 14, 1 RSC.
- A. G. Coedo, M. T. Dorado, I. Padilla and F. J. Alguacil, J. Anal. At. Spectrom., 1998, 13, 1193 RSC.
- A. K. Cheburkin and W. Shotyk, Fresenius’ J. Anal. Chem., 1996, 354, 688 CAS.
- W. Shotyk, P. Blaser, A. Grünig and A. K. Cheburkin, Sci. Total Environ., 2000, 249, 257 CrossRef CAS.
- M. Krachler, Fresenius’ J. Anal. Chem., 2001, 371, 944 Search PubMed.
- R. Djingova, J. Ivanova, G. Wagner, S. Korhammer and B. Markert, Sci. Total Environ., 2001, 280, 85 CrossRef CAS.
- A. Wyttenbach, L. Tobler, P. Schleppi and V. Furrer, J. Radioanal. Nucl. Chem., 1998, 231, 101 CAS.
- K. A. Rahn, The Chemical Composition of the Atmospheric Aerosol, Technical Report, Graduate School of Oceanography, University of Rhode Island, Narragansett, 1976, pp. 151–177 Search PubMed.
- M. Krachler, W. Shotyk and H. Emons, Anal. Chim. Acta, 2001, 432, 303 CrossRef CAS.
- M. Krachler, H. Emons, C. Barbante, G. Cozzi, P. Cescon and W. Shotyk, Anal. Chim. Acta, 2002, 458, 387 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Concentration profiles of the 14 rare earth elements in the 2F core at Etang de la Gruère as determined by microwave digestion-ICP-MS. See http://www.rsc.org/suppdata/em/b2/b208355h/ |
| This journal is © The Royal Society of Chemistry 2003 |