Rapid screening by MALDI-TOF mass spectrometry to probe binding specificity at enzyme active sites†
Michael E.
Webb
a,
Elaine
Stephens
a,
Alison G.
Smith
b and
Chris
Abell
*a
aUniversity Chemical Laboratory, University of Cambridge, Lensfield Road, Cambridge, UK CB2 1EW. E-mail: ca26@cam.ac.uk; Tel: +44 1223 336405
bDepartment of Plant Sciences, University of Cambridge, Downing Street, Cambridge, UK CB2 3EA
First published on 1st September 2003
Abstract
The binding affinity of aspartate decarboxylase has been probed using MALDI-TOF spectrometry; adducts formed covalently in the active site were detected by MALDI-TOF mass spectrometry after incubation of the enzyme with a range of potential ligands in the presence of NaCNBH3; this has highighted key structural features which will aid design of potential inhibitors.
Aspartate decarboxylase (ADC) is a small tetrameric protein (subunit size ∼ 14 kDa) in which the zymogenic protomer is autocatalytically cleaved to form the catalytically active form of the enzyme. This cleavage results in the formation of a small chain from residues 1–24 (β) and a longer chain (α) which has an N-terminal pyruvoyl group derived from Ser25. In the catalytic mechanism this pyruvoyl group forms an iminium ion (Schiff base) with the substrate L-aspartate and subsequently supports the negatively charged intermediate formed as a result of decarboxylation. (Scheme 1). Adducts corresponding to this iminium ion and that formed by the product β-alanine have previously been observed after reduction using electrospray mass spectrometry (ESMS).1 ESMS is now an established technique for the detection of covalent2 and non-covalent3 enzyme–inhibitor complexes.
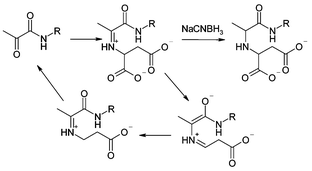 | ||
| Scheme 1 ADC mechanism. | ||
Here we present the use of this approach to probe the active site of ADC with a range of small molecules. We believe this is a convenient generalisable method for the identification of small molecules with the capacity to bind into an active site. This approach allows rapid pharmacophore development for novel protein targets in which covalent protein–ligand interactions occur. From this we can explore the substrate specificity of the enzyme and together with a knowledge of the structure of the active site (Fig. 2, see later), can develop a model of the likely binding arrangement. Previous work on this area has utilised the formation of disulfides in the active site using either a natural or introduced active site cysteine.4 With ADC it has been possible to extend that work to systems that already possess an electrophilic functionality available for tethered binding, by exploiting the reactive electrophilic carbonyl functionality in the enzyme active site in this screen.
Compounds were incubated with ADC (∼60 µM) and NaCNBH3 (0.2 mM) at 2 mM and 20 mM concentration before analysis by MALDI-TOF mass spectrometry.5 The spectrum of the unmodified protein (Fig. 1a) shows two peaks: one at m/z 11002 (α chain) and one at m/z 11018 (α′ chain).6 Ligand binding to the α chain is observed by the appearance of additional peaks at higher molecular weight. The example for β-glutamate (9) is shown in Fig. 1b. A strong peak at m/z 11134 corresponds to the enzyme with β-glutamate reductively bound; the peak for the α chain is diminished in intensity, whilst that for the α′ chain remains unchanged.
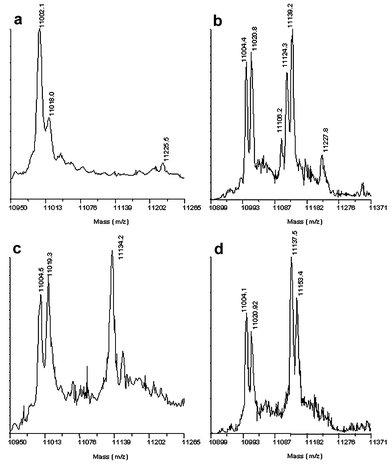 | ||
| Fig. 1 MALDI mass spectrum of a – underivatised ADC; b – ADC derivatised with L-cysteine; c – ADC derivatised with 2 mM β-glutamate; d – ADC derivatised with L-homocysteine. | ||
A set of 55 compounds bearing a primary amine functionality were selected for the binding screen (see Scheme 2). The compounds were based on size and structural similarity to the natural substrate L-aspartate, and included the previously identified inhibitor β-hydroxyaspartate.7 Only a small number of compounds were observed to bind when used at 2 mM: L-aspartate (2), β-alanine (6), L-cysteine (3), methyl L-aspartate (10), D-serine (14), β-glutamate (9), β-leucine (13), β-hydroxyaspartate (1) and glycine (5). At 20 mM, D-aspartate (7), L-glutamate (15), D-glutamate (11), S-2-aminobutyrate (8), L-homocysteine (4) and 2-aminomethylsuccinate (16) also bound. Two compounds, L-aspartate (2) and β-hydroxyaspartate (1) are detected as their reductively bound decarboxylated adducts.
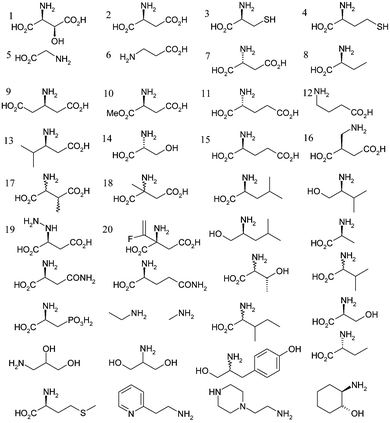 | ||
| Scheme 2 Compounds used to assay for covalent binding affinity to ADC active site. Compounds 1, 2, 3, 5, 6, 9, 10, 13 and 14 bound at 2 mM final concentration. Compounds 4, 7, 8, 11, 12, 15 and 16 bound at 20 mM concentration. | ||
Analysis of the screening results is very informative. An extension in the linker between the α-carbon and the β-carboxylate to form L-glutamate (15) is clearly unfavourable as is an extension in the chain between the α-carbon and α-amino group to form 2-aminomethylsuccinate (16). The increase in chain length between the the α-carboxylate and α-carbon to form β-glutamate (9) is tolerated and this binds at 2 mM concentrations. This compound cannot act as a substrate and so compounds based on this structure are promising as competitive inhibitors. Both α- and β-branching appears unfavourable, as is apparent from the failure to bind either α- and β-methylaspartate (18 and 17). The binding capacity of β-hydroxyaspartate (1) and D-serine (14) perhaps identifies a new binding interaction (see below).
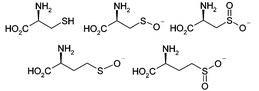
The mass spectrum obtained with L-cysteine (3) reveals a series of three peaks corresponding to covalently bound species (Fig. 1c). These correspond to L-cysteine (m/z 11106) as well as the sulfenate (m/z 11124) and sulfinate (m/z 11139) derived from oxidation of the thiol (Scheme 3). No peak is seen for the corresponding sulfonate species. Similarly homocysteine (4) is seen to bind in singly and doubly oxidised forms at high concentrations (Fig. 1d). This suggests that these intermediate oxidation state compounds may be of interest for investigation as inhibitors.
The active site has been crystallographically defined8 (see Fig. 2). Ligand binding in the pocket of the enzyme is dominated by formation of an iminium ion between the α-amine and pyruvoyl group. The key non-covalent interaction for the substrate is a salt bridge from Arg54 to the β-carboxylate of L-aspartate. This leads to the observed selectivity for β-amino acids. Other important interactions are likely to be hydrophobic packing interactions with Trp47 and hydrophilic with Thr57 which may prevent binding of α- and β-methyl derivatised compounds whilst allowing binding of the β-hydroxy compounds. The catalytic residues (Lys9, His11) would be expected to preferentially bind an α-carboxylate functionality on the other side to Arg54. This selectivity seems not to be as strong as that of Arg54 and is tolerant of chain extension.
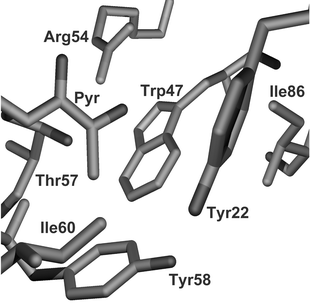 | ||
| Fig. 2 Active site of ADC determined from the crystal structure.6 The pyruvoyl group and the residue Arg54 dominate the binding affinity. Other hydrogen bonding is provided by Lys9 (not shown — occludes front of active site). Hydrophobic interactions are dominated by Trp47. | ||
This screening methodology is readily extendable to a range of other protein and ligand systems. Using reductive amination chemistry it should be feasible to isolate covalent binding complexes in PLP-dependent enzymes and in other enzymes where iminium ion chemistry is observed, in a rapid and effective manner. This approach has allowed us to gain a rapid understanding of the binding capacity of an enzyme active site. It is envisaged that in combination with site directed mutagenesis this technique will be able to pinpoint the residues responsible for selectivity and lead to rapid design of mutants with different catalytic specificity.
The authors would like to acknowledge L. M. Birch, T. M. Murray-Rust, R. de la Salud-Bea and D. B. Berkowitz for gifts of compounds. We thank the EPSRC, BBSRC and Astex Technology for financial support. E. Stephens is funded as part of the Structural Biology Initiative of the BBSRC.
Notes and references
- M. K. Ramjee, U. Genschel, C. Abell and A. G. Smith, Biochem. J., 1997, 323, 661 CAS.
- E. J. Parker, C. G. Bello, J. R. Coggins, A. R. Hawkins and C. Abell, Bioorg. Med. Chem. Lett, 2000, 10, 1 CrossRef.
- P. A. Wright, A. A. Rostom, C. V. Robinson and C. J. Schofield, Bioorg. Med. Chem. Lett., 2000, 10, 1219 CrossRef CAS.
- D. A. Erlanson, A. C. Braisted, D. R. Raphael, M. Randal, R. M. Stroud, E. M. Gordon and J. A. Wells,, Proc. Natl. Acad. Sci. U.S.A., 2000, 97, 9367 CrossRef CAS.
- The mass spectra were recorded in linear mode using a 4700 Proteomics Analyser (Applied Biosystems) fitted with a Nd:YAG laser radiating at 355 nm. All spectra were obtained by averaging 7500 laser shots and were externally calibrated using a standard protein mixture. Typically, 2.5 µl of protein sample was mixed with 10 µl of matrix solution (saturated sinapinic acid in aqueous 50% MeCN–0.1%TFA) and 0.5 µl was directly spotted onto the MALDI target and allowed to air dry.
- The α′ chain corresponds to the same peptide as the α chain with an N-terminal serine residue rather than a pyruvoyl group.1.
- J. M. Williamson and G. M. Brown,, J. Biol. Chem., 1979, 254, 8074 CAS.
- A. Albert, V. Dhanaraj, U. Genschel, G. Khan, M. K. Ramjee, R. Pulido, B. L. Sibanda, F. von Delft, M. Witty, T. L. Blundell, A. G. Smith and C. Abell, Nat. Struct. Biol., 1998, 5, 289 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: details of suppliers of chemicals used in MALDI-TOF mass spectrometry screening assay. See http://www.rsc.org/suppdata/cc/b3/b308182f/ |
| This journal is © The Royal Society of Chemistry 2003 |