Direct catalytic sulfonation of methane with SO2 to methanesulfonic acid (MSA) in the presence of molecular O2
Sudip
Mukhopadhyay
and
Alexis T.
Bell
*
Department of Chemical Engineering, University of California, Berkeley, CA 94720, USA. E-mail: bell@cchem.berkeley.edu; Fax: 1 510 642 4778; Tel: 1 510 642 1536
First published on 3rd June 2003
Abstract
Methane is transformed selectively to methanesulfonic acid at low temperature by liquid-phase sulfonation of methane with SO2 and O2 in the presence of Pd- and Cu-salts as the catalysts.
The selective catalytic functionalization of methane to value added products is a subject of considerable contemporary interest. Because of favorable thermodynamics, many authors have investigated the oxidation and oxidative carbonylation of methane.1 By contrast, the sulfonation of methane has not received as much attention despite its commercial importance.2 It has been shown3a–f that in the presence of a free radical initiator methane can be sulfonated with SO3 in fuming sulfuric acid to methanesulfonic acid (MSA) under very high methane pressure. Most recently we have shown that methane can be sulfonated to MSA by SO2 in the presence of an excess amount of K2S2O8.3g The same approach, however, does not work if O2 is used instead of K2S2O8 as the oxidant. Thus, there is incentive to identify a catalytic system that would enable the use of molecular O2. While Ishii and coworkers have reported success in the vanadium-catalyzed sulfonation of adamantane to the corresponding sulfonic acids using SO2 and O2, methane did not undergo sulfonation to methanesulfonic acid.3h The question therefore arises whether SO2 and O2 can be used for methane sulfonation. In this communication, we show that methane will undergo liquid-phase sulfonation to MSA with SO2 and O2 in acid solvents, with catalytic amounts of Pd- and Cu-salts (Scheme 1).
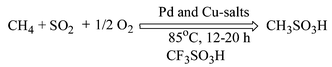 | ||
| Scheme 1 Direct sulfonation of methane to methanesulfonic acid. | ||
In a typical reaction4 (Scheme 1) methane was reacted with SO2 in CF3SO3H to form MSA in presence of Pd- and Cu-salts in a high-pressure, glass-lined, Parr autoclave. Reactions were carried out for 12 h at 85 °C and the MSA thus formed was identified and quantified by 1H NMR.3b,c Use of 13C enriched methane and 1H, 13C NMR of the reaction mixture confirmed that MSA is the only liquid-phase product generated from methane in presence of SO2. The conversions are reported on the basis of the limiting reagent, SO2, and defined as the ratio of the moles of SO2 converted to MSA to the moles of SO2 fed initially to the reactor.
Table 1 shows the effect of different catalyst combinations on the rate of methane sulfonation. In the absence of any catalyst or co-catalyst, no conversion of SO2 to MSA was achieved (Table 1, entry 1). The use of PdCl2 in the absence of CuCl2 gives only 6% conversion of SO2 to MSA, whereas use of CuCl2 in the absence of PdCl2 gives only 1% conversion of SO2 to MSA (Table 1, entries 2, 3). SO2 conversions of 12–20% to MSA were obtained when PdCl2 and CuCl2 were used together (Table 1, entries 4, 5). A nearly identical conversion was obtained after 12 h of reaction when CuCl2 was replaced by Cu2Cl2 (Table 1, entry 6). Similar levels of SO2 conversion to MSA were achieved with acetate, trifluoroacetate, or triflate salts of Pd(II) and CuCl2 or with PdCl2 and acetate, trifluoroacetate, or triflate salts of Cu(II) (Table 1, entries 7–12). Chloride salts of Rh(III), Hg(II), Co(II), Pt(II), Ru(III), Al(III), Ag(I), Ca(II), Fe(III), together with CuCl2 showed very little or no activity, as did VOCl3 and VO(acac)2 with CuCl2 (Table 1, entries 13–23). Pd(acac)2 and Cu(acac)2 also gave very little product (Table 1, entry 24).
| Entry | Catalyst | Co-catalyst | t/h | MSA/mmol | % SO2 to MSAb |
|---|---|---|---|---|---|
| a Reaction conditions: methane, 1200 psig (321 mmol); SO2, 30 psig (11.82 mmol); molar ratio of methane to SO2, 27; O2, 30 psig (11.82 mmol); PdCl2, 0.2 mmol; CuCl2, 0.3 mmol; solvent, CF3SO3H, 5 ml; temperature, 85 °C. b This is the ratio of the moles of SO2 converted to MSA to total moles of SO2 taken initially in this reaction. | |||||
| 1 | None | None | 16 | 0 | 0 |
| 2 | PdCl2 | None | 12 | 0.71 | 6 |
| 3 | None | CuCl2 | 14 | 0.12 | 1 |
| 4 | PdCl2 | CuCl2 | 12 | 1.42 | 12 |
| 5 | PdCl2 | CuCl2 | 40 | 2.36 | 20 |
| 6 | PdCl2 | Cu2Cl2 | 12 | 1.18 | 10 |
| 7 | Pd(CH3COO)2 | CuCl2 | 12 | 1.42 | 12 |
| 8 | Pd(CF3COO)2 | CuCl2 | 12 | 1.18 | 10 |
| 9 | Pd(CF3SO3)2 | CuCl2 | 12 | 1.3 | 11 |
| 10 | PdCl2 | Cu(CH3COO)2 | 18 | 1.18 | 10 |
| 11 | PdCl2 | Cu(CF3COO)2 | 17 | 1.3 | 11 |
| 12 | PdCl2 | Cu(CF3SO3)2 | 16 | 1.42 | 12 |
| 13 | RhCl3 | CuCl2 | 12 | 0.35 | 3 |
| 14 | HgCl2 | CuCl2 | 12 | 0.24 | 2 |
| 15 | CoCl2 | CuCl2 | 12 | 0.12 | 1 |
| 16 | PtCl2 | CuCl2 | 12 | 0 | 0 |
| 17 | RuCl3 | CuCl2 | 12 | 0 | 0 |
| 18 | AlCl3 | CuCl2 | 12 | 0 | 0 |
| 19 | AgCl | CuCl2 | 16 | 0 | 0 |
| 20 | CaCl2 | CuCl2 | 12 | 0 | 0 |
| 21 | FeCl3 | CuCl2 | 14 | 0 | 0 |
| 22 | VOCl3 | CuCl2 | 12 | 0 | 0 |
| 23 | VO(acac)2 | CuCl2 | 12 | 0 | 0 |
| 24 | Pd(acac)2 | Cu(acac)2 | 12 | 0.35 | 3 |
Table 2 shows the effect of different process parameters on the rate of methane sulfonation using PdCl2 salts as the catalyst and CuCl2 as the co-catalyst. Reactions were performed to study the effect of methane pressure on the rate of MSA formation. Increasing the CH4 pressure from 200 to 1200 psig increased the conversion of SO2 to MSA from a barely detectable level to 12% (Table 2, entries 1–5).
| Entry | CH4/psig | SO2/psig | O2/psig | PdCl2/mmol | CuCl2/mmol | T/°C | % SO2 to MSA |
|---|---|---|---|---|---|---|---|
| a Reaction conditions: time, 12 h; solvent, CF3SO3H, 5 ml. | |||||||
| 1 | 200 | 30 | 30 | 0.2 | 0.3 | 85 | tr |
| 2 | 400 | 30 | 30 | 0.2 | 0.3 | 85 | 1 |
| 3 | 650 | 30 | 30 | 0.2 | 0.3 | 85 | 2 |
| 4 | 1000 | 30 | 30 | 0.2 | 0.3 | 85 | 8 |
| 5 | 1200 | 30 | 30 | 0.2 | 0.3 | 85 | 12 |
| 6 | 1200 | 0 | 30 | 0.2 | 0.3 | 85 | 0 |
| 7 | 1200 | 10 | 30 | 0.2 | 0.3 | 85 | 3 |
| 8 | 1200 | 20 | 30 | 0.2 | 0.3 | 85 | 7 |
| 9 | 1200 | 30 | 0 | 0.2 | 0.3 | 85 | 0 |
| 10 | 1200 | 30 | 10 | 0.2 | 0.3 | 85 | 6 |
| 11 | 1200 | 30 | 20 | 0.2 | 0.3 | 85 | 9 |
| 12 | 1200 | 30 | 40 | 0.2 | 0.3 | 85 | 10 |
| 13 | 1200 | 30 | 30 | 0.05 | 0.3 | 85 | 3 |
| 14 | 1200 | 30 | 30 | 0.1 | 0.3 | 85 | 7 |
| 15 | 1200 | 30 | 30 | 0.3 | 0.3 | 85 | 12 |
| 16 | 1200 | 30 | 30 | 0.2 | 0.05 | 85 | 7 |
| 17 | 1200 | 30 | 30 | 0.2 | 0.1 | 85 | 8 |
| 18 | 1200 | 30 | 30 | 0.2 | 0.2 | 85 | 10 |
| 19 | 1200 | 30 | 30 | 0.2 | 0.3 | 65 | 2 |
| 20 | 1200 | 30 | 30 | 0.2 | 0.3 | 75 | 8 |
| 21 | 1200 | 30 | 30 | 0.2 | 0.3 | 100 | 13 |
The rate of sulfonation reaction depends on the SO2 pressure. No MSA was detected in the absence of SO2; however, approximately 0.1 mmol of CF3SO3CH3 was formed. With an increase in SO2 pressure from 0 to 30 psig, the conversion of SO2 to MSA increased from 0 to 12% (Table 2, entries 5–8).
No MSA was formed in the absence of O2. With an increase in O2 pressure from 0 to 30 psig, the conversion of SO2 to MSA increased from 0 to 12%. However, a further increase had no effect on MSA production (Table 2, entries 9–12).
Increasing the amount of PdCl2 from 0.05 to 0.2 mmol, the conversion of SO2 to MSA increased from 3 to 12%. A further increase in the amount of PdCl2 had no effect on the MSA conversion (Table 2, entries 13–15).
When the amount of CuCl2 was increased from 0.05 to 0.3 mmol, the SO2 conversion to MSA increased from 7 to 12% (Table 2, entries 16–18). In the absence of CuCl2, Pd-black particles were observed in the reaction mixture after 4 h of reaction, whereas in presence of CuCl2 the appearance of Pd-black particles was not so prominent. This suggests that CuCl2 enhances the rate of oxidation of Pd(0) to Pd(II) species.
The conversion of SO2 to MSA increased from 2 to 12% when the temperature was raised from 65 to 85 °C. At 100 °C, a 13% conversion of SO2 to MSA was achieved and a trace amount of CF3SO3CH3 was also detected (Table 2, entries 19–21).
The reaction requires a highly acidic solvent. When performed in H2SO4, 5% conversion of SO2 to MSA was observed; however, no reaction was observed using acetic acid as the solvent. A 12% conversion of SO2 to MSA was achieved using CF3SO3H as the solvent. To verify that the solvent CF3SO3H does not react with CH4 to give CH3SO3H and CHF3 (CH4 + CF3SO3H → CH3SO3H + CHF3), a controlled reaction was performed in presence of O2 and catalysts in CF3SO3H. No SO2 was added. Under these conditions, MSA was not detected after 12 h of reaction. Likewise, no CHF3 was detected by 19F NMR. A small amount of CF3SO3CH3 was observed as the sole product.
The mechanism by which Pd(II) and Cu(II) promote the sulfonation of CH4 to MSA is not understood. It seems plausible to suggest, though, that the reaction proceeds via an electrophilic substitution of high valent Pd-species with CH41h–j,5,6 and subsequent SO2 insertion and oxidation3h to form MSA and Pd(0). Cu(II) then promotes the reoxidation of Pd(0) to Pd(II) in presence of O2.7
In conclusion, we have developed a highly selective low-temperature reaction protocol to sulfonate methane to methanesulfonic acid using SO2 as the sulfonating agent and O2 as the oxidant in the presence of a redox catalyst system comprising Pd(II) and Cu(II) salts. The reaction is highly selective, and as much as 20% of the SO2 charged is converted to MSA with only 30 psig SO2, the maximum available pressure. The product MSA can be isolated from the reaction mixture by distillation under reduced pressure.
ATOFINA Chemicals, Inc., North America, funded this study.
Notes and references
- (a) C. L. Hill, Activation and functionalization of Alkanes, Wiley, New York, 1989 Search PubMed; (b) M. G. Axelrod, A. M. Gaffney, R. Pitchai and J. A. Sofranko, Natural Gas Conversion II, Elsevier, Amsterdam, 1994, p. 93 Search PubMed; (c) G. A. Olah and A. Molnar, Hydrocarbon Chemistry, Wiley, New York, 1995 Search PubMed; (d) G. J. Hutchings, M. S. Scurrell and J. R. Woodhouse, Chem. Soc. Rev., 1989, 18, 251 RSC; (e) R. M. Ormerod, Chem. Soc. Rev., 2003, 32, 17 RSC; (f) K. Otsuka and Y. Wang, Appl. Catal., 2001, 222, 145 CrossRef CAS; (g) A. Ueno, Catalysis, 2000, 15, 185 Search PubMed; (h) G. Dyker, Angew. Chem., Int. Ed., 1999, 38, 1698 CrossRef; R. A. Periana, D. J. Taube, E. R. Evitt, D. G. Loffer, P. R. Wentrcek, G. Voss and T. Masuda, Science, 1993, 259, 340 CAS; (i) R. A. Periana, D. J. Taube, S. Gamble, H. Taube, T. Satoh and H. Fujii, Science, 1998, 280, 560 CrossRef CAS; (j) R. A. Periana, O. Mirinov, D. J. Taube and S. Gamble, Chem. Commun., 2002, 2376 RSC.
- (a) Ullmann's Encyclopedia of Industrial Chemistry, VCH, Weinheim, 1994, Vol. A25, pp. 503–506 Search PubMed; (b) F. M. Beringer and R. A. Falk, J. Am. Chem. Soc., 1959, 81, 2997 CrossRef CAS; (c) H. A. Young, J. Am. Chem. Soc., 1937, 59, 811 CrossRef CAS; (d) R. C. Murray, J. Chem. Soc., 1933, 739 RSC.
- (a) N. Basickes, T. E. Hogan and A. Sen, J. Am. Chem. Soc., 1996, 118, 13111 CrossRef CAS; (b) L. J. Lobree and A. T. Bell, Ind. Eng. Chem. Res., 2001, 40, 736 CrossRef CAS; (c) S. Mukhopadhyay and A. T. Bell, Ind. Eng. Chem Res., 2002, 41, 5901 CrossRef CAS; (d) S. Mukhopadhyay and A. T. Bell, Org. Process. Res. Dev., 2003, 7, 161 Search PubMed; (e) S. Mukhopadhyay and A. T. Bell, Angew. Chem., Int. Ed., 2003, 42, 1019 CrossRef CAS; (f) S. Mukhopadhyay and A. T. Bell, Angew. Chem., Int. Ed., 2003, in press; (g) S. Mukhopadhyay and A. T. Bell, J. Am. Chem. Soc., 2003, 125, 4406 CrossRef CAS; (h) Y. Ishii, K. Matsunaka and S. Sakaguchi, J. Am. Chem. Soc., 2000, 122, 7390 CrossRef CAS.
- In a 100-ml glass lined high pressure Parr autoclave reactor, 0.2 mmol PdCl2, 0.3 mmol CuCl2, and 5 ml of trifluoromethanesulfonic acid were charged together with a small Teflon coated magnetic stir bar. The reactor was then pressurized with 30 psig SO2, 30 psig O2, and then ultimately with 1200-psig methane from the adjacent connecting cylinders. The reactor was then heated to 85 °C under stirring and kept at that temperature for 12 h. After the stipulated period of time, the reactor was cooled to room temperature and opened to collect the reaction mixture. The mixture was then added slowly to 1.0 g of water and then taken for 1H NMR analysis. D2O and methanol were used in a capillary as the lock references. The corresponding chemical shift for MSA was 2.78 to 2.98 ppm, depending on the concentration of MSA in the mixture.
- Pd and Cu catalyst combination is used for the carbonylation of methane to acetic acid in CF3COOH as the solvent, see (a) T. Nishiguchi, K. Nakata, K. Takaki and Y. Fujiwara, Chem. Lett., 1992, 1141 CAS; (b) A. Sen, Platinum Met. Rev., 1991, 35, 126 Search PubMed; (c) L.-C. Kao, A. C. Hutson and A. Sen, J. Am. Chem. Soc., 1991, 113, 700 CrossRef CAS; (d) Carbene based Pd-catalyst has been used recently for methane oxidation, see M. Muehlhofer, T. Strassner and W. A. Herrmann, Angew. Chem., Int. Ed., 2002, 41, 1745 Search PubMed.
- Pt-salt based methane activation by Shilov chemistry, see (a) A. E. Shilov and G. B. Shul'pin, Activation and Catalytic Reactions of Saturated Hydrocarbons in the Presence of Metal Complexes, Kluwer Academic Publishers: Dordrecht, 2000 Search PubMed; (b) For a nice mechanistic recent review see J. A. Labinger and J. E. Bercaw, Nature, 2002, 417, 507 Search PubMed.
- For the role of co-catalysts in PdCl2 catalyzed C–H bond activation in benzene, see S. Mukhopadhyay, G. Rothenberg, G. Lando, K. Agbaria, M. Kazanci and Y. Sasson, Adv. Synth. Catal., 2001, 343, 455 Search PubMed.
| This journal is © The Royal Society of Chemistry 2003 |