‘Template-switching’: a supramolecular strategy for the quantitative, gram-scale construction of a molecular target in the solid state
Tomislav
Friščič
and
Leonard R.
MacGillivray
*
Department of Chemistry, University of Iowa, Iowa City, IA 52242-1294, USA. Fax: +319-335-1270; Tel: +319-335-3504E-mail: len-macgillivray@uiowa.edu
First published on 6th May 2003
Abstract
A template–switching strategy based on the modularity of template-directed solid-state synthesis has been successfully utilised to achieve a quantitative, stereospecific solid-state synthesis of a p-[2.2]-cyclophane target in gram quantities.
The efficient synthesis of molecular targets is one of the central goals of synthetic chemistry.1 The solid state is an attractive medium for such a goal, since it offers a highly organized environment for stereoselective reactions.2 However, the solid state has remained largely unexploited as a medium for synthesizing molecular targets, even in the case of the well-studied [2+2] photodimerisation.3 The reason for this has been twofold. First, chemists have been largely unable to deliberately orient molecules in arrangements suitable for reaction, owing to the sensitivity of crystal structure to molecular structure. Second, the yields are often unsatisfactory and, furthermore, are difficult to improve owing to the inability to ‘adjust’ the crystal packing environment of the reactants.4 Overcoming these difficulties may enable the solid state to be used for the stereocontrolled syntheses of molecular targets in high yield.5
We have introduced a supramolecular6 approach for controlling the [2+2] photodimerization in the solid state that employs bifunctional molecules as linear templates. The templates control the positioning of reactant carbon–carbon double (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) bonds within hydrogen-bonded assemblies. Consequently, the approach allows us to deliberately orient CC bonds in solids such that we have been able to conduct a target-oriented solid-state synthesis, the first target being a p-cyclophane 3.7 Using 5-methoxyresorcinol (5-OMe-res) as a linear template, we achieved the construction of 3 from diolefin 1 in 60 % yield (Scheme 1). In addition to 3, other products formed, which we ascribed to short contacts between neighbouring assemblies in the solid.
C) bonds within hydrogen-bonded assemblies. Consequently, the approach allows us to deliberately orient CC bonds in solids such that we have been able to conduct a target-oriented solid-state synthesis, the first target being a p-cyclophane 3.7 Using 5-methoxyresorcinol (5-OMe-res) as a linear template, we achieved the construction of 3 from diolefin 1 in 60 % yield (Scheme 1). In addition to 3, other products formed, which we ascribed to short contacts between neighbouring assemblies in the solid.
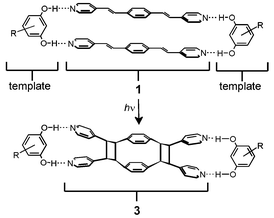 | ||
| Scheme 1 Schematic representation of template-directed solid-state synthesis of the p-cyclophane 3. R = 4-benzyl (2) or 5-OMe.7 | ||
With a target-oriented synthesis in the solid state realized, it has occurred to us that we may achieve the construction of 3 in quantitative yield (i.e. 100%). Specifically, we anticipated that switching the template to a different resorcinol would, de facto, lead to a different crystal packing environment (i.e. a reaction cavity) which, in contrast to 2(1)·2(5-MeO-res), could accommodate the generation of 3, as the sole product, in quantitative yield.8 Indeed, the modular nature of the approach permits the template and, hence, the crystal packing environment, to be changed, while such template-switching has been used to direct the formation of discrete assemblies in solids.9 The ability to generate 3 in quantitative yield may also provide ready access to gram quantities of the target.10
We now report template-switching as a supramolecular strategy for the quantitative construction of 3 in 2(1)·2(2) (where 2 = 4-benzylresorcinol). This strategy has made gram quantities of 3 readily available such that we are able to confirm the structure of our first target by way of single crystal X-ray diffraction.
Co-crystals of composition 2(1)·2(2) were prepared by mixing 1 and 2 in 1∶1 ratio in nitromethane.† X-ray crystal structure analysis of 2(1)·2(2)‡ reveals that, similar to 2(1)·2(5-OMe-res), the solid consists of discrete four-component molecular assemblies, each of which sits around a crystallographic centre of inversion, held together by four O–H⋯N hydrogen bonds (Fig. 1). The olefin groups of the assemblies lie parallel and organized with C⋯C separation distances of 3.91 Å
(C6⋯C15) and 3.77 Å
(C7⋯C14). Notably, in contrast to 2(1)·2(5-OMe-res), the CC bonds are ordered.
 | ||
| Fig. 1 ORTEP3 representation of the structure of the assembly 2(1)·2(2). Thermal ellipsoids of nonhydrogen atoms are shown at 30% probability level. Symmetry operator i: −1−x, 1−y, −z. Selected hydrogen bond distances (Å): O1⋯N1 2.765(2), O2⋯N2 2.778(2). | ||
UV-irradiation (500 W Hg lamp) of a powdered crystalline sample of 2(1)·2(2) resulted in the quantitative conversion of 1 to target 3.§ This conversion is evidenced from the 1H NMR spectra (solvent: DMSO-d6) of 2(1)·2(2) before and after photoreaction. The disappearance of olefin proton signals (7.32 and 7.57 ppm) of 1 is coincident with the appearance of cyclobutane proton signals (4.60 and 4.75 ppm) (Fig. 2). Thus, switching of the template has facilitated a stereospecific, as well as a quantitative, conversion of 1 to the target 3. To our knowledge, 2(1)·2(2) represents the first example in which template-switching has been exploited as a means to achieve the quantitative construction of a molecular target.12
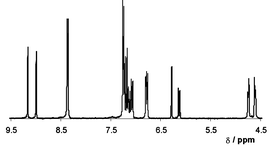 | ||
| Fig. 2 1H NMR spectrum of 2(1)·2(2) following UV-irradiation. | ||
The generation of 3 within 2(1)·2(2) has provided ready access to gram quantities of the target. This, in turn, has enabled us to prepare single crystals of 3 suitable for X-ray analysis.‡
The crystal structure of 3 reveals (Fig. 3) that the central aromatic rings of the cyclophane, which sits around a crystallographic centre of inversion, assume a boat conformation,13 with p-carbon atoms (C8 and C12ii) deviating 0.14 Å away from the plane of the remaining four benzene atoms. Notably, the cyclobutane ring C–C single bonds perpendicular to the planes of the central aromatic rings of 3 are longer [C6–C15, 1.582(3) Å and C7–C14, 1.609(3) Å] than the remaining two C–C bonds [C6–C7, 1.539(3) Å and C14–C15, 1.542(3) Å].2
 | ||
| Fig. 3 An ORTEP representation of 3. Symmetry operator ii: 2−x, −y, 2−z. | ||
The quantitative generation of the target 3 in 2(1)·2(2) can be rationalized by the switching of the template. A view of the extended structure of 2(1)·2(2)
(Fig. 4) reveals that the benzyl groups of the templates participate in offset, face-to-face π–π interactions that connect the assemblies into strands. The strands are arranged into layers parallel to the z-axis (Fig. 4a). Neighbouring strands are displaced such that, in contrast to 2(1)·2(5-OMe-res), there are no short (i.e. < 4.2 Å) contacts between CC bonds of adjacent 2(1)·2(2) assemblies, the closest contact being 5.4 Å
(Fig. 4b). Consequently, according to Schmidt's topochemical postulates,14
[2 + 2] photodimerization is unlikely to occur between neighbouring assemblies. Thus, in changing the crystal-packing environment of the organized olefins, the switching of the template has largely prohibited [2 + 2] photodimerization from occurring between the assemblies. This condition, presumably, favours the generation of 3 in quantitative yield within the solid 2(1)·2(2).¶
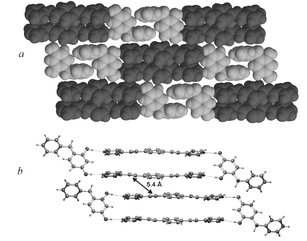 | ||
| Fig. 4 a) Space-filling representation of three strands of 2(1)·2(2) within a layer; b) ball-and-stick representation of assemblies in neighbouring layers. | ||
In summary, we have demonstrated template-switching as a supramolecular strategy for the stereospecific, quantitative construction of a p-cyclophane 3 in gram quantities. Application of 3 for the construction of metal-organic frameworks15 is now being investigated. We are also exploring template-switching as a means to develop a library of linear templates for the solid-state syntheses of targets of different size, shape, and functionality. Such systematic investigations may enable template-switching to be developed as a construction principle for directing reactivity in molecular solids. Recognition and development of such construction principles may lead to routine use of the solid state as a medium for efficient syntheses of diverse molecular targets.
We are grateful to the National Science Foundation (CA-REER Award, L. R. M., DMR-0133138) and the University of Iowa for support of this work. Acknowledgment is also made to the donors of the Petroleum Research Fund, administered by the American Chemical Society, and the Research Corporation (Research Innovation Award).
Notes and references
- K. C. Nicolaou, D. Vourloumis, N. Winssinger and P. S. Baran, Angew. Chem. Int. Ed., 2000, 39, 44 CrossRef CAS.
- Y. Maekawa, S. Kato and M. Hasegawa, J. Am. Chem. Soc., 1991, 113, 3867 CrossRef CAS.
- A. E. Keating and M. A. Garcia-Garibay, Molecular and Supramolecular Photochemistry, Vol. 2, V. Ramamurthy and K. Schanze, Ed., Marcel Dekker, New York, 1998, 195 Search PubMed; L. R. MacGillivray, Cryst. Eng. Comm., 2002, 4, 37 Search PubMed.
- H. Zitt, I. Dix, H. Hopf and P. G. Jones, Eur. J. Org. Chem., 2002, 2298 CrossRef CAS.
- K. S. Feldman and R. F. Campbell, J. Org. Chem., 1995, 60, 1924 CrossRef CAS; D. G. Amirsakis, M. A. Garcia-Garibay, S. J. Rowan, J. F. Stoddart, A. J. P. White and D. J. Williams, Angew. Chem. Int. Ed., 2001, 40, 4256 CrossRef CAS and references therein.
- J.-M. Lehn, Science, 2002, 2400 CrossRef CAS.
- L. R. MacGillivray, J. L. Reid and J. A. Ripmeester, J. Am. Chem. Soc., 2000, 122, 7817 CrossRef CAS.
- V. Ramamurthy, Tetrahedron, 1986, 42, 5753 CrossRef CAS; H. E. Zimmerman and E. E. Nesterov, Acc. Chem. Res., 2002, 35, 77 CrossRef CAS.
- G. S. Papaefstathiou and L. R. MacGillivray, Org. Lett., 2001, 3, 3835 CrossRef CAS.
- G. Kaupp, J. Schmeyers and J. Boy, Chemosphere, 2001, 43, 55 CrossRef CAS.
- V. Enkelmann, G. Wegner, K. Novak and K. B. Wagener, J. Am. Chem. Soc., 1993, 115, 10390 CrossRef CAS; D. B. Varshney, G. S. Papaefstathiou and L. R. MacGillivray, Chem. Commun., 2002, 1964 RSC.
- K. Tanaka, F. Toda, E. Mochizuki, N. Yasui, Y. Kai, I. Miyahara and K. Hirotsu, Angew. Chem. Int. Ed., 1999, 38, 3523 CrossRef CAS; Y. Ito, Synthesis, 1998, 1 CrossRef.
- D. J. Cram and J. M. Cram, Acc. Chem. Res., 1971, 4, 204 CrossRef CAS.
- G. M. J. Schmidt, Pure Appl. Chem., 1971, 27, 647 CAS.
- G. S. Papaefstathiou and L. R. MacGillivray, Angew. Chem. Int. Ed., 2002, 41, 2070 CrossRef CAS.
Footnotes |
| † In a typical experiment a solution of 2 (450 mg) in nitromethane (10 cm3) was added to a suspension of 1 (600 mg) in nitromethane (15 cm3). After 20 minutes reflux, the cooled mixture was filtered to give yellow crystals of 2(1)·2(2) (990 mg, 97%). On UV-irradiation the crystalline powder turned white and was subsequently heated with 1 M NaOH (10 cm3), followed by extraction in CH2Cl2 to give 3 (510 mg, 88% yield). The procedure has been repeated with up to 1 g of 1. |
‡ Crystal data for 2(1)·2(2): triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a
= 7.577(2), b
= 10.951(2), c
= 15.687(3)
Å, α
= 77.47(3)°, β
= 87.47(3)°, γ
= 84.38(3)°, Z
= 2, μ(MoKα)
= 0.079 mm−1, 6456 reflections measured, 3298 unique (Rint
= 0.014). The final wR(F2) was 0.132 (all data); crystal data for 3: monoclinic, P21/c, a
= 15.489(2), b
= 8.048(1), c
= 11.523(1)
Å, β
= 96.991(2)°, Z
= 2, μ(MoKα)
= 0.078 mm−1 , 8705 reflections measured, 3432 unique (Rint
= 0.040). The final wR(F2) was 0.158 (all data). CCDC 203760 and 203761. See http://www.rsc.org/suppdata/cc/b3/b301726p/ for crystallographic data in .cif or other electronic format. , a
= 7.577(2), b
= 10.951(2), c
= 15.687(3)
Å, α
= 77.47(3)°, β
= 87.47(3)°, γ
= 84.38(3)°, Z
= 2, μ(MoKα)
= 0.079 mm−1, 6456 reflections measured, 3298 unique (Rint
= 0.014). The final wR(F2) was 0.132 (all data); crystal data for 3: monoclinic, P21/c, a
= 15.489(2), b
= 8.048(1), c
= 11.523(1)
Å, β
= 96.991(2)°, Z
= 2, μ(MoKα)
= 0.078 mm−1 , 8705 reflections measured, 3432 unique (Rint
= 0.040). The final wR(F2) was 0.158 (all data). CCDC 203760 and 203761. See http://www.rsc.org/suppdata/cc/b3/b301726p/ for crystallographic data in .cif or other electronic format. |
| § Analyses of X-ray powder diffraction patterns suggest that the crystallinity of 2(1)·2(2) is retained upon UV-irradiation. Further investigations are underway to determine whether the reaction proceeds by way of a single-crystal-to-single-crystal transformation.11 |
| ¶ A detailed discussion of 2(1)·2(2) may also require the reaction cavity model.8 |
| This journal is © The Royal Society of Chemistry 2003 |