Cation–π interactions as a tool to enhance the power of a chiral auxiliary during asymmetric photoreactions within zeolites†
Lakshmi S.
Kaanumalle
,
J.
Sivaguru
,
N.
Arunkumar
,
S.
Karthikeyan
and
V.
Ramamurthy
*
Department of Chemistry, Tulane University, New Orleans, LA 70118, USA. E-mail: murthy@tulane.edu
First published on 3rd December 2002
Abstract
Owing to the existence of cation–π interactions, aryl chiral auxiliaries perform far better than alkyl chiral auxiliaries during asymmetric photoreaction.
Based on an analysis of three independent systems undergoing different types of photoreactions we show here that the best asymmetric induction in photoreactions within zeolites is achieved when chiral auxiliaries contain an aromatic group. Ab initio computations at the Hartree–Fock level suggest that interactions (cation–π) between an alkali metal ion and the aromatic group of the chiral auxiliary is likely to be the primary cause for the high asymmetric induction with chiral auxiliaries containing aryl groups. In the systems we discuss here the chiral auxiliary (a–d, Scheme 1) was placed at a center remote from the reaction site via an amide linkage and the asymmetric induction during a photoreaction was measured by the diastereomeric excess (de) in the photoproduct.1 We did not identify the absolute stereochemistry of the two diastereomers. For the current discussion this information is not absolutely essential.
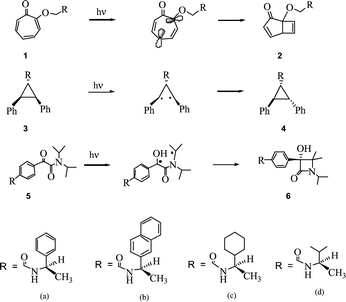 | ||
| Scheme 1 | ||
Tropolone derivatives 1a–d upon irradiation in solution under conditions of reduced secondary photoreactions (by <20 min irradiation) gave a diastereomeric mixture of the bicyclo[3.2.0] products 2a–d (Scheme 1) with de varying between 0 and 10%.2 The same cyclized products were obtained with de varying between 45 and 88% (Table 1) on irradiation of 1a–d included in NaY. As seen in Table 1, the two chiral auxiliaries (a and b) containing an aryl group gave de >85% while those substituted with only alkyl groups (c and d) gave lower de (45%). Consistent with this, tropolone derivatives containing auxiliaries derived from (−)-norephedrine and L-phenylalanine methyl ester, both containing phenyl groups, gave the bicyclo[3.2.0] product within NaY in 78% and 86% de respectively.
| Percentage diastereomeric excess | ||||
|---|---|---|---|---|
| Chiral auxiliary attached to the reactant | Si/Al ratio of the zeolite used | 1 (NaY) | 3 (LiY) | 5 (NaY) |
| a | 2.4 | 85 | 85 | 62 |
| a | 6 | 22 | 25 | 0 |
| a | 15 | 16 | 12 | 0 |
| a | 40 | 4 | 10 | 7 |
| b | 2.4 | 88 | 70 | 54 |
| c | 2.4 | 45 | 29 | 22 |
| c | 6 | 30 | 27 | 30 |
| c | 15 | 28 | 13 | 28 |
| c | 40 | 20 | 21 | 20 |
| d | 2.4 | 45 | 7 | 30 |
Enhanced performance of aryl chiral auxiliary within zeolites is not restricted to the above reaction. Excitation of trans,trans-2,3-diphenylcyclopropane-1-carboxamides 3a–d in solution and MY zeolites (M = alkali metal ion) resulted in geometric isomerization to yield cis,trans-2,3-diphenylcyclopropane-1-carboxamides 4a–d (Scheme 1).3 While in solution the product was obtained in less than 2% de, within LiY the extent of de obtained was dependent on the nature of the chiral auxiliary. Irradiation of trans,trans-2,3-diphenylcyclopropane-1-carboxamides 3a and 3b that contain aryl chiral auxiliaries included in LiY gave products in 85% and 70% de while 3c and 3d that contain alkyl chiral auxiliaries gave products in 29% and 7% de (Table 1). α-Oxoamides 5a–d upon excitation yielded β-lactams 6a–dvia a sequence of electron and proton transfers (Scheme 1).4 Upon irradiation in NaY zeolite, α-oxoamides that contain aryl chiral auxiliaries (5a and 5b) gave higher de than those with alkyl chiral auxiliaries (5c and 5d) (Table 1).
A number of observations suggested that alkali metal ions present in zeolites play an important role in the asymmetric induction process in the above three systems. We highlight results from one system as representative of the behavior of all the molecules investigated. (a) The de was dependent on the nature of the alkali metal ion (e.g., de in the cases of 1a in LiY, NaY, KY, RbY and CsY are 64, 85, 26, 5 and 5 respectively). (b) The de varied with water content of NaY used (1a: dry 85% and wet 3%). (c) The de upon irradiation of 1a adsorbed on silica gel, a surface that does not contain cations, was only 3%. (d) The diastereomeric excess in the case of 1a decreased from 85% to 4% when the Si/Al ratio of NaY zeolite was changed from 2.4 to 40 (fewer aluminium atoms in the framework mean fewer Na+ ions). In Table 1, de obtained upon irradiation of 1a, 1c, 3a, 3c, 5a and 5c in Y zeolites with different Si/Al ratios are listed. Clearly in every case the de was dependent on the number of alkali metal ions present in a zeolite. Especially for systems containing aryl chiral auxiliaries there was at least an eight-fold reduction in de between the zeolites with Si/Al ratios of 2.4 and 40. Alkali ions dissolve in solvents where they would be solvated by solvent molecules and such solvated ions are not expected to form strong complexes with the reactant molecules. Under such conditions most molecules would remain uncomplexed.
The important question concerning the reason for the differential behavior of the aryl and the alkyl chiral auxiliaries was answered with the help of ab initio computations at the Hartree–Fock level. Ab initio computations of energies and geometries of alkali metal ion bound 1a, 1c, 3a, 3c, 5a and 5c were performed with the Gaussian 98 package.5 All the above molecules interacted strongly with Li+ and Na+ ions with binding affinities >60 kcal mol−1. Since alkali metal ions are bound to the surface of a zeolite, the binding interaction between the cation and the guest molecules within a zeolite are expected to be smaller than the values computed here for free cations. It is quite likely that the computed structures may not be the most preferred structure within the confined cages of a zeolite.
The relevant optimized structures and binding affinities in the cases of 1a and 1c are presented in Fig. 1 and the rest are presented as electronic supplementary information (ESI†). In the two structures shown for 1a, Na+ is bound to the aryl ring present in the chiral auxiliary part as well as to the carbonyl group of the amide linkage. In the case of 1c, the cation is bound either to the tropolone or amide oxygens and does not interact with the cyclohexyl ring. Similar features were found in stable cation bound structures for 3a, 3c, 5a and 5c. In the structurally similar cation bound 1a, 3a and 5a the cation interacts simultaneously with both phenyl group and the amide carbonyl oxygen. Such an interaction is expected to reduce the rotational freedom of the chiral auxiliary and thus make it ‘rigid’. On the other hand, in the cases of 1c, 3c and 5c the cation primarily interacts only with the amide carbonyl oxygen via an ion–dipolar type interaction and does not interact with the chiral auxiliary part that contains the cyclohexyl group. Such a type of interaction would have no effect on the rotational mobility of the chiral auxiliary.
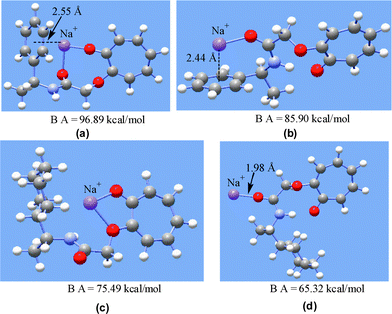 | ||
| Fig. 1 HF/3-21G* optimized structures of Na+ bound 1a (a and b) and 1c (c and d). Binding affinities are included at the bottom of each structure. | ||
A model based on the difference in flexibility of the chiral auxiliary parts due to differences in cation binding between aryl and alkyl chiral auxiliaries accounts for the observed variation in de between the two classes of chiral auxiliaries within MY zeolites. The prominent interaction between the alkali metal ion and the aryl group could be characterized as a cation–π6 interaction that helps to restrict the freedom of the chiral auxiliary. While one might question the relevancy of the computed ‘free-space’ cation–organic structures to those within a ‘confined space’, the fact that the photochemical behavior of several molecules could be rationalized on the basis of a model developed with computed structures suggests that simple ab initio computations are valuable to understand reactions within zeolites.
Consistently, chiral auxiliaries containing an aryl group give higher de than those with only alkyl groups. Our studies suggest that in order to achieve asymmetric induction within zeolites one needs to think beyond the ‘shape selective features’ of a zeolite and use chiral auxiliaries that would interact with the active sites of a zeolite, namely cations.
The authors thank the National Science Foundation for support of the research (CHE-9904187 and CHE-0212042) and the Tulane Center for Computational Science for providing time on their computers.
Notes and references
- All zeolites used in this study were procured from Zeolyst International and were activated at 500 °C prior to use. Reactants were introduced into the zeolite by stirring the zeolite (300 mg) and the reactant (∼5 mg) together in hexane. The loading level was generally one molecule per four supercages. By GC analysis of the supernatant, it was made sure that all the reactants were adsorbed onto the zeolite. Following irradiation of the zeolite–hexane slurry the products were isolated by extraction of the zeolite with either tetrahydrofuran, acetonitrile or ether and analyzed by GC or HPLC. The analytical columns used were the following: 1a and 1c: β-dex-350 (GC); 1b and 1d: Chiralpack ADRH (HPLC); 3a, 3c and 3d SE-30 (GC); 3b: Chiralpack ADRH (HPLC); 5a: Chiralcel OD (HPLC); 5b and 5c: Chiralpack ADRH (HPLC); 5d: Chiralcel OJ (HPLC).
- A. Joy and V. Ramamurthy, Chem. Eur. J., 2000, 6, 1287–1293 CrossRef CAS.
- J. Sivaguru, J. R. Scheffer, J. Chandrasekhar and V. Ramamurthy, Chem. Commun., 2002, 830–831 RSC.
- N. Arunkumar, K. Wang, V. Ramamurthy and J. R. Scheffer, Org. Lett., 2002, 4, 1443–1446 CrossRef.
- Gaussian 98, Revision A.9, Gaussian Incorporation, Pittsburgh, PA, 1998.
- J. C. Ma and D. A. Dougherty, Chem. Rev., 1997, 97, 1303–1324 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: HF/3-21G optimized structures of Li+ bound 3a and 3c, Na+ bound 5a and 5c and enlarged Fig. 1. See http://www.rsc.org/suppdata/cc/b2/b209810e/ |
| This journal is © The Royal Society of Chemistry 2003 |