From Lobry de Bruyn to enzyme-catalyzed ammonia channelling: molecular studies of D-glucosamine-6P synthase
Alexey Teplyakova, Caroline Lericheb, Galya Obmolovaa, Bernard Badet*c and Marie-Ange Badet-Denisotc
aCenter for Advanced Research in Biotechnology, University of Maryland, 9600 Gudelsky Drive, Rockville, MD 20850, U.S.A.
bDepartment of Medicinal Chemistry, Merck, 4 avenue du Président F. Mitterrand, F91380, Chilly-Mazarin, France
cInstitut de Chimie des Substances Naturelles-CNRS, 91198, Gif-sur-Yvette Cedex, France. E-mail: badet@icsn.cnrs-gif.fr
First published on 7th November 2001
Abstract
Covering: up to June 2001
This review summarizes the state of knowledge on D-glucosamine-6P synthesis catalyzed by glucosamine-6P synthase. The mechanisms of L-glutamine hydrolysis, ammonia transfer and fructose-6P conversion into D-glucosamine-6P are analyzed with the E. coli enzyme in light of recent X-ray structures. With 92 references this paper covers the literature up to June 2001 and emphasizes the potential implication of the mammalian glucosamine-6P synthase in type 2 diabetes.
 Alexey Teplyakov | Alexey Teplyakov is Senior Research Associate at the Center for Advanced Research in Biotechnology of the University of Maryland. Following his postdoctoral work with Professor W. Hol at the University of Groningen, he spent seven years as a staff scientist in the European Molecular Biology Laboratory in Hamburg. His research is focused on structure–functional studies of proteins with potential medical applications using X-ray crystallography. He is also interested in developing methods for structural analysis. |
 Caroline Leriche | After completing her PhD degree in 1995 with Dr B. Badet, Caroline Leriche was a 2-year post-doctorate fellow at the University of Minnesota, USA under the guidance of Professor H-W. Liu. She is now head of laboratory at Merck (France) in the Medicinal Chemistry department where she also pursued a High-Tech training at Merck KGaA (Germany) and in Professor S. Schreiber's laboratory at Harvard Medical School (USA). Her research interests are mainly focused on bio-organic chemistry directed toward type-2-diabetes. |
 Galya Obmolova | Galina Obmolova is Research Associate at the Center for Advanced Research in Biotechnology of the University of Maryland. She applies crystallographic and biochemical techniques to studies of proteins and macromolecular assemblies, with particular interest in proteins involved in DNA repair, pyruvate dehydrogenase complex, and peptidoglycan biosynthesis. |
 Bernard Badet | Bernard Badet (born 1949) graduated in 1973 from the Engineer School of Chemistry in Caen. He received his PhD degree in chemistry (1980) from Paris 6 University under the guidance of Professor Marc Julia. After two years of postdoctoral work (1983–1985) with Professor Christopher T. Walsh (MIT, USA) as a CNRS scientist, he worked at Ecole Nationale Supérieure de Chimie de Paris (1985–1992). In 1996 he moved to the Chemical Institute of Natural Products in Gif-sur-Yvette where he is CNRS Research Director. His research interests are the study of enzyme mechanisms and the elaboration of enzyme inhibitors. |
 Marie-Ange Badet-Denisot | Marie-Ange Badet-Denisot got her PhD degree in 1990 from University Paris 6 under the guidance of Dr Badet. Following postdoctoral work with Professor H. Zalkin (Purdue, USA), she joined the Gif-sur-Yvette team as a CNRS scientist. Her research interests focus on the comprehension of the various aspects of the catalysis performed by glucosamine-6P synthase. |
1 Introduction
Subsequent to its entry into the cell and its rapid phosphorylation to glucose-6-phosphate, glucose is primarily utilized by the pathways of glycogen synthesis and glycolysis. The hexosamine pathway receives part of incoming glucose via the conversion of fructose-6-phosphate (Fru-6P) into D-glucosamine-6-phosphate (GlcN-6P) by the rate-limiting enzyme glucosamine 6-phosphate synthase. GlcN-6P is then isomerized into GlcN-1P by GlmM, and further converted into uridine diphosphoglucose-N-acetyl D-glucosamine (UDP-GlcNAc) by the bifunctional enzyme GlmU.1 The latter is an important structural building block for bacterial peptidoglycan, lipopolysaccharide of Gram-negative bacteria, chitin and mannoproteins, which are the essential constituents of fungal cell wall. Moreover, mannosamine and galactosamine derivatives used in the synthesis of a variety of bacterial surface polysaccharides or mammalian structural polymers, such as chondroitin and keratan sulfates of connective tissues, are also obtained from epimerization at C-2 or C-4 of this substrate. Indeed, it also serves as a substrate for virtually all glycosylation pathways, which could be implicated in the regulation of glucose.This considerable involvement of glucosamine-6-phosphate synthase in a number of biological processes, as well as its potential as a therapeutic target in a disease such as type-2 diabetes, shows that this enzyme should be understood at a molecular level. We review here the progress we have made in the study of glucosamine-6-phosphate synthase from Escherichia coli on a chemical and enzymological basis, and more specifically concerning the aspects of the nitrogen transfer reaction.
2 A brief history of the chemistry of sugar amination
In 1893, reaction of glucose (or fructose) with methanolic ammonia revealed the direct transformation of carbohydrates into amino derivatives.2 The formation of 2-amino-2-deoxy-D-glucose (GlcNol) from D-fructose was deduced by Lobry de Bruyn who called these first so-characterized derivatives “osamines.”3 Over five decades later when D-glucosamine was identified from short time reaction between D-fructose and ammonia under mild conditions4 Heyns gave his name to this corresponding 1,2-oxido-reduction reaction.5 Ammonia addition on the ketose carbonyl functionality provides the corresponding carbinolamine, which in turn undergoes a 1,2 rearrangement similar to the Amadori rearrangement previously observed with aldoses.6 The mechanism of the reaction catalyzed by glucosamine-6P synthase (Scheme 1) was then postulated.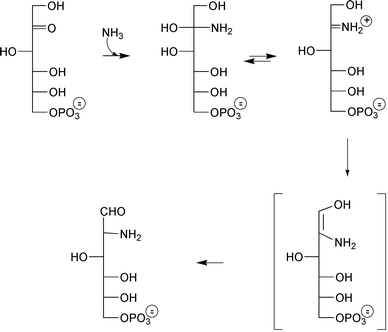 | ||
| Scheme 1 Initially proposed mechanism for the reaction of ammonia with fructose-6P catalyzed by GlmS. | ||
3 The family of L-glutamine-dependent amidotransferases
3.1 Two pathways for NH3 assimilation
In prokaryotes, there are two alternatives for the assimilation of ammonia, depending on its available concentration. When in excess (>1 mM), L-glutamate is directly obtained by L-glutamate dehydrogenase catalyzed amination of 2-ketoglutarate (eqn. (1)) (KmNH3 ∼ 100 mM). When limited (< 0.1 mM), the L-glutamate is obtained by an alternative two-step process requiring L-glutamine synthetase catalyzed amination of L-glutamate (eqn. (2), KmNH3 ∼ 0.1 mM) followed by L-glutamate synthase catalyzed amino transfer from L-glutamine to 2-ketoglutarate (eqn. (3)).| NH3 + 2-ketoglutarate + NADPH → L-glutamate + NADP | (1) |
| NH3 + L-glutamate + ATP → L-glutamine + ADP + Pi | (2) |
| L-glutamine + 2-ketoglutarate → 2 L-glutamate | (3) |
In bacteria, free ammonia, L-glutamate α-amino group or L-glutamine δ-amino group are the sole sources of nitrogen for the synthesis of macromolecules. Indeed, L-glutamate essentially provides nitrogen for the synthesis of amino acids whereas, more widely, the L-glutamine δ-amino group is a source of nitrogen for the synthesis of purines, pyrimidines, histidine, tryptophan, asparagine, L-glutamate, p-aminobenzoate, NAD, Glu-tARNGln and aminosugars. About 8 times more L-glutamate than L-glutamine is required for cellular biosyntheses.7L-Glutamine is the most abundant amino acid in human blood and in the free amino acid pool of the body. Certain tissues only are able to release significant amounts of L-glutamine even though the plasma L-glutamine concentration in the healthy adult human is approximately 0.6 mM. For example, its concentration of about 20 mM in skeletal muscle (similar to what is present in E. coli) contributes to approximately 60% of the total free amino acid pool.8
3.2 L-Glutamine as an important nitrogen donor: the L-glutamine dependent amidotransferases
L-Glutamine-dependent amidotransferases are a class of enzymes that catalyze the transfer of the δ-amino group of L-glutamine (amide function) to a wide variety of acceptors. Among this family of close to 20 enzymes9,10 some (involved in the biosynthesis of L-glutamate, histidine or tryptophan for example) are restricted to microorganisms. Other amidotransferases (involved in de novo nucleotides and aminosugar) are ubiquitous in all cells except for parasites. Analyses of cloned genes, high level of production of recombinant enzymes and determination of several structures by X-ray crystallography11,12 have considerably improved our understanding of this particular class of enzymes. Comparisons within the amidotransferase family mainly focused on the mechanism of ammonia generation since each member uses a different acceptor. In these studies, two sub-classes were found: class I enzymes, which use a catalytic triad to perform the L-glutamine amide bond cleavage and class II enzymes which use a different mechanism. Glucosamine-6P synthase belongs to the second subfamily.Glucosamine-6P synthase (L-glutamine:D-fructose-6P amidotransferase, EC 2.6.1.16) converts fructose-6P into D-glucosamine-6P using L-glutamine as a nitrogen donor according to eqn. (4):
| D-fructose-6P + L-glutamine → L-glutamate + D-glucosamine-6P | (4) |
This is the sole biosynthetic route to D-glucosamine-6P known to date. The latter can be inversely deaminated into fructose-6P and ammonia by the allosteric glucosamine-6P deaminase (EC 5.3.1.10).13 Depending on its prokaryotic, eukaryotic or mammalian origin, glucosamine-6P synthase has been termed GlmS, Gfa or Gfat.
Despite its apparent simplicity, this reaction raises a number of basic questions concerning the mechanism of the catalysis: (i) How does the enzyme perform amide bond cleavage? (ii) How is the released nitrogen transferred to fructose-6P and in which form? (iii) What is the nature of the reaction occurring on the sugar? (iv) Is the mechanism similar among all species?
4 The kinetics and mechanism of sugar amination
4.1 Modular organisation of GlmS
The native enzyme is a homo-dimer and, unlike most other amidotranferases, cannot use exogenous ammonia as nitrogen donor.14 The GlmS monomer (608 aa) is composed of two structurally and functionally distinct domains situated on the same polypeptide chain. Controlled chymotrypsin hydrolysis cleaves the native protein into a catalytically N-terminal 30 kDa glutaminase domain (NGlmS, responsible for hydrolysis of L-glutamine in L-glutamate and ammonia) and a C-terminal 40 kDa isomerase domain (in charge of ammonia utilization for the conversion of fructose-6P into D-glucosamine-6P).15 The two domains overexpressed separately16 retain their ability to bind substrates and to catalyze, respectively, L-glutamine hydrolysis and sugar isomerization.17 The three-dimensional structures of both glutaminase and isomerase domains of E. coli GlmS in complex with substrate (L-glutamate, γ-glutamyl hydroxamate 3, Glc-6P, GlcN-6P) or transition-state (GlcNol-6P 6) analogues have been determined by X-ray crystallography.18,19 The crystal structure of the native protein, which has recently been solved,20 is essential for understanding how the ammonia produced at the L-glutamine site is delivered to the sugar phosphate site. Moreover, it will be very precious for the design of specific inhibitors of this enzyme.4.2 The mechanism of L-glutamine hydrolysis
The observed binding of L-glutamate and γ-glutamyl hydroxamate in NGlmS allowed us to propose a detailed mechanism of L-glutamine hydrolysis by the enzyme. The domain has a four-layer structure (Fig. 1) with two antiparallel β-sheets sandwiched between layers of α-helices. This fold is the signature of the recently discovered superfamily of N-terminal nucleophile hydrolases (Ntn-hydrolases).21 The catalytic mechanism of L-glutamine hydrolysis discussed below therefore appears to be similar to that of the other Ntn hydrolases, although the catalytic amino acid varies: it is cysteine in amidotransferases, serine in penicillin acylase or threonine in the 20S proteasome and aspartylglucosaminidase.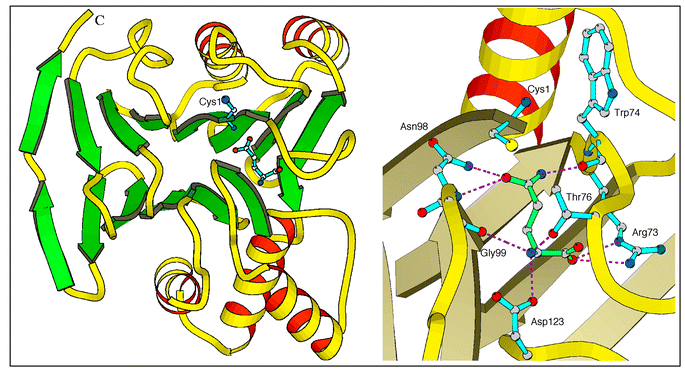 | ||
| Fig. 1 1.8 Å X-ray structure of the 240 aa NGlmS domain (PDB accession number 1GDO). Left: overall organization as a signature of the Ntn-hydrolase superfamily. Right: close-up view of the glutamine active site in complex with glutamine. | ||
The N-terminal catalytic cysteine in GlmS is located at the edge of the central β-sheet in a shallow cleft on the surface of the molecule. Other amino acids involved in substrate binding and catalysis reside in the loops connecting β-strands. One of the loops (residues 73–80, the Q-loop) is of particular importance as it covers the active site upon binding of L-glutamine and thus shields it from the solvent. The crystal structure of the glutaminase domain of GlmS does not indicate any residues that could enhance the nucleophilic character of Cys-1, except its own α-amino group. The proposed mechanism is as follows (Scheme 2). The N-terminal α-amino group of Cys-1 serves as a general base catalyst and abstracts the proton from the thiol group, through a bridging water molecule. The activated nucleophile attacks the amide carbon of the substrate to form a tetrahedral intermediate that is stabilized in the oxyanion hole formed by Asn-98 and Gly-99. The intermediate then collapses to form a γ-glutamylthioester and to release ammonia, which may receive a proton from the bridging water. Deacylation is accomplished by the nucleophilic attack through another water molecule that is activated through the same general base mechanism. This mechanism does not rule out the existence of an ammonium-thiolate ion pair in the resting state.
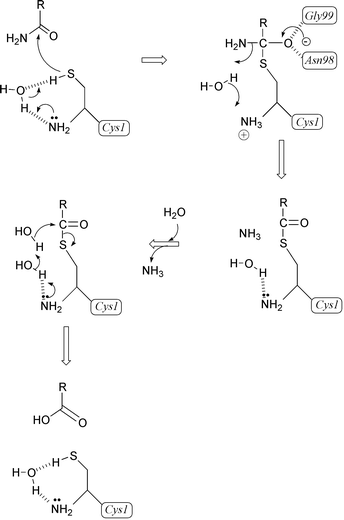 | ||
| Scheme 2 Mechanism accounting for glutamine hydrolysis by GlmS. | ||
The absolute requirement for the presence of the cysteine has been demonstrated by utilization of thiol-specific reagents and affinity label. Iodoacetamide was first recognized as an inhibitor of GlmS activity by a time-dependent irreversible process, which occurred in the minute range.14 The decrease in the pseudo-first-order rate constant in the presence of L-glutamine suggested the covalent derivation of a critical cysteine residue of the L-glutamine binding site. This was confirmed by DTNB titration of available SH groups before (4 accessible thiols) and after (3.5 accessible thiols) inactivation. Similar behaviour was observed using the L-glutamine-like affinity label reagent 6-diazo-5-oxo-L-norleucine (DON 1, Scheme 3), which reacted much faster than iodoacetamide (600 times faster from the ratio of kcat/Km). The incorporation of only 0.5 mole of reagent per mole of enzyme subunit in the absence of Fru-6P, rather than the expected stoichiometry of 1 reached in its presence, reflected the reactivity of only half of the site.22 It is worth noting that, despite the fact the reaction obeyed an ordered mechanism with fructose-6P binding first, the covalent modification of the L-glutamine site with an electrophilic substrate analogue could occur in the absence of fructose-6P. The amino acid responsible for covalent fixation of 6-[14C]-DON was identified as the N-terminal cysteine, suggesting possible participation of this thiol group in the catalysis. These experiments demonstrated the catalytic role of the N-terminal cysteine in GlmS. The activity vs. pH profile of the enzyme isolated from T. thermophilus identified two ionizable groups with pK values of 8.6 and 7.6, which are in agreement with the intervention of a SH group and of the N-terminal amine, respectively.23 Such a mechanism could explain the drastic (900-fold) drop in catalytic activity of the purified GST-GlmS fusion protein.24
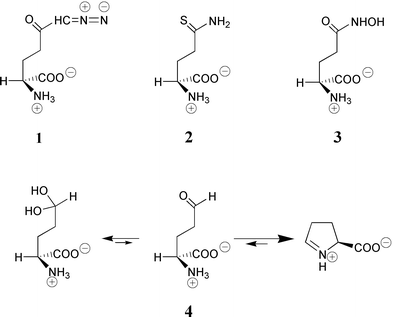 | ||
| Scheme 3 Structure of the glutamine analogues used in this work. | ||
The isolation of the acyl-enzyme intermediate, depicted in Scheme 2, from incubation experiments of GlmS with L-glutamine was not possible using conditions reported for class I amidotransferases. Furthermore, 5-thioglutamine (2, Scheme 3) did not form the expected dithioester adduct (analogous to the thioester of Scheme 2) that would have been detected by its characteristic UV absorption25 at ∼315 nm as was observed with papain using a thionoester substrate.26 Isolating a glutamyl-enzyme with class II amidotransferases is obviously much more difficult than with class I enzymes whose L-glutamine domains are of triad type. The absence of accumulation of the intermediate might explain these failures. However, the strong inhibition of GlmS by L-glutamate γ-semi-aldehyde (4, Scheme 3) is fully consistent with a γ-glutamyl enzyme intermediate.27 Compound 4 is mainly present in neutral solution as a cyclized imine form, Δ1-pyrroline-5-carboxylate (P5C) and is supposed to act as a transition state analogue as do aldehydes with cysteine proteases. Racemic P5C competitively inhibited GlmS with respect to L-glutamine with an apparent Ki = 23 μM. The complete lack of inhibition observed with proline or pyrrolidone carboxylic acid indicated that L-glutamate semi-aldehyde 4 may be responsible for the observed inhibition. From the evaluation of the equilibrium constant P5C/L-glutamate γ-semi-aldehyde (∼25) and the percentage of free aldehyde (∼1.4%) at pH 7.228 the observed dissociation constant underestimated the binding affinity by a factor ranking the aldehyde potency in the nanomolar range.
The structures of the complexes with the reaction product, L-glutamate, and the competitive inhibitor 3, reveal amino acids residues involved in substrate binding and catalysis (Fig. 1, right-hand side). The invariant residues Arg-73 and Asp-123 play a key role in substrate recognition, anchoring the α-COO− and α-NH3+ of L-glutamine. Substrate binding is probably accompanied by an induced-fit closure of the active centre that involves main-chain shifts of up to 10 Å. No evidence for the participation of histidine residue could be drawn from the inspection of the structure. The covalent, active site-directed modification of a single histidine residue reported upon use of diethyl pyrocarbonate29 in slight excess remains to be explained since the lability of the carbethoxyhistidine adduct to peptide mapping conditions precluded its localization in protein sequence.
Binding of a substrate at the nitrogen acceptor site increases by 130-fold (kcatsynthase/kcathydrolase) the rate of L-glutamine hydrolysis by GlmS. The central element of the mechanism that couples the glutaminase and synthase half-reactions is the invariant residue Arg-26. Conformational changes at the acceptor site are transferred to the L-glutamine site through Arg-26 (not shown), which locks catalytic Cys-1 in the active conformation, so that every L-glutamine molecule that binds to the enzyme is hydrolyzed. At low concentrations of the nitrogen acceptor, L-glutamine hydrolysis is grossly depressed and this may serve to save L-glutamine in the cell.
4.3 The mechanism of D-glucosamine-6P formation
The isomerase domain of GlmS encompasses residues 241–608. It is responsible for the binding of fructose-6P and its conversion into D-glucosamine-6P. This domain was crystallized in the presence of the reaction product D-glucosamine-6P, which is also a competitive inhibitor of GlmS with Ki = 0.35 mM.30 The crystal structure of this domain has been determined at 1.57 Å resolution (Fig. 2).19 It is bilobal in shape and consists of two topologically identical subdomains of equal size, each of which has a nucleotide-binding fold of a flavodoxin type. This fold is characterized by a five-strand parallel β-sheet flanked by α-helices on both sides. Amino acids implicated in catalysis are distributed over a number of secondary structural elements, which belong to both halves of the domain. One residue, His-504*, which is believed to play a key role in the sugar ring opening, is on a different polypeptide chain (therefore marked with an asterisk). Hence, for the active site to be assembled, dimerization of the protein is required.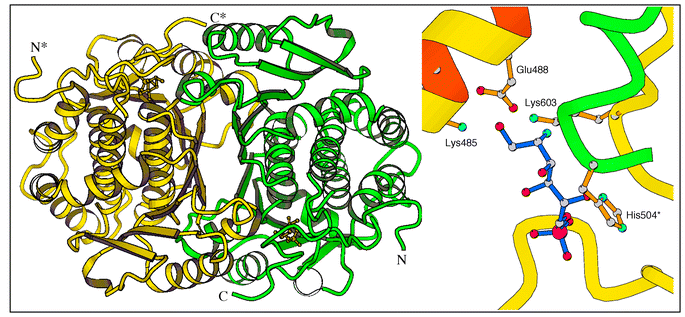 | ||
| Fig. 2 1.6 Å X-ray structure of the 367 aa CGlmS domain in complex with 6 (PDB accession number 1MOS). Left: overall organization of the dimer. Right: close-up view of the active site. Amino acid numbered with an asterisk belongs to the other subunit. | ||
D-Glucosamine-6P is likely to occupy the catalytic site of the isomerase domain in the crystal. Lys-603 located in the pocket has been implicated, through borohydride trapping experiments, as playing a catalytic role.31 In addition, the proximity of Cys-300 to the bound D-glucosamine-6P is consistent with the results of peptide mapping studies using the affinity label anhydro-1,2-hexitol 6P (see below, section 5.2).25
Numerous hydrogen bonds and salt bridges suggest that the framework of the active site is quite rigid. Conformational freedom required for substrate binding and transformation may be ensured by the water molecules that mediate the enzyme–substrate interactions. Relative flexibility in binding of the sugar moiety is compensated by the tight anchoring of the phosphate group that defines the proper orientation of the substrate with respect to catalytic elements. Such a mode of substrate binding may be typical for all sugar-phosphate isomerases and has implications for the catalytic mechanism.
There was initially no evidence that GlmS could catalyze the opening of the sugar ring as the first step of the reaction. The rate of spontaneous ring opening of Fru-6P is32 about 20 s−1 (KmFru-6P = 0.4 mM) which is comparable to the catalytic rate of GlmS. The first argument in favour of this recognition comes from the behaviour of the carbocyclic analogue of Fru-6P33 (Scheme 4).
 | ||
| Scheme 4 Structures of hexose-phosphate analogues, reversible inhibitors of GlmS. | ||
This compound which is frozen in a cyclopentyl configuration is recognized by the enzyme, exhibiting a mixed type inhibition behaviour in the millimolar range.34 The second argument in favour of the occurrence of this step is provided by the crystal structures with Glc-6P and GlcN-6P, which reveal complementarity of the binding site with the cyclic form of the sugar and indicate His-504 as a suitable base to perform the reaction. Ring opening has been considered to occur in other sugar isomerases such as phosphoglucose isomerase PGI,35D-xylose isomerase,36L-arabinose isomerase37 and GlcN-6P deaminase.13 In both enzyme:product complexes of GlmS with Glc-6P and with GlcN-6P, His-504 is H-bonded to the hydroxy group O-1 of the pyranose ring whereas in the complex with the transition state analogue GlcNo1-6P, the same imidazole Nδ binds the hydroxyl at C-5. We propose that upon binding of Fru-6P, the imidazole group of His-504 abstracts the O-2 hydrogen of the cyclic substrate and then returns it to O-5, thus completing the ring-opening step. The substrate molecule in an extended conformation is then ready to be processed further. The structure of the active-site and the mode of substrate binding through a number of bridging water molecules would allow the transition of the substrate from the cyclic to the extended form to occur without significant conformational change in the protein. Ring closure may be catalyzed as the reverse of the ring-opening step though it is not really required as the product cyclizes spontaneously under physiological conditions.
The reaction catalyzed by GlmS is thought to proceed via stereospecific abstraction of the C-1 HR hydrogen of fructosimine-6P to form a cis-enolamine intermediate which, upon protonation of C-2, gives rise to the product D-glucosamine-6P39 (Scheme 5). A similar mechanism could occur, albeit much less efficiently, with Fru-6P in the absence of ammonia, accounting for the phosphoglucose isomerase activities of CGlmS and GlmS (kcat/Km = 0.9 and 0.4 M−1 s−1 respectively). Identification of residues involved in catalysis is not so straightforward. As mentioned above, Lys-603 probably plays a role in amination reaction. Trapping of a radiolabelled adduct showed the formation of a Schiff base between Lys-603 and a substrate molecule.31 Replacement of Lys-603 by arginine, using site-directed mutagenesis, resulted in a 40-fold decrease in kcat for D-glucosamine-6P synthesis.22 The crystal structure shows that Lys-603 is indeed located in the active centre close to D-glucosamine-6P. The possibility of formation of a Schiff base with the substrate is compatible with the flexibility of the lysine side chain. As there is no other residue in the active site that could function as a general base in the isomerization step, Glu-488 is likely to be responsible for transferring a proton from C-1 to C-2 of a substrate. Early tritium transfer experiments demonstrated the participation of a single enzyme base with an estimated pKa of 7.3, which is in good agreement with a carboxylic residue with reduced acidity.38 In the crystal structure, the carboxylate group of Glu-488 interacts with the C-1 hydroxy group of 6, an analogue of the open form of the product.39Mutagenesis studies are underway to determine what role individual residues play in catalysis.
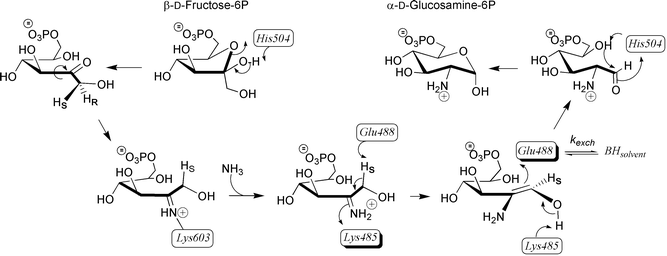 | ||
| Scheme 5 Proposed mechanism accounting for isomerisation of Fru-6P into GlcN-6P catalysed by GlmS. | ||
4.4 Ammonia channelling in dimeric GlmS
The ammonia produced by L-glutamine hydrolysis should remain trapped in the enzyme, to avoid protonation and to retain its nucleophilic character. This is consistent with the fact that GlmS cannot use exogenous ammonia for D-glucosamine-6P synthesis.14 There are two possible ways of transferring NH3 to the amination site located in the isomerase domain; either the two active sites merge together upon binding of substrates to allow a direct interaction, or they form an intramolecular channel that could isolate the L-glutamine-derived ammonia from bulk solvent. Inactivation studies using N-iodoacetyl-D-glucosamine-6P40 (14, Scheme 6) have shown that either of the substrates, fructose-6P or L-glutamine, could protect the enzyme from inactivation, suggesting the proximity of the active sites. In a similar experiment, inactivation with a radiolabelled sugar-phosphate analogue, 1,2-anhydro-hexitol-6P 15,25 was shown to affect Cys-1, the catalytic residue of the L-glutamine site, providing another argument in favour of a united active centre. On the other hand, lack of protection by L-glutamine25 and the inability of GlmS to bind bisubstrate analogues that consist of L-glutamine coupled by various spacers to 2-amino-2-deoxyglucitol-6P 627 corroborate the possibility of separate sites. This is also consistent with the observation that GlmS retains isomerase activity upon inactivation by 6-diazo-5-oxo-L-norleucine (DON, 1), a specific affinity label of Cys-1.17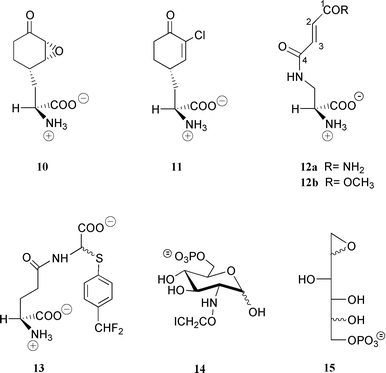 | ||
| Scheme 6 Structure of some irreversible inhibitors of GlmS. | ||
The 3D structures of the glutaminase and isomerase domains of GlmS indicate that the merging of the active sites would require substantial conformational changes, supporting the idea of a channel. The crystal structure of GlmS presents an internal channel between the two active sites, which are separated by 18 Å. The walls of the channel are lined with hydrophobic side chains that allow diffusion of NH3 between the two sites. Both isomerase and glutaminase domains contribute to the formation of the channel. The central portion of the channel resides in the C-terminal loop 601–607 (Fig. 3). Leu-601, Ala-602, Val-605, and Val-607 of this loop form a hydrophobic entrance to the sugar binding-site. The glutaminase “contribution” to the channel is constituted by the side chains of Trp-74 and Arg-26 (not shown). A third party involved is the histidine loop (H-loop) of another protein subunit that bears the catalytic His-504*. The dipeptide 504*–505* forms one wall of the channel. All these residues are strictly conserved in the ∼50 reported GlmS/Gfa/Gfat sequences.
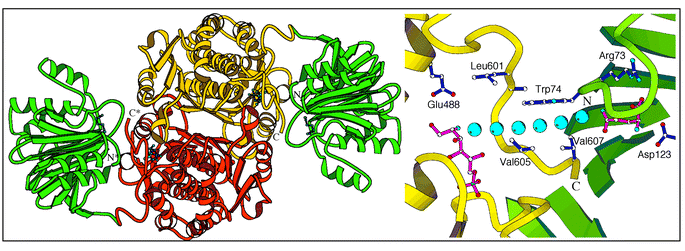 | ||
| Fig. 3 3.1 Å X-ray structure of native GlmS. Left: overall organization of the dimer. Right: suggested ammonia transfer between the two sites within a single polypeptide chain. | ||
In the initial state of the protein, prior to substrate binding, (i) the isomerase site is open due to the flexibility of the C-tail (residues 599–608), (ii) the glutaminase site is open due to the interaction between the Q-loop (residues73–80) and Arg-539*, and (iii) the intramolecular channel is formed although Trp-74 blocks it.
Binding of fructose-6P causes the closure of the site by the C-tail (Fig. 4).41 An ordered binding of substrates with the sugar binding prior to the amino acid substrate was deduced from the product and dead-end inhibition studies using DON. The sugar ring opening is catalyzed by His-504*. As a result of the conformational change in the sugar, H-bonds to the C-tail are lost and instead a Schiff base with Lys-603 is formed.
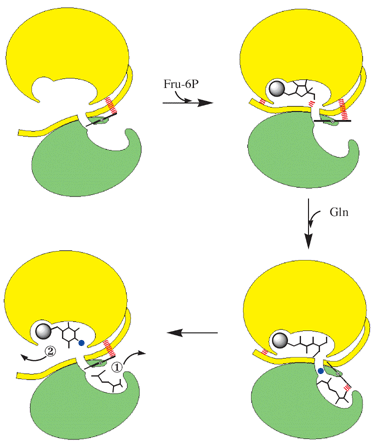 | ||
| Fig. 4 Schematized catalytic cycle of GlmS. For clarity only one monomer is shown. Green, glutamine binding domain; yellow, Fru-6P binding domain. The spheres represent phosphate (grey) and ammonia (blue). | ||
Upon binding of L-glutamine the Q-loop closes the active site, and the indole ring of Trp-74 is displaced from the channel. Ammonia produced by the hydrolysis of L-glutamine is transferred to the sugar-phosphate site to replace Lys-603 in the Schiff base. Following the amination at C-2, the sugar undergoes isomerization to GlcN-6P catalyzed by Glu-488.
Upon cyclisation of the reaction product, perhaps with the help of His-504*, the sugar-phosphate site opens, allowing a water molecule to come in and reach the L-glutamine site through the same “ammonia” channel. The glutamyl thioester adduct breaks down to L-glutamate, which is released from the active site following the displacement of the Q-loop. Remarkably, a similar gateway mechanism coupling the two reactions was observed in tryptophan synthase.42
5 Inhibitors and inactivators of sugar amination
The amount of GlcN-6P synthesized by bacterial GlmS during a single E. coli generation time corresponds to about 2 × 107 molecules per cell (as calculated from the amount of enzyme recovered after purification from large-scale culture of wild-type E. coli), which is only 2–3 times higher than the amount required for peptidoglycan biosynthesis. The inhibition of the enzyme could have important consequences on cell life, a conclusion that is corroborated by the fact that mutation inside the glms gene is lethal for the bacteria. Moreover, the concentration of the glmS encoded protein in E. coli is in the micromolar range, rendering this enzyme a very attractive target for the design of new antibacterial and/or antifungal agents. Knowledge concerning a lead structure would then be very helpful in the search for GlmS inhibitors.5.1 Inhibitors
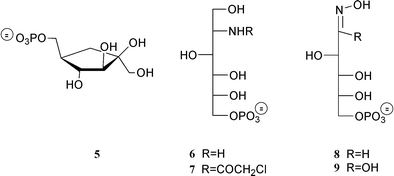
As discussed in section 4.2 L-glutamate semi-aldehyde 4 (Scheme 3) is a very potent reversible inhibitor of the GlmS L-glutamine site with an estimated Ki of 10 nM. It is by far superior to γ-glu-hydroxamate 3 (Ki ∼ 100 μM) and 2 orders of magnitude more efficient than DON 1 (Ki ∼ 1 μM) or anticapsine and fumaroyldiaminopropionate (Ki ∼ 0.5 μM) which are discussed below.Investigations of the fructose-6P binding site using substrate (Fru-6P) or product (GlcN-6P) analogues have met with less success. The observation that 6 (i.e. the reduced form of GlcN-6P) behaved as a potent competitive inhibitor (Ki ∼ 20 μM)43 offered a means of designing active site directed inactivators by incorporation of a chemically reactive haloacetyl function on the nitrogen. However, 2-N-chloroacetylglucitol-6P, 7 (Scheme 4), behaved as a simple competitive inhibitor of the sugar-binding site of GlmS with a Ki of 58 μM. Bearne28 was more successful in investigating the behaviour of N-iodoacetyl-D-glucosamine-6P 14 (Scheme 6), which inactivated the enzyme with a first-order rate constant kinact = 0.22 min−1 and a Kirr = 0.22 mM. The potency of 6, which can be considered as a tetrahedral analogue of the Schiff base between fructose-6P and ammonia (or of the cis-enolamine resulting from H-1 proton abstraction), validates the initial hypothesis that the reaction occurs via the imine intermediate. This molecule was used as the Fru-6P binding moiety in the construction of bisubstrate analogues. They were synthesized by coupling, directly or via a spacer, the amino group of 2-amino-2-deoxyglucitol-6P 6 to the γ-carboxyl function of L-glutamic acid.27 None of these derivatives exhibited significant inhibition up to 3 mM, which may be due to the high flexibility of these bisubstrate analogues preventing the correct orientation required to enter the binding pockets and the substantial conformational change occurring in the protein upon binding of its substrates.
Arabinose-5P oxime 8 (Scheme 4) was found to be the most potent competitive inhibitor of the GlmS sugar site (Ki = 14 μM). The Km/Ki value of ∼30 is consistent with this molecule being a high energy intermediate analogue of the GlmS-catalyzed keto/aldose isomerization.44 The poor recognition (Ki = 0.8 mM, unpublished) of the corresponding hydroxamate 9, the best reported inhibitor of the mechanistically related enzyme phosphoglucose isomerase (Ki ∼ 0.2 μM),45 illustrates the specificity of GlmS. Replacement of the phosphate group in 8, 9 and related compounds by the stable and isosteric methylenephosphonate and difluoromethylene phosphonate function might generate interesting tools to study the enzyme in action.
5.2 Inactivators
Four naturally occurring inhibitors were observed to exhibit antibacterial and antifungal activity: Bacilysin,46 A19009,47 its epoxide derivative48 and chlorotetain.49,50 All of these compounds are produced and transported inside the microorganism as dipeptides. After proteolytic cleavage they release L-alanine and the corresponding GlmS inhibitor, i.e. anticapsin 10, 12a or its epoxide (not shown) and 11. Both 10 and 12a were found to be potent time-dependent irreversible inhibitors of the bacterial enzyme with Kirr of 2.3 and 33 μM and first-order inactivation rate constants of 1.7 and 0.15 min−1, respectively.51 Good inhibition properties were also found with the fungal enzyme.52The mechanism of inactivation with compound 12b, a synthetic analogue of 12a exhibiting better inhibitory properties (Kirr=5.5 μM, kinact=1.04 min−1), proceeds via a Michael addition of the SH group of the N-terminal cysteine residue on C-3 of the fumaroyl moiety of the inhibitor, resulting in covalent modification of the enzyme. This conclusion was drawn from studies with a model peptide based on the N-terminal sequence of the protein, which showed that, after inactivation, a cyclization reaction occurs with the free α-amino group of the N-terminal cysteine at slightly alkaline pH, resulting in a blocked N-terminus.53 A similar mechanism occurs with the N3-haloacetyl derivatives of L-diaminopropionate54 and is likely to apply for GlmS inhibition by anticapsine.
Peptide conjugates of compound 12b such as Nva-12b or Lys-Nva-12b exhibit good to excellent antifungal properties with minimal inhibitory concentrations ranging from 0.01 to 0.5 μg ml−1.55,56 Despite the lack of cytotoxicity of these derivatives, the spontaneous reactivity of the fumaroyl moiety towards nucleophiles (which might explain the absence of antibacterial properties) precludes further developments as therapeutic agents. The need to find more specific inhibitors prompted us to design L-glutamine derivatives bearing a latent electrophilic function. As a first result of this approach the first mechanism-based inhibitor, L-γ-glutamyl-2-{[(p-difluoromethyl)phenyl]thio} glycine 13,57 was described. Enzyme-catalyzed hydrolysis of its peptide bond (Scheme 7) led to the formation of L-glutamate and N-deacylated α-arylthioglycine, which is known to be highly unstable.58,59 Its decomposition generates ammonia and glyoxylate on the one hand as well as HF and 4-thioquinone fluoromethide on the other hand. This latter powerful electrophile reacts with any active-site nucleophilic residue, resulting in enzyme inactivation. Reversible enzyme/inhibitor complex formation occurred much faster than the inactivation step itself (Kinact = 0.054 min−1, kirr = 36 mM), which occurred with a partition ratio of 7.57
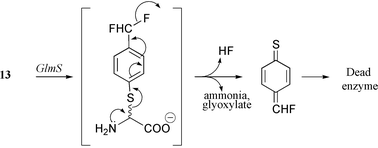 | ||
| Scheme 7 Mechanism-based inhibition of GlmS by 13. | ||
In an effort to investigate the fructose-6P binding site, 1,2-anhydro-hexitol-6P 15, previously known as affinity label of phosphoglucoisomerase, was synthesized.25 The mixture of the four diastereoisomers (C-2 + C-5) irreversibly inactivated the bacterial GlmS with a second-order rate constant for inactivation kinact/Kirr of 430 M−1 min−1, 2.4-fold lower than the value reported for inactivation by N-iodoacetyl-D-glucosamine-6P.40 Enzyme inactivation with tritiated 15 followed by tryptic digestion and purification of the radioactive fragments allowed identification of three peptides. Two of them, accounting for 65% of the recovered radioactivity, resulted from the nucleophilic attack of side-chain thiols of Cys-300 of the Fru-6P binding site (see section 4.3) and of Cys-1 of the Gln binding site on the epoxide functionality of the inhibitor. These results provide evidence for the proximity of L-glutamine and fructose-6P binding sites which was corroborated by the structure of the native enzyme.
6 Gfat: possible implication in type-2 diabetes and its complications
Glucose metabolism through the hexosamine biosynthesis pathway (HBP), which accounts for 2–3% of cellular glucose flux, has been hypothesized to mediate many adverse effects of hyperglycaemia and to be involved in the pathogenesis of type-2 diabetes and its complications, and glucose toxicity.60–68 When exposed to D-glucosamine, adipocytes,60 fibroblasts62,69 and even normal rodents63,64,70 develop insulin resistance. Moreover, inhibition of the HBP targeted on the mammalian glucosamine-6P synthase, Gfat, prevents glucose-induced insulin resistance in vitro.60 An amazing work on transgenic mice overexpressing Gfat in the liver71 or in skeletal muscle and fat,66 revealed development of hyperlipidemia, obesity and insulin resistance among these animals. The implication of this pathway on insulin-sensitive tissues would involve a decreased translocation of Glut4 glucose transporter70 which is reversed by the antidiabetic drug troglitazone at a point distal to the generation of hexosamines,72 thus acting as a “satiety sensor” for insulin-dependent tissues. The implication of the HBP concerning insulin secretion was also postulated, in 1998, following D-glucosamine infusion in rats.73 The severe impairment observed in glucose-induced insulin secretion, similar to that observed in patients with non-insulin dependent diabetes mellitus (NIDDM), suggested that D-glucosamine may participate in the pathogenesis of glucose toxicity at the level of the β-cell. Three years later, transgenic mice overexpressing Gfat specifically in the β-cells were shown to exhibit hyperinsulinemia and peripheral insulin resistance. The accumulation of UDP-GlcNAc would lead to the O-linked glycosylation of proteins, a phenomenon responsible for β-cells failure.74,75Apart from the implication of the HBP in insulin resistance and hyperinsulinemia, it has recently been proposed that Gfat blocks hyperglycemia-induced increases in the transcription of TGFα and TGFβ1,76 pathways shown to be implicated in diabetic complications and obesity.67,68 The hexosamine flux in fat was also shown to regulate the satiety hormone leptin, through its synthesis and secretion.77
In humans, Gfat is expressed in most major target tissues of insulin resistance (adipose tissues and skeletal muscle tissues) and also tissues involved in the development of diabetic late complications (vascular muscle cells, nerve sheath cells, mesangial cells).78 Indeed, patients with NIDDM have a skeletal muscle Gfat activity increased and sensitised to feedback inhibition by UDP-GlcNAc. In this elegant work the authors also correlated glycated haemoglobin with basal Gfat activity in these patients and control subjects.79
A second type of Gfat mostly expressed in the central nervous system was reported in 1999 by Oki et al.80,81 Since the discovery in 199160 of the possible implication of the HBP in the adverse effects of high glucose, an extraordinary regain of interest in this enzyme appeared. The scientific community has made a considerable effort to understand the possible implication of Gfat in type-2 diabetes and its complications. Moreover, it has become clear that the pharmaceutical industry is also getting involved in the utilization of this enzyme for the screening of new anti-diabetic drugs on human brain Gfat,82 on cells expressing an HBP enzyme83 on human, mouse and rat Gfat284 and on transgenic models targeted on β-cell.85
In the light of these possible therapeutic applications, studies on the catalytic mechanism of Gfat appear to be of importance. Taking into account the conservation of the crucial catalytic residues among the known glucosamine-6P synthase sequences, it is very likely that the mechanism of action of the mammalian enzyme is similar to that of the E. coli enzyme. The mammalian enzyme has two unique properties: it is subjected to feedback inhibition by UDP-GlcNAc86–90 and is regulated by phosphorylation.91,92 These features, together with the structural differences associated with the presence of a ∼50 amino acid hinge region connecting the two domains are important trails for exploring specific targeting of Gfat.
7 Summary
The incorporation of a nitrogen atom from L-glutamine amide at the C-2 position of fructose-6P is a reaction of tremendous importance since it provides D-glucosamine-6P to all living systems. Besides its crucial biological role, the enzyme exhibits remarkable features in that it performs two reactions: L-glutamine amide bond cleavage using the basic machinery of a single amino acid without intervention of a catalytic triad and utilization of the “nascent” ammonia at a site remote from its generation.In this review we have focused on the different aspects of our approach which have led to the actual understanding of the enzyme catalysis. In the light of the elegant studies performed on other L-glutamine-dependent enzymes we can now refine the different schemes proposed in this review by mutagenesis and by a chemical approach. The impact of glucosamine-6P synthase in fungal metabolism and possibly in type-2 diabetes related diseases emphasizes the need for new, potent and specific inhibitors. In this context transition state and protein–protein interaction inhibitors are the concepts we are actively trying to illustrate. Future research on glucosamine-6P synthase promises to be fruitful and exciting!
Acknowledgements
Our work on glucosamine-6P synthase was supported by a grant from CNRS (PCV 1998). We thank all the dedicated co-workers whose names appear in the references and Dr P. Durand for helpful comments concerning the manuscript.References
- In eucaryotes, GlcN-6P acetylation precedes phosphate migration from C-6 to C-1.
- C. A. Lobry de Bruyn and A. P. N. Franchimont, Recl. Trav. Chim. Pays-Bas, 1893, 12, 286 Search PubMed.
- C. A. Lobry de Bruyn, Chem. Ber., 1895, 28, 3082 Search PubMed.
- K. Heyns and W. Koch, Z. Naturforsch., 1952, 7, 486 Search PubMed.
- K. Heyns, H. Pausen, R. Eichstedt and M. Rolle, Chem. Ber., 1957, 90, 2039 Search PubMed.
- J. E. Hodge, Adv. Carbohydr. Chem., 1955, 10, 169 Search PubMed.
- E. coli and S. typhimurium: Cellular and Molecular Biology, ed. F. C. Neidhardt, American Society for Microbiology, Washington, DC, 1987 Search PubMed.
- Glutamine and Glutamate in Mammals, ed. E. Kvamme, CRC Press, Boca Raton, FL, 1988 Search PubMed.
- J. M. Buchanan, Adv. Enzymol. Relat. Areas Mol. Biol., 1973, 39, 91 Search PubMed.
- H. Zalkin, Adv. Enzymol. Relat. Areas Mol. Biol., 1993, 66, 203 Search PubMed.
- H. Zalkin and J. Smith, Adv. Enzymol. Relat. Areas Mol. Biol., 1998, 72, 87 Search PubMed.
- F. Massiere and M-A. Badet-Denisot, Cell. Mol. Life Sci., 1998, 54, 205 CrossRef CAS.
- G. Oliva, M. R. M. Fontes, R. C. Garratt, M. M. Altamirano, M. L. Calgagno and E. Horjales, Structure, 1995, 3, 1323 CrossRef CAS.
- B. Badet, P. Vermoote, P-Y. Haumont, F. Lederer and F. Le Goffic, Biochemistry, 1987, 26, 1940 CrossRef CAS.
- M-A. Denisot, F. Le Goffic and B. Badet, Arch. Biochem. Biophys, 1991, 288, 225 CrossRef CAS.
- G. Obmolova, M-A. Badet-Denisot, B. Badet and A. Teplyakov, J. Mol. Biol., 1994, 242, 703 CrossRef CAS.
- C. Leriche, M-A. Badet-Denisot and B. Badet, J. Am. Chem. Soc., 1996, 118, 1797 CrossRef CAS.
- M. N. Isupov, G. Obmolova, S. Butterworth, M-A. Badet-Denisot, B. Badet, I. Polikarpov, J. A. Littlechild and A. Teplyakov, Structure, 1996, 4, 801 CrossRef CAS.
- A. Teplyakov, G. Obmolova, M-A. Badet-Denisot, B. Badet and I. Polikarpov, Structure, 1998, 6, 1047 CrossRef CAS.
- A. Teplyakov, G. Obmolova, B. Badet and M.-A. Badet-Denisot, J. Mol. Biol., 2001, 313, 1095 CrossRef.
- J. A. Brannigan, G. Dodson, H. J. Duggleby, P. C. E. Moody, J. L. Smith, D. R. Tomchick and A. G. Murzin, Nature, 1995, 378, 416 CrossRef CAS.
- M-A. Badet-Denisot and B. Badet, Bull. Soc. Chim. Fr., 1993, 130, 249 Search PubMed.
- M-A. Badet-Denisot, L. A. Fernandez-Herrero, J. Berenguer, T. Ooi and B. Badet, Arch. Biochem. Biophys., 1997, 337, 129 CrossRef CAS.
- J. Stevens, unpublished data. We thank Professor Kudlow (University of Alabama, USA) for the generous gift of GST-GlmS plasmid.
- C. Leriche, M-A. Badet-Denisot and B. Badet, Eur. J. Biochem., 1997, 245, 418 CrossRef CAS.
- G. Lowe and A. Williams, Biochem. J., 1965, 96, 189 CAS.
- M. A. Badet-Denisot, C. Leriche, F. Massière and B. Badet, Bioorg. Med. Chem. Lett., 1995, 5, 815 CrossRef CAS.
- S. L. Bearne and R. Wolfenden, Biochemistry, 1995, 34, 11515 CrossRef CAS.
- M-A. Badet-Denisot and B. Badet, Arch. Biochem. Biophys., 1992, 292, 475 CrossRef CAS.
- B. Badet, P. Vermoote and F. Le Goffic, Biochemistry, 1988, 27, 2282 CrossRef CAS.
- B. Golinelli-Pimpaneau and B. Badet, Eur. J. Biochem., 1991, 201, 175 CAS.
- J. Pierce, A. S. Serianni and R. Barker, J. Am. Chem. Soc., 1985, 107, 2448 CrossRef CAS.
- C. Wilcox and J. Gaudino, J. Am. Chem. Soc., 1986, 108, 3102 CrossRef CAS.
- Unpublished data; Kislope = 2.8 mM and Ki intercept = 0.54 mM. We thank Professor Wilcox (University of Pittsburg, USA) for providing us with a sample of 5.
- F. Malaisse-Lagae, V. Liemans, B. Yayali, B. Sener and W. J. Malaisse, Biochim. Biophys. Acta, 1989, 998, 118 CAS.
- A. Lavie, K. N. Allen, G. A. Petsko and D. Ringe, Biochemistry, 1994, 33, 5469 CrossRef CAS.
- S. Banerjee, F. Anderson and G. K. Farber, Protein Eng., 1995, 8, 1189 CAS.
- B. Golinelli-Pimpaneau, F. Le Goffic and B. Badet, J. Am. Chem. Soc., 1989, 111, 3029 CrossRef CAS.
- A. Teplyakov, G. Obmolova, M-A. Badet-Denisot and B. Badet, Protein Sci., 1999, 8, 596 CAS.
- S. L. Bearne, J. Biol. Chem., 1996, 271, 3052 CAS.
- It is therefore no surprise that the activity of C-terminal (His)6-tagged GlmS exhibit only 0.5% the activity of the wild-type enzyme (J. Boetzel, unpublished work).
- S. Rhee, K. D. Parris, S. A. Ahmed, E. W. Miles and D. R. Davies, Biochemistry, 1996, 35, 4211 CrossRef CAS.
- M. A. Badet-Denisot, C. Leriche and B. Badet, in Chitin Enzymology, ed. R. Muzzarelli, European Chitin Society, Ancona, 1993, p. 161 Search PubMed.
- C. Le Camus, M-A. Badet-Denisot and B. Badet, Tetrahedron Lett., 1998, 39, 2571 CrossRef CAS.
- R. Hardré, C. Bonnette, L. Salmon and A. Gaudemer, Bioorg. Med. Chem. Lett., 1998, 8, 3435 CrossRef CAS.
- J. E. Walker and E. P. Abraham, Biochem. J., 1970, 118, 563 CAS.
- B. B. Molloy, D. H. Lively, M. Gorman, L. D. Boeck, C. E. Higgins, R. E. Kastener, L. L. Huckstep and N. Neuss, J. Antibiot., 1972, 25, 137 Search PubMed.
- R. Copper, A. C. Horan, F. Gentile, V. Gullo, D. Loebenberg, J. Marquez, M. Patel, M. S. Puar and I. Truumees, J. Antibiot., 1988, 41, 13 Search PubMed.
- C. Rapp, G. Jung, W. Katzer and W. Loeffler, Angew. Chem., Int. Ed. Engl., 1988, 27, 1733 CrossRef.
- H. Wild, J. Org. Chem., 1994, 59, 2748 CrossRef CAS.
- P. Vermoote, PhD Thesis, Université Paris 6, 1988.
- S. Milewski, H. Chmara, R. Andruszkiewicz and E. Borowski, Biochim. Biophys. Acta, 1985, 828, 247 CAS.
- N. Kucharckzyk, M-A. Denisot, F. Le Goffic and B. Badet, Biochemistry, 1990, 29, 3668 CrossRef.
- S. Auvin, O. Cochet, N. Kucharczyk, F. Le Goffic and B. Badet, Bioorg. Chem., 1991, 19, 143 CrossRef CAS.
- N. Kucharczyck, PhD Thesis, Université Paris 6, 1989.
- R. Andruszkiewicz, S. Milewski, T. Zieniawa and E. Boroswski, J. Med. Chem., 1990, 33, 132 CrossRef CAS.
- F. Massière, M-A. Badet-Denisot, L. René and B. Badet, J. Am. Chem. Soc., 1997, 119, 5748 CrossRef CAS.
- W. D. Kingsbury, J. C. Boehm, R. J. Mehta, S. F. Grappel and C. Gilvarg, J. Med. Chem., 1984, 27, 1447 CrossRef CAS.
- W. D. Kingsbury, J. C. Boehm, D. Perry and C. Gilvarg, Proc. Natl. Acad. Sci, U.S.A., 1984, 81, 4573 CAS.
- S. Marshall, V. Bacote and R. R. Traxinger, J. Biol. Chem., 1991, 266, 4076.
- E. D. Crook, M. C. Daniels, T. M. Smith and D. A. McClain, Diabetes, 1993, 12, 1289 Search PubMed.
- E. D. Crook, J. Zhou, M. C. Daniels, J. L. Neidigh and D. A. McClain, Diabetes, 1995, 44, 314 Search PubMed.
- A. Giaccari, L. Morviducci, D. Zoretta, P. Sbraccia, F. Leonetti, S. Caiola, A. Buongiorno and R. C. Bonadonna, Diabetologia, 1995, 38, 518 CrossRef CAS.
- L. Rossetti, M. Hawkins, W. Chen, J. Gindi and N. Barzilai, J. Clin. Invest., 1995, 96, 132 Search PubMed.
- D. A. McClain and E. D. Crook, Diabetes, 1996, 45, 1003 Search PubMed.
- L. F. Hebert, M. C. Daniels, J. Zhou, E. D. Crook, R. L. Turner, S. T. Simmons, J. L. Neidigh, J. S. Zhu, A. D. Baron and D. A. McClain, J. Clin. Invest., 1996, 98, 930 Search PubMed.
- P. P. Sayeski and J. E. Kudlow, J. Biol. Chem., 1996, 271, 15237 CrossRef CAS.
- V. Kolm-Litty, U. Sauer, A. Nerlich, R. Lehmann and E. Schleicher, J. Clin. Invest., 1998, 101, 160 Search PubMed.
- E. D. Crook and D. A. McClain, Diabetes, 1996, 45, 322 Search PubMed.
- A. D. Baron, J. S. Zhou, M. L. Weldon and W. T. Garvey, J. Clin. Invest., 1995, 96, 132 Search PubMed.
- G. Veerababu, J. Tang, R. T. Hoffman, M. C. Daniels, L. F. Hebert Jr., E. D. Crook, R. C. Cooksey and D. A. McClain, Diabetes, 2000, 49, 2070 Search PubMed.
- R. C. Cooksey, L. F. Hebert, J. S. Zhu, P. Wofford, W. T. Garvey and D. A. McClain, Endocrinology, 1999, 140, 1151 Search PubMed.
- R. R. Shankar, J. S. Zhu and A. D. Baron, Metabolism, 1998, 47, 573 CrossRef CAS.
- D. A. McClain, Curr. Opin. Endocrinol. Diabetes, 2001, 8, 186 Search PubMed.
- K. Liu, A. J. Paterson, E. Chin and J. E. Kudlow, Proc. Natl. Acad. Sci. U.S.A., 2000, 97, 2820 CrossRef CAS.
- X. L. Du, D. Edelstein, L. Rossetti, I. G. Fantus, H. Goldberg, F. Ziyadeh, J. Wu and M. Brownlee, Proc. Natl. Acad. Sci. U.S.A., 2000, 97, 12222 CrossRef CAS.
- D. A. McClain, T. Alexander, R. C. Cooksey and R. V. Considine, Endocrinology, 2000, 141, 1999 Search PubMed.
- A. G. Nerlich, U. Sauer, V. Kolm-Kitty, E. Wagner, M. Koch and E. D. Schleicher, Diabetes, 1998, 47, 170 Search PubMed.
- H. Yki-Järvinen, M. C. Daniels, A. Virkamäki, S. Mäkimattila, R. A. DeFronzo and D. A. McClain, Diabetes, 1996, 45, 302 Search PubMed.
- T. Oki, K. Yamazaki, J. Kuromitsu, M. Okada and I. Tanaka, Genomics, 1999, 57, 227 CrossRef CAS.
- K. Yamazaki, Y. Mizui, T. Oki, M. Okada and I. Tanaka, Gene, 2000, 261, 329 CrossRef CAS.
- Takeda Chem. Ind. Ltd, Pat., EP824149, 1998 Search PubMed.
- University of Washington, Pat. WO99/63101, 1999 Search PubMed.
- Searle, Pat. WO00/37617, 2000 Search PubMed.
- UAB Research foundation, Pat WO00/10389, 2000 Search PubMed.
- R. Kornfeld, J. Biol. Chem., 1967, 3135 CAS.
- P. J. Winterburn and C. F. Phelps, Biochem. J., 1971, 121, 711 CAS.
- H. Kikuchi and S. Tsukui, Biochim. Biophys. Acta, 1976, 422, 241 CAS.
- G. L. McKnight, S. L. Mudri, S. L. Mathewes, R. R. Traxinger, S. Marshall, P. O. Sheppard and P. J. O'Hara, J. Biol. Chem., 1992, 267, 25208 CAS.
- Q. K. Huynh, E. A. Gulve and T. Dian, Arch. Biochem. Biophys., 2000, 379, 307 CrossRef CAS.
- J. Zhou, Q. K. Huynh, R. T. Hoffman, E. D. Crook, M. C. Daniels, E. A. Gulve and D. A. McClain, Diabetes, 1998, 47, 1836 Search PubMed.
- Q. Chang, K. Su, J. R. Baker, X. Yang, A. J. Paterson and J. E. Kudlow, J. Biol. Chem., 2000, 29, 21981 CrossRef.
| This journal is © The Royal Society of Chemistry 2002 |