The oxylipin chemistry of attraction and defense in brown algae and diatoms
Georg
Pohnert
* and
Wilhelm
Boland
Max Planck Institute for Chemical Ecology, Winzerlaer Str. 10, D-07745 Jena, Germany
First published on 15th November 2001
Abstract
This review covers the research on brown algal pheromones from the first structural characterisation of an active principle in 1971 to the recent detailed insight into their biosynthesis. Development of analytical methods and bioassays that lead to the identification of a structural variety of different fatty acid-derived pheromones are reported. Special emphasis is focused on the inactivation of initially released pheromones through pericyclic reactions. The impact of pheromone-research on the defensive chemistry of brown algae and diatoms is discussed. 121 references are cited.
 Georg Pohnert Georg Pohnert | Georg Pohnert was born in Gelnhausen, Germany, in 1968. He attended the University of Karlsruhe where he received his Diploma in Chemistry in 1994. He moved to the University of Bonn where he started his work on algal pheromones under the guidance of Wilhelm Boland. After a short period at the laboratory of Heinz G. Floss in Seattle he received his PhD in 1997. He then joined the groups of Bruce Ganem and David B. Wilson at the Cornell University, where he spent one year with biophysical investigations of phenylalanine binding to the E. coli P-protein. He returned to Germany in 1998 where he currently heads a group working on algal defensive mechanisms at Max Planck Institute for Chemical Ecology in Jena. |
 Wilhelm Boland Wilhelm Boland | Wilhelm Boland, born 1950 in Rheinberg, Germany, studied Chemistry at the Universities of Münster and Cologne. He completed his PhD work in 1978 under the guidance of Professor Dr L. Jaenicke in the Institute of Biochemistry at the University of Cologne. Subsequently he carried out his habilitation, dealing with “Chemical communication between sexual cells of brown algae” at the same institute. In 1987 he was appointed to Associate Professor for Organic Chemistry at the University of Karlsruhe and in 1994 to Full Professor of Bioorganic Chemistry at the University of Bonn. In 1996 he accepted his present position as a director at the Max Planck Institute for Chemical Ecology in Jena. His research is focused on the induction of the plant's defense pathways, secondary metabolism, insect defenses and chemical communication. |
1 Introduction
Communication through chemical signals is an old phenomenon on an evolutionary time scale. Even bacteria show the ability to recognise chemical signals leading to food sources and optimum environmental conditions. For compounds that coordinate activities between individuals within one species, Karlson and Lüscher in 1959 introduced the term pheromone.1 Despite this broad definition, pheromones are most often associated with the animal kingdom, and, indeed, some of the most spectacular examples of specificity and sensitivity of chemical signals have been discovered among insects. In the case of sessile organisms such as plants, fungi, and algae, the need for such signals might not be directly obvious. However, certain phenomena and observations clearly demonstrate that even for these organisms chemical communication also plays an important role. The pheromone-based attraction between gametes of different sex, for example, is rather common and has long been known in fungi2 and algae.3–6 The first description of a “chemotaxis” in algae dates back to 1854, when the French scientist Thuret described how male gametes of the brown alga Fucus vesiculosus find their conspecific eggs.7“Il est difficile, quand on observe ces phénomènes avec attention, de ne pas se laisser aller à croire qu'une impulsion particulière dirige les anthérozoides vers les corps qu'ils doivent féconder.” [During observation of these phenomena it is difficult not to believe in a particular matter that leads the antherozoides to the bodies they have to fertilise.]
Today we know that this pheromone-mediated mating process is common for most brown algae during sexual reproduction. These algae, or kelps, are widespread in the coastal regions, and their vegetative stages are dominant in certain regions. Fig. 1 displays the typical aspects of the life cycle of brown algae. During sexual reproduction the vegetative sporophytes form gametophytes, which then produce haploid gametes. Some of the approximately 900 species of brown algae regularly undergo a generation cycle involving the fusion of gametes, whereas only under stress do others enhance their ability to adapt through sexual reproduction.
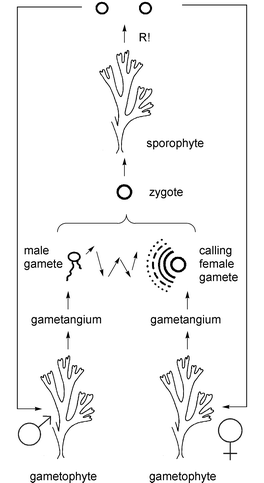 | ||
| Fig. 1 Schematic representation of the life cycle of a dioecious brown alga. After release from the female gametangia, the haploid gamete settles and releases pheromones that guide the male gametes towards the pheromone source. Fusion of a male and female gamete results in the formation of a diploid zygote which develops to the vegetative sporophyte. The cycle is finished after formation of meiospores and their development to the gametophytes. This general scheme can vary strongly from species to species. Adapted after E. Strassburger, Lehrbuch der Botanik, Gustav Fischer Verlag, Stuttgart, Germany. | ||
In oogamous algae male gametes and eggs differ significantly in size and shape. The immobile female eggs attract a very large number of much smaller male spermatozoids to form a diploid zygote that develops into a macroscopic vegetative sporophyte. Depending on the species, the pheromones may have two clearly discernible functions. They may guide – as Thuret observed – the flagellated male gametes along a gradient towards the calling female. The permanently changing local concentration of signal molecules along the trail results in specific movement patterns of the flagella of male gametes, forcing them into directed movements towards the pheromone source; for example, by performing characteristic U-turns when heading away from a pheromone source.5,6,8,9 In the higher evolved orders such as Laminariales, Desmarestiales, and Sporochnales, the female sexual pheromones initially act as “release factors”, inducing spermatozoid release from fertile male antheridia. These flagellated male spermatozoids are then guided by the same compounds that also act as attractants to the “calling” female.10,11 This release on demand synchronises the female and male gametes, and has the additional advantage of providing an original and continuous pheromone gradient to the female. The mass release is an impressive phenomenon (Fig. 2) and represents one of the fastest signal/response actions known in the plant kingdom.12 For example, the release of male gametes from the macroalga Laminaria digitata occurs within only 8–12 s after application of the signal.
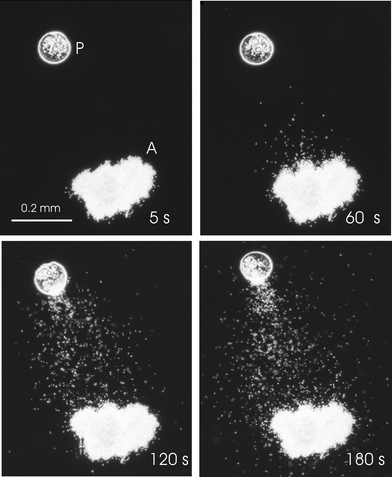 | ||
| Fig. 2 Mass release of male gametes from fertile antheridia (A) of Laminaria digitata. 60 s after addition of a lamoxirene (4) loaded florisil bead as a pheromone source (P) the release of gametes becomes obvious. During the 180 s observation period attraction of gametes leads to an accumulation of gametes in the vicinity of the pheromone source. Figure kindly provided by Dr Ingo Maier, Amtzell, Germany. | ||
Since Thuret's initial observation, it took ca. 100 years until Cook et al. proved the existence of a female-released diffusible attractant for male gametes.13 Another 20 years were required to unravel the structure of the first brown algal pheromone by Müller and Jaenicke.14 In this study, gynogametes (female eggs) of the filamentous brown alga Ectocarpus siliculosus from more than 1000 culture dishes were continuously extracted by a stream of air passing a cold trap for condensation of the active principle. At the end, 7 mg of the active principle were obtained by bioassay guided chromatographic fractionation. The compound was identified by NMR and mass spectroscopy as 6-(1Z-butenyl)-cyclohepta-1,4-diene 1 and was called “ectocarpene”, due to its origin from female gametes of E. siliculosus.14
Today we know that Müller and colleagues isolated a moderately active thermal rearrangement product of the genuine thermolabile pheromone with ca. 10000-fold higher activity (see section 4.4). Currently 12 pheromones (Scheme 1) and many structural isomers are known.
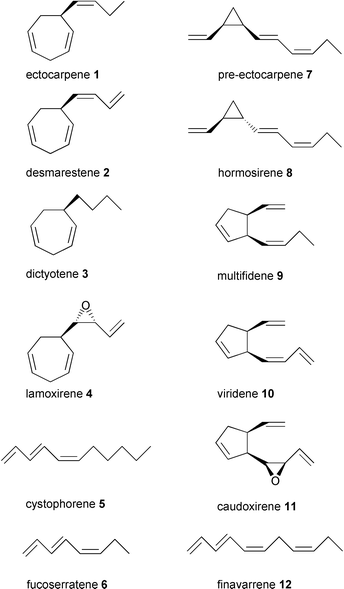 | ||
| Scheme 1 Identified brown algal pheromones (references given in Table 1). | ||
Crucial for their identification was the development of highly effective extraction techniques in conjunction with sensitive modern spectroscopic techniques.
1.1 Laboratory techniques
![Droplet bioassay according to Maier.5 For the determination of accumulation factors the number of gametes above pheromone loaded (nb, nd) and blank solvent droplets (na, nc) is determined and the accumulation factor Q is calculated after: Q
=
[(nb/na) + (nb/nc) + (nd/na) + (nd/nc)]/4.5 Figure kindly provided by Dr Ingo Maier, Amtzell, Germany.](/image/article/2002/NP/a806888g/a806888g-f3.gif) | ||
| Fig. 3 Droplet bioassay according to Maier.5 For the determination of accumulation factors the number of gametes above pheromone loaded (nb, nd) and blank solvent droplets (na, nc) is determined and the accumulation factor Q is calculated after: Q = [(nb/na) + (nb/nc) + (nd/na) + (nd/nc)]/4.5 Figure kindly provided by Dr Ingo Maier, Amtzell, Germany. | ||
In a typical set-up four micro-droplets (0.1 μl), two with a defined quantity of pheromone (b,d; Fig. 3) and two solvent blanks (a,c; Fig. 3), are placed on the bottom of a polystyrene Petri dish. Then the droplets are covered by a dense suspension of male gametes. After 3 to 4 min in the dark (gametes are phototactic),19,20 their distribution is documented by a flashlight microphotograph. The number of cells above a standard area of droplets with or without pheromones is determined. The chemotactic potential of a compound is expressed by the accumulation factor Q, representing the arithmetic mean of the quotients of the number of gametes above pheromone loaded and blank droplets. The threshold concentration is defined as the lowest attractive pheromone concentration in seawater (calculated with the distribution coefficient of the pheromone between water and the fluorocarbon).21,22 Threshold concentrations for chemo-accumulation in brown algae are usually in the concentration range of 1–1000 pM and impressively demonstrate the sensitivity and specificity of brown algal chemoreceptors.
The spermatozoid release is quantified by covering fertile male gametophytes with culture medium containing known amounts of the pheromones or test compounds. The threshold concentration is defined as the concentration at which microscopic observation shows a statistical significant spermatozoid release. Thresholds for gamete release usually cover the same range as observed for attraction.12 The ease and reliability of the bioassays are demonstrated by the fact that they have even been adapted in laboratory training classes on an undergraduate level, giving reproducible and pedagogically valuable results.23
2 Pheromone structures
2.1 Pheromones
Since the isolation of ectocarpene 1, eleven other pheromones along with many structural isomers or structurally related compounds have been obtained from other species. 1 proved to be a widespread constituent of algal pheromone blends, albeit often as a less active or even inactive byproduct (Scheme 1).Table 1 compiles the currently known brown algal pheromones, which have been isolated and identified during the last three decades. Except for 1 and 7, the structure elucidation of most other pheromones and byproducts relied entirely on GLC/MS methods and use of synthetic references. These were designed after the results of hydrogenation experiments provided information on the principal alicyclic structures along with biosynthetic considerations. Today a large body of literature on (stereo)-selective syntheses exists, which will not be reviewed here.
| Pheromone | Species |
|---|---|
a
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Both structures show activity, re-examination with pre-ectocarpene 7 required. Both structures show activity, re-examination with pre-ectocarpene 7 required.
|
|
| Ectocarpene 1 | Scytosiphon sp.24 |
| Ectocarpus fasciculatus 25 | |
| E. siliculosus (revised to 7)14 | |
| Ectocarpene 1/Hormosirene 8a | Analipus japonicus (1S,2S)-8 (>90% ee);26 (1R,2R)-8 (64% ee)27 |
| Adenocystis utricularis 28 | |
| Sphacelaria rigidula 28 | |
| Desmarestene 2 | Desmarestia acculeata (6R)-3 (87% ee)29,30 |
| D. viridis 29,30 | |
| Cladostephus spongiosus (6R)-3 (96% ee)31,29 | |
| D. firma (6R)-3 (28% ee)29 | |
| Dictyotene 3 | Dictyota dichotoma 32 |
| D. diemensis (6R-3 and 6S-3 have identical threshold concentration as pheromone)33 | |
| D. prolifera 34 | |
| Lamoxirene 4 | Laminaria angustata 35 |
| L. japonica 35 | |
| L. saccharina 25 (6R)-(1′S,2′R) and (6S)-(1′S,2′R) most active in bioassays36 | |
| L. sinclarii 35 | |
| L. digitata (6R)-(1′S,2′R) (71%), (6S)-(1′S,2′R) (29%),17 (6R)-(1′S,2′R) most active in bioassays36 | |
| L. hyperborea (6R)-(1′S,2′R) most active in bioassays36 | |
| Pleurophydus gardneri 35 | |
| Alaria crassifolia 35 | |
| A. esculenta 35 (6R)-(1′S,2′R) most active in bioassays36 | |
| A. marginata 35 | |
| Ecklonia radiata 35 | |
| Eisenia arborea 35 | |
| Pterygophora californica 35 | |
| Undaria pinnatifida 35 (6R)-(1′S,2′R) most active in bioassays36 | |
| Dictyoneuropsis reticulata 35 | |
| Lessonia variegata 35 | |
| Lessoniopsis littoralis 35 | |
| Macrosystit integrifolia 35 | |
| Macrocystis pyrifera 35 (6R)-(1′S,2′R) most active in bioassays36 | |
| Nereocystis luetkeana 35 | |
| Pelagophycus porra 35 | |
| Agarum cribrosum 35 | |
| Cymmathere triplicata 35 | |
| Hedophyllum sessile 35 | |
| Kjellmaniella gyrata 35 | |
| Cystophorene 5 | Cystophora siliquosa 37 |
| Fucoserratene 6 | Fucus serratus 38 |
| F. spiralis 39 | |
| F. vesiculosus 38 | |
| Pre-ectocarpene 7 | Ectocarpus siliculosus 40 |
| Hormosirene 8 | Hormosira banksii 37,41,42 |
| Xiphophora chondrophylla ((−)-enantiomer more active)37,42 | |
| X. gladiata 37,42 | |
| Durvillaea antarctica 37 | |
| D. potatorum 37,42 | |
| D. willana 37 | |
| Colpomenia peregrina 37 | |
| C. bullosa (1R,2R)-8 (93% ee)24,27 | |
| Ascoseira mirabilis (1R,2R)-8 (>85% ee)43 | |
| Myelophycus simplex 44 | |
| Scytosiphon lomentaria (1R,2R)-8 (95% ee)37,24 | |
| Multifidene 9 | Cutleria multifida (3S,4S)-9 (>99% ee)45,46,47 |
| Zonaria angustata (3S,4S)-9 (>98% ee)48 | |
| Chorda tomentosa (3S,4S)-9 (>99% ee)49,46 | |
| Viridiene 10 | Syringoderma phinneyi (3R,4S)-10 (>98.5% ee)50 |
| Syringoderma sp.51 | |
| Caudoxirene 11 | Perithalia caudata (2R,3R,1′S,5′S)-11 (>98% ee)52,53 |
| Finavarrene 12 | Dictyosiphon foeniculaceus 54 |
| Ascophyllum nodosum 55 | |
Given the large number of brown algal species that has been investigated, the rather small number and the conservative structure of the pheromones is surprising. The active compounds principally possess either eight or eleven carbon atoms. They can be classified into monosubstituted cycloheptadienes (1–4), linear unsaturated hydrocarbons (5, 6, 12), divinylcyclopropanes (7, 8), and 3,4-disubstituted cyclopentenes (9–11). Only two pheromones from the higher developed Laminariales (lamoxirene 4 and caudoxirene 11) bear an epoxide function that is important for bioactivity.21 The close structural similarities of the pheromones suggests a common biosynthetic origin from fatty acids with only a few different tailoring enzymes involved in the production of the active compounds.
2.2 Enantiomeric composition of C11 hydrocarbons
In analogy to the communication systems of insects the diversity of the algal signal molecules is enhanced by using different stereoisomers. All alicyclic pheromones possess at least one stereocentre. The cis-disubstituted cyclopentenes 9–11 generally are of high optical purity (> 95% ee)46,56 and only the enantiomers released by eggs or female gametes display the highest activity in bioassays with conspecific males. Unlike the cyclopentenes, the divinylcyclopropane- and cycloheptadiene-pheromones are most often released as enantiomeric mixtures. This becomes particularly obvious with the cyclopropyl pheromone hormosirene 8. Algal gametes or thalli of brown algae produce 8 with often low but species-characteristic enantiomeric excess (ee).42 The ee and the configuration varied depending on the species or the geographic region, and values ranging from almost pure (−)-(1R,2R) 8 in Dictyopteris undulata (92% ee) over moderately pure (−)-(1R,2R) 8 from Durvilleaea potatorum (51% ee) to the opposite enantiomer (+)-(1S,2S) 8 (90% ee) in Analipus japonicus have been reported.57,42 In A. japonicus the ee of 8 varied even locally between two collection sites in Japan.57 Such a habitat-linked release of enantiomeric mixtures may serve as a means to individualise pheromone blends of sympatric species,4 but this hypothesis still lacks experimental verification. The origin of this highly variable ee may be due to insufficient control of the fatty acid substrate in the active site of a hydroperoxide lyase (see section 4.3).582.3 Hydrocarbon byproducts
In general female gametes or eggs of brown algae secrete a blend of compounds rather than a single pheromone, but the strongest biological activity was found to be associated with single components albeit not always with the major product. For example, female gametes of Cutleria multifida release a complex blend of linear and cyclic C8, C9 and C11 hydrocarbons with different degrees of saturation besides the major biologically active cyclopentene 9 (Fig. 4).In a few cases the byproducts were shown to be able to modulate the gamete response,6 but for the majority of byproducts no biological function is known.
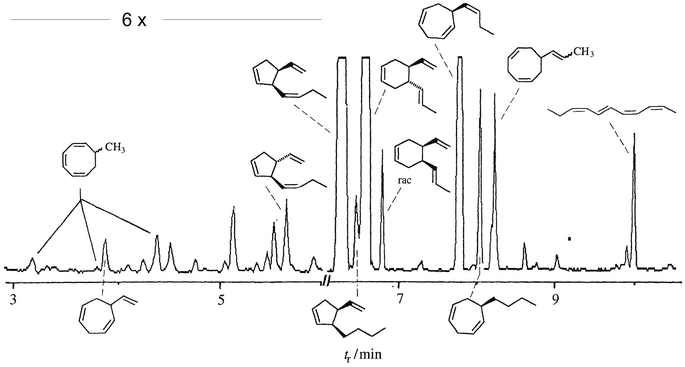 | ||
| Fig. 4 Gas chromatographic separation of the collected volatiles from fertile gynogametophytes of Cutleria multifida. | ||
In most species minor components are made up of stereoisomers of bioactive pheromones or comparable structures with varying degree of unsaturation. Some compounds display different carbon skeletons, such as, for example, the monosubstituted cyclopentenes 21–23, the disubstituted cyclohexene aucantene(s) 19 and 20, or the propenylcyclooctadiene 31. Only two C9 compounds methylcyclooctatriene 30 and vinylcycloheptadiene 16 have been detected so far (Scheme 2).59,60
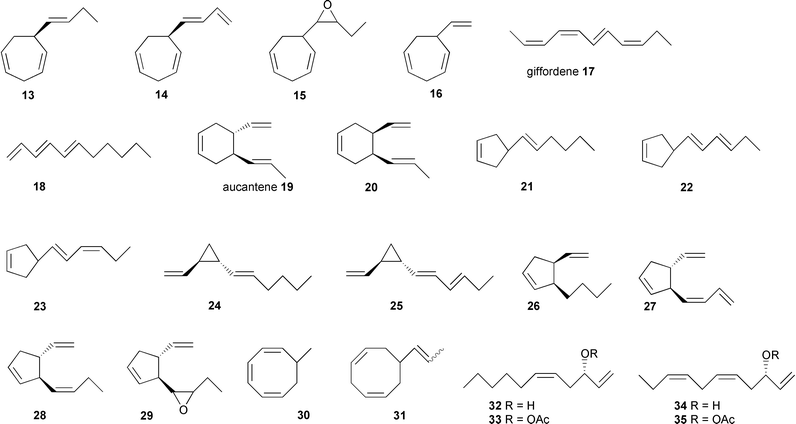 | ||
| Scheme 2 Pheromone related hydrocarbons identified from brown algal gametes and thalli: 13, 14,2615,6116,6017,6218,6019,4620,6021,6322, 23,6424–28,6426, 29,5230, 31,6032–35.65 | ||
The rather uniform pattern of the structures suggests a common biosynthetic origin for all compounds. Whether the complex bouquets have a biological function in the environment or simply reflect the lack of specificity of the pheromone producing enzymes remains to be established.
3 Pheromone perception
In the first place it is surprising that pheromones of organisms living in an aqueous environment are non-polar volatile hydrocarbons of very low water solubility. However, owing to the very high activity, only low amounts of the pheromones are released from female gametes, and, hence, solubility is not a limiting factor. For example, in one hour Hormosira banksii released only 75 fmol of hormosirene 8 per egg.15 The amount of hydrocarbons released from female gametes of E. siliculosus was even lower and did not exceed ca. 0.6 fmol per individual cell and hour.66 On the other hand, the non-polar nature of the pheromones contrasts strongly with the highly polar carrier that is water, which secures a high signal-to-noise ratio. Perception of the signals starts with simple partition of the hydrocarbon from water into the gamete′s outer membrane and is governed by the characteristic partition coefficient of the compound between water and the lipid membrane.22,67 Selectivity of recognition is linked to the subsequent interaction of the membrane-dissolved compounds with the macromolecular receptors, which show a high to very high preference for the genuine pheromones. Several studies on structure–activity relationships with different pheromones and synthetic pheromone analogues demonstrated that both geometry and electronic structure of the pheromones are essential for the precise recognition that warrants the high sensitivity and selectivity of communication.21,22,68 This preference for the genuine pheromones does not, however, prevent algae from attracting gametes of different and even competing species.22 This is due to the fact that female gametes usually release a blend of structurally related hydrocarbons;60 as a result, competing species might rely on the same pheromone or minor components of the blend. In contrast to insects that use an immense variety of volatile airborne signals, the development of very specific communication systems based on the synergistic action of individual components of a blend has apparently not occurred in brown algae. This might partly not be required because long-range interactions, which are typical for the insect world, are uncommon in brown algal communication. Because of rapid dispersion processes that interfere with the reliable detection of pheromone gradients by the microscopically small gametes, the effective range of a calling female is limited to about one millimeter.22 The same is true for the “gamete-release” that occurs locally as soon as the required threshold is reached. This ensures that male gametes are released only when the pheromone gradient is stable and significant enough to facilitate the finding of the sexual partners. The lack of species-specificity of the pheromonal communication is compensated for by immunological barriers on the cell surface.69,70 For example, no cross fertilisation occurs between the two sympatric species of the Northern Atlantic Fucus serratus71 and F. vesiculosus,38 although both use fucoserratene 6 as their pheromone. Accordingly, gametes of the two species are mutually attracted but cross fertilisation does not occur. As demonstrated with female gametes of E. siliculosus, this specificity of fertilisation is linked to complementary carbohydrate signatures on the surfaces of the male and female gametes.724 Pheromone biosynthesis
4.1 Fatty acids as sources for pheromone production
The investigation of the biosynthesis of algal pheromones has long been hampered by the limited access to large amounts of algal gametes. Culturing and fertilisation of macroalgae is often difficult, and induction of gamete release in amounts sufficient for biosynthetic studies is still not easily accomplished. Thus, it might not be surprising that initial biosynthetic studies have been performed on other model systems that produce the same hydrocarbons. The C11-hydrocarbons 1 and 12 have been detected as components in the odour of ripening mango and pineapple.73,74 Ectocarpene 1 is the major metabolite in leaves of the asteraceae, Senecio isatideus.75,76 Due to the ready availability of S. isatideus, initial biosynthetic studies on the origin of ectocarpene 1 have been performed using this flowering plant.The lack of methyl branches and the double bond pattern of the hydrocarbons 1–12 suggests unsaturated fatty acids as precursors of these metabolites. Experiments administering isotopically labelled fatty acids to S. isatideus verified their incorporation into the skeleton of ectocarpene (Scheme 3).76
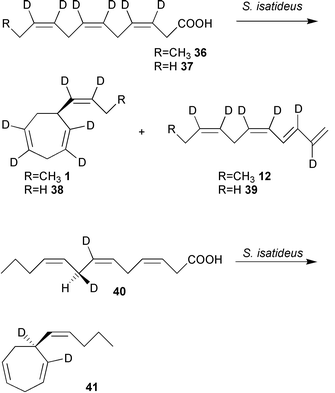 | ||
| Scheme 3 Biosynthesis of ectocarpene 1 in the asteracea S. isatideus occurs from dodecatrienoic acid. Re-face attack at C8 can be proven by administration of the chirally deuterated C13-fatty acid 40 resulting in homo-ectocarpene 41. | ||
When [3,4,6,7,9,10-2H6]dodeca-3,6,9-trienoic acid 36 was administered to freshly excised plantlets of S. isatideus, all six deuterium markers were detected in ectocarpene 1 and finavarrene 12 at their original positions. Remarkably, this uncommon fatty acid was well accepted as a substrate, while the more abundant early precursor, linolenic acid, was hardly incorporated into 1. To obtain insight into the stereochemical course of the hydrogen abstraction of C8 chiral [7,8-2H2]-(8R) and [7,8-2H2]-(8S)-tridecatrienoic acid 40 were administered to S. isatideus (Scheme 3). The higher boiling point of the resulting homo-ectocarpene 41 compared to natural 1 facilitated GC/MS analysis of the labelled product. While [7,8-2H2]-(8S) 40 was transformed into homo-ectocarpene 41 with retention of the two deuterium atoms, monolabelled 41 resulted from [7,8-2H2]-(8R) 40 (Scheme 3).77,78 Thus the decarboxylation (?) – cyclisation sequence to 41 proceeds via a re-faced attack of the enzyme onto the substrate acid 40.78
Surprisingly, all attempts to achieve a successful transformation of dodecatrienoic acid 36 to 1 by brown algal gametes of E. siliculosus failed, suggesting that an alternative pathway towards 1 may exist in brown algae. In contrast to higher plants, brown algae are rich in eicosanoids such as arachidonic and eicosapentaenoic acids, the latter often present as major fatty acid.79–82 Therefore labelled C20-precursors have been administered to the gametes which transformed them into ectocarpene 1 and related C11 hydrocarbons.83,84 [2H8]arachidonic acid 42, externally applied to female gametes of E. siliculosus or Sphacelaria rigidula was readily converted into deuterium labelled 6-butylcycloheptadiene (dictyotene) 3 and undeca-(1,3E,5Z)-triene 5 (Scheme 4).
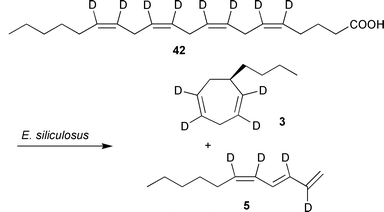 | ||
| Scheme 4 Biosynthesis of C11 hydrocarbons 1 in E. siliculosus. | ||
The ease and efficiency of incorporation is demonstrated by the relative amount of [2H4]-3 that exceeded the unlabelled compound by ca. 800%. Other labelled eicosanoic- and nor-eicosanioc fatty acids showed that the hydrocarbons were formed from the C10–C20 terminus of the precursors. Linolenic acid and other shorter chain fatty acids were not converted into 1.83,84 This demonstrates that the intact framework of the eicosanoic precursors is essential to the formation of C11-hydrocarbons. Only minor modifications of the fatty acids are tolerated by the pheromone-producing enzymes. Degradation via β-oxidation to shorter chain fatty acids as demonstrated for higher plants (Scheme 3) does not occur beforehand in algae.
4.2 Fatty acid activation and cleavage
Since limited access to female gametes of brown algae hampered a detailed biochemical study of the pathway, other model organisms had to be chosen. Following reports of the occurrence of C8- and C11-hydrocarbons such as ectocarpene 1 from diatom cultures and during algal blooms in freshwater lakes,85–88 diatom cultures were screened for their ability to produce brown algal pheromone-like structures. This led to the identification of the freshwater diatom Gomphonema parvulum (Bacillariophyceae) as a highly productive source of hormosirene 8, along with minor quantities of dictyopterene A 24 and finavarrene 12.89 When labelled [2H8]arachidonic acid 42 was administered to cell-free preparations of G. parvulum, high amounts of [2H4]dictyopterene A 24 were detected in the volatile blend (Scheme 5). Thus, it is clear that the biosynthesis of C11-hydrocarbons in both diatoms and brown algae relies on eicosanoid precursors. As in the brown alga E. siliculosus eicosapentaenoic acid 43 is the precursor of the predominant C11H16 hydrocarbons in G. parvulum but administration of external arachidonic acid 42 strongly increased the production of the more saturated C11H18 hydrocarbons. Due to the similarities found in these incubation experiments, it is likely that the related heterocontophytic brown algae and diatoms follow similar, if not identical, biosynthetic routes to the C11 hydrocarbons.89 The rapidly reproducing diatoms thus offer a unique chance to study the metabolism of eicosanoids in more detail.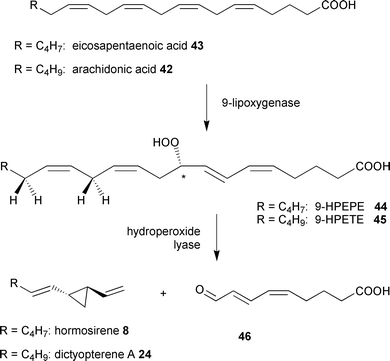 | ||
| Scheme 5 Conversion of eicosanoic acids by cell free solutions of G. parvulum. | ||
Kinetic experiments with cell-free extracts of G. parvulum showed a strong correlation between an increasing UV absorption at 277 nm and the production rate of the C11-hydrocarbons. Subsequent treatment of the medium with NaBH4 shifted the UV maximum to 235 nm.89 Following the hypothesis that a coupled lipoxygenase/hydroperoxide lyase mechanism is responsible for activation and cleavage of eicosanoic acids, the conjugated oxoacid 46 was suggested as the second fragment of the fatty acid precursor (Scheme 5).
Using a synthetic reference of the 9-oxononadienoic acid 46, this polar fragment was readily identified as a natural byproduct of hydrocarbons because of its identical HPLC-retention volume and UV-absorption. Administration of [2H8]arachidonic acid 42 led to the two fragments 24 and 46, each of which showed the presence of four deuterium atoms. This is consistent with a direct cleavage of the eicosanoid precursor (Scheme 5).89 If the transformation of [2H8]arachidonic acid is conducted in the presence of 18O2, the oxygen isotope was detected in the C(9)-aldehyde group. Nevertheless, the putative precursor 9-hydroperoxyeicosatetraenoic acid (HPETE) 45 could not be detected in cell-free solutions of G. parvulum, suggesting this hydroperoxide converts rapidly into downstream metabolites. Evidence for the involvement of 9-HPETE 45 was obtained by trapping the corresponding alcohol using a system of glutathion and glutathion peroxidase, which is known to reduce hydroperoxides fast and efficiently.90 The resulting alcohol 9-hydroxyeicosatetraenoic acid (9-HETE) accumulated since it was not converted into the downstream metabolites 24 and 46 and was fully characterized by RP-HPLC and LC-MS.
The biosynthesis of C8-hydrocarbons such as fucoserratene 6 could also be studied with freshwater diatoms as model organisms. As reported by Jüttner et al. the planktonic alga Asterionella formosa produces fucoserratene 6 together with some other volatiles.87 Following the general concept of lipid peroxidation and subsequent oxidative cleavage of the carbon skeleton, the biosynthesis of C8-hydrocarbons involves 12-hydroperoxyeicosapentaenoic acid 49 (12-HPEPE) as intermediate. Besides the hydrocarbon 6, 12-oxododeca-(5Z,8Z,10E)-trienoic acid 56 would result as the polar metabolite out of the cleavage of 49 (Scheme 6).91 The mechanism was proven using synthetic 12-oxo-acid 56 and LC/MS comparison with the natural product.92 The enzymes of A. formosa show a remarkable substrate tolerance. Synthetic eicosanoids with different degrees of unsaturation ranging from a butyl to a highly unsaturated butenynyl terminus have been incorporated into the nonnatural C8-hydrocarbons 53–55 (Scheme 6).91
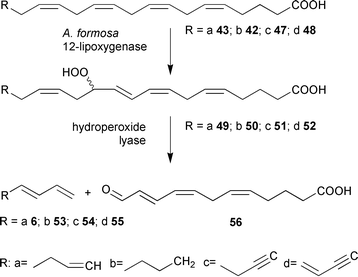 | ||
| Scheme 6 Conversion of eicosanoic acids by cell free solutions of A. formosa. | ||
A recent report on the formation of volatiles by Dictyopteris prolifera may suggest a third pathway for the production of dictyotene 3, dictyopterene A 24, hormosirene 8, and ectocarpene 1.93 Yamamoto et al. isolated larger amounts of dictyoprolenol 32 and neodictyoprolenol 34 in the essential oils of several Dictopteris species. These alcohols had been previously discussed as precursors for the C11-hydrocarbons 1 and 3 by Moore et al.65 When racemic mixtures of 32 and 34 were administered to homogenates of D. prolifera, a significant decrease of the S-enantiomeric alcohols was observed, and chiral hydrocarbons were present after the experiments (Scheme 7). Accordingly, another pathway could lead from the initial 9-HPETE 45 to the secondary alcohol dictyoprolenol 32 and the 9-oxo acid 46.
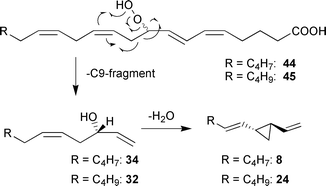 | ||
| Scheme 7 Suggested biosynthesis for C11-hydrocarbons in thalli of Dictyopteris prolifera. | ||
Application of such precursors to gametes of E. siliculosus failed to produce labelled C11-hydrocarbons,83,84 while eicosanoid precursors were readily incorporated. The suggested third pathway thus seems to differ from that in brown algal gametes, diatoms and higher plants.
4.3 Cyclisation mechanisms
Studies on the stereochemistry of fatty acid oxygenation and hydrogen abstraction offer insight into the cyclisation mechanism leading to divinyl cyclopropane hydrocarbons such as dictyopterene A 24. After reduction of the intermediate hydroperoxy fatty acid 9-HPETE 45 the configuration of the hydroxy acid 9-HETE reflected the configuration and the ee of the unstable precursor and was established as (9S)-HETE (with >71% ee; competing autoxidation reduces the ee).58G. parvulum produces hormosirene 8 as the major product (56% ee) and (1S,2R)-dictyopterene A 24 (24% ee); in both cases the ee of the hydrocarbons is significantly lower than that of the precursors 44 and 45.58,89 The rather high ee of 9-HPETE 45 demonstrates that the hydroperoxy group at C9 does not control the product-stereochemistry alone. In principle, the configuration and the ee of cyclopropanes 8 and 24 can be controlled either by the configuration of the hydroperoxide or by selective abstraction of one of the two enantiotopic hydrogen atoms from the methylene group C16. The two alternative modes of folding the fatty acid substrate within the enzyme's active site may contribute to the low ee. To identify the hydrogen atom that is lost from C(16) during olefin formation, the enzyme preparation was incubated with chirally deuterium labelled [15,16-2H2]-(16S)-42 or [15,16-2H2]-(16R)-42 (Scheme 8).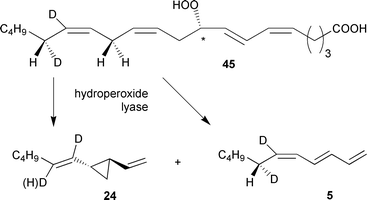 | ||
| Scheme 8 Product spectrum after administration of chiral labelled arachidonic acid 42 to G. parvulum. | ||
After administration of [15,16-2H2]-(16S)-arachidonic acid 42, the hydrogen atom was abstracted from C16, resulting in [1′,2′-2H2]dictyopterene A 24. Both enantiomers of [1′,2′-2H2]-24 displayed the same ee as the natural, unlabelled 24. Moreover, since both enantiomers showed the same degree of deuterium labelling, the mode of hydrogen abstraction at C16 does not govern the stereochemistry of the product; hence, the alternative modes of folding the substrate in the enzyme-active site may determine the ee. Administration of the enantiomeric [15,16-2H2]-(16R)-arachidonic acid 42 led to an unexpected result. Owing to the kinetic isotope effect related to the removal of a deuterium atom the enzyme not only attacked the C16-HR of 42 leading to [1′-2H]dictyopterene A 24, but also to a certain extent the C16-HS. This demonstrates the lack of a precise control of the transition state geometry. Moreover, the kinetic isotope effect additionally shifted the product spectrum to the linear hydrocarbon 5 by removing a hydrogen atom from C13 instead of C16 of the hydroperoxide precursor. The observed dynamic product channelling of labelled and unlabelled precursors is highly indicative of a single hydroperoxide lyase exerting only limited control of the orientation of the substrate at the active site (Scheme 9). This might result in the attack of either C16-HR, C16-HS, or even a hydrogen atom from C13, depending on the ease of hydrogen abstraction.58
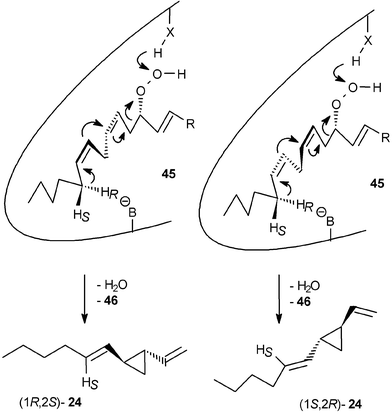 | ||
| Scheme 9 Proposed folding of the hydroperoxy fatty acids in the enzyme active site. | ||
4.4 Pericyclic reactions
According to the isotope pattern of olefinic products from labelled fatty acid precursors, all C11-hydrocarbons are generated from the C10–C20 terminus of eicosanoid fatty acids in algae. In principle, cycloheptadienes can originate by different routes. A hydroperoxide lyase could cleave the reactive intermediate 9-HPEPE 44 and mediate a direct cyclisation between C10 and C16, yielding the cycloheptadiene ring of ectocarpene 1 (Scheme 10 path B). On the other hand, a thermally labile cis-disubstituted cyclopropane such as (1R,2S)-7 could represent the first product, which then rearranges via a [3.3]-sigmatropic process94,95 to the cycloheptadiene 1 (Scheme 10 path A). Since all previous pheromone identifications relied entirely on GC-methods, only the rearranged products could be detected.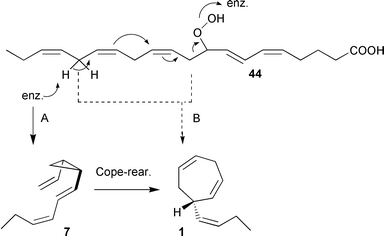 | ||
| Scheme 10 Proposed cyclisation mechanisms to ectocarpene 1. | ||
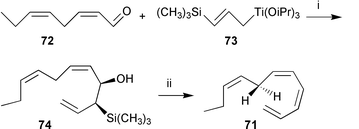
To test the route involving thermally unstable cis-disubstituted bis-alkenylcyclopropanes as intermediates, alternative methods of extraction, accumulation, and analysis of the volatile pheromones had to be developed. Low-temperature syntheses provided the references 7, 67, and 70 that allowed kinetic investigations of the rearrangements, and helped to prove the identity of the naturally produced thermolabile pheromones.
A particular versatile synthon for the thermolabile cycloheptadiene-precursors 7, 67, and 70 is the mono acetylated diol 59. Both enantiomers are available by two complementary enzymatic transformations. The diol 58 can be transformed to (1S,2R)-59 by enzymatic esterification using porcine pancreatic lipase in vinyl acetate. The enantiomeric (1R,2S)-59 results after hydrolysis of the corresponding bis-acetate 60 using porcine liver esterase.29,96 Both products were of high ee and served as starting materials for pheromone synthesis (Scheme 11).
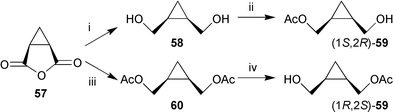 | ||
| Scheme 11 Reagents: i, LiAlH4; ii, porcine pancreatic lipase/vinylacetate; iii, LiAlH4, Ac2O, pyridine; iv, porcine liver esterase. | ||
Subsequent oxidation and reaction of the resulting aldehyde 61 with CHI3 and CrCl3/Zn according to the protocol of Utimoto and Takai97 provided the central vinyliodide 62 in high purity and good yield (Scheme 12).40,98 The hexadienyl side chain of the cyclopropane 63 was introduced by a Pd(0)- and CuI-promoted carbon–carbon bond formation with cis-butenylcuprate derived from ethylmagnesium bromide and acetylene. Further manipulation of the C2-side chain involved deacetylation, Swern-oxidation, and a low-temperature Wittig-reaction using the dimsyl anion (NaH/DMSO) as the base. Isolation of (1R,2S)-7 at low temperature (0 °C) provided material for bioassays and the determination of the kinetic parameters of the spontaneously occurring Cope-rearrangement to ectocarpene 1 (Scheme 12).40,98
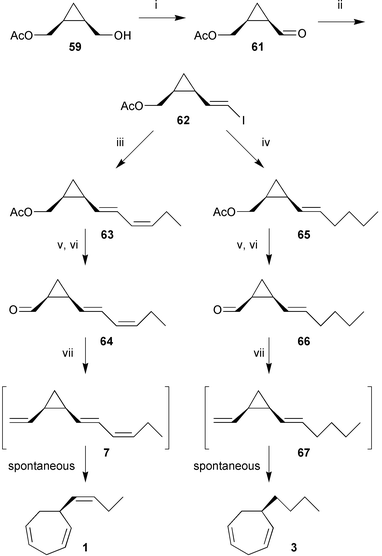 | ||
Scheme 12
Reagents: i, Swern oxidation; ii, CrCl3/Zn, CHI3; iii, EtMgBr, CuBr, acetylene, ZnBr2, Pd(PPh3)4; iv, BuLi, CuI; v, K2CO3; vi, Swern oxidation; vii NaH/DMSO, H2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) PPh3. PPh3. | ||
The synthesis of pre-dictyotene 67 followed the same overall strategy utilising a copper-mediated coupling of butyllithium to the vinylic iodide 62 to establish the hexenyl side chain. The hexatrienyl side chain of pre-desmarestene 70 was assembled via a Pd(0) mediated coupling of 62 with propargylic alcohol, followed by reduction of the triple bond and completion of the side chain by oxidation and Wittig-olefination (Scheme 13).
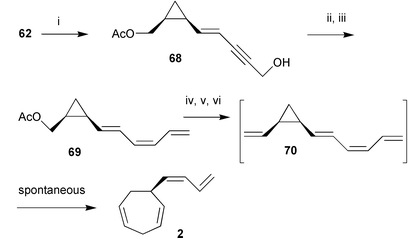 | ||
| Scheme 13
Reagents: i, Propargylic alcohol, Pd(PPh3)4; ii, 1) Zn/Cu/Ag, 2) MnO2; iii, NaH/DMSO, H2CPPh3; iv, K2CO3; v, Swern oxidation; vi NaH/DMSO, H2CPPh3. | ||
Pre-desmarestene 70 proved to rearrange readily, and special care had to be taken during the final Wittig reaction and work-up. Half-life times of the labile hydrocarbons at two typical environmental temperatures of marine brown algae (spring temperature in Arctic and Mediterranean regions) and the Arrhenius parameters of the rearrangements are given in Table 2.40,98
| Rearrangement | R | E a/kJ mol−1 | ln a | t ½ (8 °C)/min | t ½ (18 °C)/min |
|---|---|---|---|---|---|
| 7 → 1 | -CHCH-C2H5 |
63.8 ± 1.2 | 18.9 ± 0.4 | 56 | 21 |
| 70 → 2 | -CHCH-CHCH2 |
62.6 ± 1.5 | 18.5 ± 0.5 | 45 | 18 |
| → 16 | -CHCH2 |
64.9 ± 1.2 | 19.0 ± 0.4 | 77 | 30 |
| 67 → 3 | -C4H9 | ca. 73.8 | 155 | 49 |
With the synthetic standard in hand, the identity of the pheromone released by female gametes of E. siliculosus could be proven. A dense suspension of freshly released female gametes of E. siliculosus was extracted at 0 °C, and the HPLC retention time along with a transient UV-signal identical to that of synthetic pre-ectocarpene 7 was observed.40 The half-life time of both the UV-signals from the separated natural product and the synthetic reference were identical and demonstrated the thermolability of the secreted pheromone 7. Comparative bioassays with male gametes of E. siliculosus using pre-ectocarpene 7 and 1 demonstrated that only 7 exhibited a very high attraction potential. The threshold concentrations reported after the original identification of 1 as an active pheromone were found at 10 nmol l−199 while pre-ectocarpene 7 was active down to 5 pmol l−1 and resulted in a significantly higher accumulation factor (Fig. 5). Since only a few minutes are required between pheromone-release by the females and attraction of the first male gametes, the biological function of the Cope-rearrangement is evident. The thermolabile cyclopropyl-pheromones degrade rapidly, controlled only by the temperature of the environment. This represents the fastest and most effective mechanism known for a spontaneous inactivation of attractant and/or release factors.
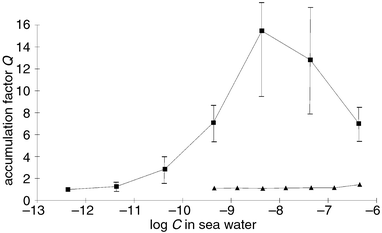 | ||
| Fig. 5 Biological activity of ectocarpene 1 (filled triangles) and pre-ectocarpene 7 (filled squares) in the droplet bioassay with male gametes of E. siliculosus. The diagram shows a plot of the accumulation factor Q (determined according to Fig. 3) against the logarithm of the pheromone concentrations in sea water.40 | ||
Since the half-life of the divinylcyclopropanes varies significantly, depending on the degree of unsaturation of the C6-side-chain, one might speculate that some algae use this molecular property to regulate the inactivation mechanism.
Besides the thermal Cope-rearrangement, other pericyclic and electrocyclic rearrangements were postulated to occur in the biosynthesis of algal pheromones. For example, from mature gynogametophytes of Giffordia michellae the linear hydrocarbon giffordene 17 was isolated as the major volatile. Administration of labelled substrates clearly showed that giffordene 17 is derived from eicosapentaenoic acid 43via the hydroperoxide HPEPE 44.83 However, the stereochemistry of the conjugated tetraene cannot directly result from the previously discussed cleavage of an intermediate fatty acid hydroperoxide (Scheme 14).
![[1,7]-H-Shift in the biosynthesis of giffordene 17.](/image/article/2002/NP/a806888g/a806888g-s14.gif) | ||
| Scheme 14 [1,7]-H-Shift in the biosynthesis of giffordene 17. | ||
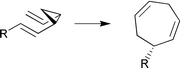
The plausible 1,7-sigmatropic hydrogen shift, which readily accounts for the peculiar geometry of giffordene 17, could be demonstrated by the ease of the rearrangement of the synthetic tetraene 71. According to Scheme 15, the unstable tetraene 71 is available from the dienal 72 by a modified version of the Peterson-reaction. The hydroxysilane 74 could be selectively transformed into E- or Z-olefins at −30 °C.100
 | ||
| Scheme 15 Reagents: i, BuLi; ii, KH. | ||
Determination of the activation parameters and half-life times at different temperatures showed that the 1,7-hydrogen shift is a spontaneous reaction that proceeds within minutes (Table 3).100 Compared to the activation parameters of the previously studied [1,7]-hydrogen shift from (1,3Z,5Z)-octatriene to (2Z,4Z,6E)-octatriene101,102 or those of the isomerisation of previtamin D3 to vitamin D3,103,104 the reaction 71 → 17 has the lowest activation energy due to the formation of the extended π-system of 17 (Table 3). The short half-life of e.g. 148 min for 71 at 18 °C suggests that the [1,7]-hydrogen shift occurs spontaneously under environmental conditions and during the usual enrichment of algal volatiles. Since neither 71 nor any of its stereoisomers proved to be the genuine pheromone, the attractive principle of female gametes of G. mitchellae remains open.
| Reaction | E a/kJ mol−1 | ΔH#298 /kJ mol−1 | ΔS#298/J mol−1 K−1 | ι1/2/min |
|---|---|---|---|---|
| 71 → 17 | 67.4 | 64.9 | −91.9 | 148 (18 °C) |
| 77 → 30 | 59.4 | 57.1 | −89.7 | 33 (−5 °C) |
| 3.4 (18 °C) |
To account for the presence of 7-methylcycloocta-1,3,5-triene 30 and other C9-hydrocarbons in the pheromone blend of Cutleria multifida,60 the assumption of an 8π-electrocyclic ring closure of (1,3Z,5Z,7E)-nonatetraene 77 (Scheme 16) appeared to be reasonable. The fatty acid derived precursor 77 could rearrange to 30 and its valence tautomers 78 and 79 which were detected in the GC-profile of the pheromone blend of C. multifida.100
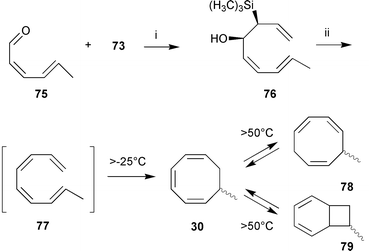 | ||
| Scheme 16 Reagents: i, BuLi; ii, KH. | ||
Again, the low-temperature Peterson-olefination proved to be ideally suited for the synthesis of the labile conjugated tetraene 77 (Scheme 16). The kinetic data show that 77 is very unstable and possesses a half-life of only a few minutes at the average spring temperature of the Mediterranean environment of the algae (Table 3). Analysis of the products of the electrocyclic rearrangement of 77 proved that all C9-hydrocarbons 30, 78, and 79, detected in the Culteria-bouquet could be derived from 77.
5 Algal defense
5.1 (A)biotic degradation of algal pheromones
Owing to their high degree of unsaturation and pronounced susceptibility to oxidative degradation, the C11 hydrocarbons do not accumulate in the environment. Even the rather high contents in the thalli of Dictyopteris sp. have been found to readily degrade in relatively short time spans. The extent and the mechanistic basis of the abiotic degradation of the alicyclic pheromones have been studied with synthetic 6-butylcyclohepta-1,4-diene 3 (dictyotene) and molecular oxygen in the presence of Fe3+, mimicking the conditions of the natural environment (sea water).4 Since these model reactions can be carried out at a large scale, even the minor compounds of Scheme 17 became available as pure compounds for spectroscopy and bioassays.105–107 The structures fit into a general scheme of an oxidative sequence starting with the endocyclic cycloheptadienyl radical 80 as depicted in Scheme 17.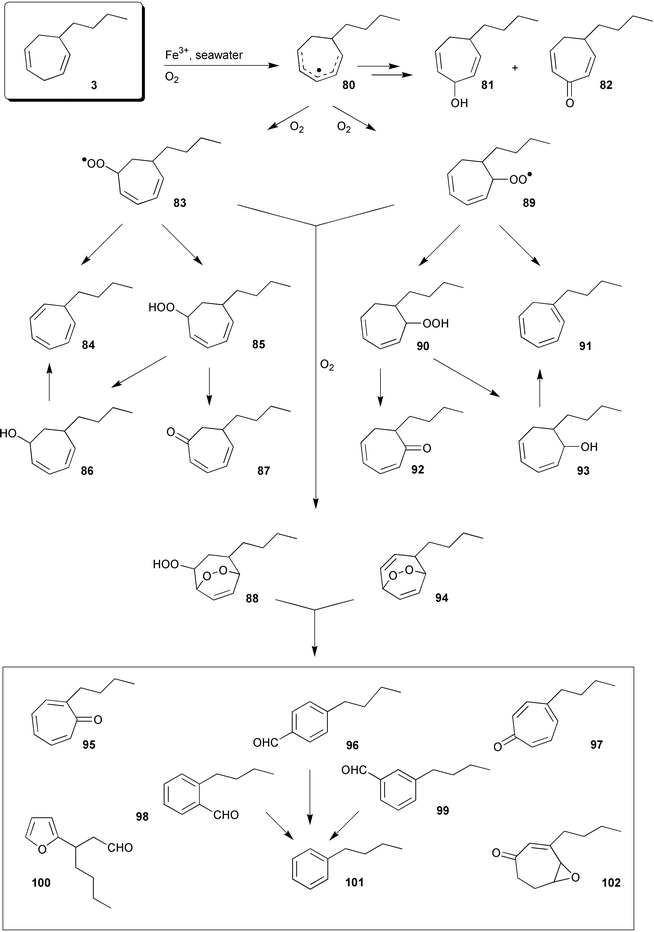 | ||
| Scheme 17 Oxidative degradation of 3. | ||
Once generated, this radical reacts with molecular oxygen at all possible positions and yields three isomeric hydroperoxides as the primary products. Their subsequent decomposition is responsible for the formation of ketones 82, 87, and 92, alcohols 81, 86, and 93 (minor compounds), and the fragmentation of the carbon framework. The stereoisomeric butylbenzaldehydes 96, 98, and 99, along with the substituted furan 100, fit into such reaction channels. The significance of this metal-assisted (Fe3+) model reaction is strongly supported by the observation that dictyotene 3 is stable over several days in de-ionised water and even in sea water, if radical scavengers such as BHT are present.105
The major product(s) of the abiotic degradation (up to 70%) are, however, isomers of the epoxyketone 102. This compound probably originates from a primary endo-hydroperoxide 88, analogous to the thermal decomposition of tropilidene endoperoxide.108 The furan 100 is assumed to result from an Fe2+-catalyzed reduction of the endo-peroxide 88 to the intermediate radical-anion 103, which decomposes to an acyclic hydroxydialdehyde 104 that in turn cyclises to the furan 100 with concomitant elimination of water.109
The intermediate 104 may also lead to the isomeric aromatic aldehydes 96, 98, and 99via intramolecular aldol-reaction and subsequent loss of two molecules of water (Scheme 18). The hydrocarbon butylbenzene 101 stems from free radical decarbonylation of the alkylbenzaldehydes.106 The trace compounds butylbenzene and the alkyltropilidenes 84 and 91 are remarkably attractive to male gametes of E. siliculosus (unpublished results); the alcohols and the ketones are not. The isomeric cycloheptadienones 82, 87, and 92 were first isolated from thalli of the Hawaiian Dictyopteris plagiogramma and D. australis, and could be synthesized by Sarret-oxidation of dictyotene.110
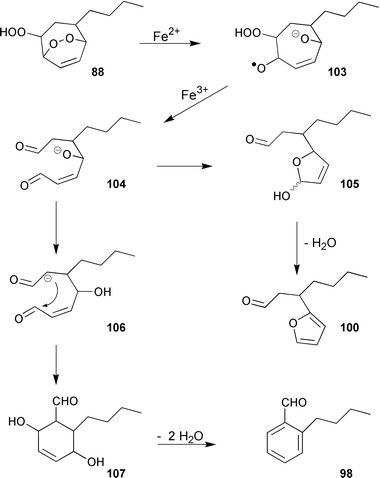 | ||
| Scheme 18 Degradation of 88. | ||
When dictyotene 3 was exposed to a dense population of male gametes of E. siliculusus (ca. 108 cells ml−1 of sea water) or thalli of the Mediterranean brown alga Dictyopteris membranacea, the three alcohols 81, 86, and 93 dominated the reaction mixture, accompanied by small amounts of the tropolones 95 and 97.106,4 A similar spectrum of products was obtained upon oxidation of dictyotene with activated porphinato complexes (e.g. TPPMnCl; iodosylbenzene) which served as biomimetics for natural P450-type enzymes.111 Due to the rather close similarity of product profiles, enzymes of this type might be involved in the biotic degradation of the pheromones by male gametes and those algae that produce significant amounts of the C11 hydrocarbon in their thalli.106 Especially, algae of the genus Dictyopteris continuously release C11 hydrocarbons along with the cycloheptadienones 82, 87, and 92 into the surrounding seawater.64 While the release of large amounts of hydrocarbons may interfere with the communication system of other brown algae (natural “mating disruption”22) sharing the same habitat, the α,β-unsaturated ketones are directly toxic for certain herbivores.112 In this respect dictyotene may have a dual function for the gametes by first acting as a pheromone and then providing chemical defense against sperm-feeding mezo-grazers (copepods, amphipods), especially after oxidative transformation into the isomeric cycloheptadienones. In the presence of gametes of E. siliculosus or intact thalli of D. membranacea, within the first ten hours the biotic degradation of dictyotene is much faster than the competing abiotic oxidation which becomes dominant after about 12–15 h (predominant formation of isomeric epoxyketones of type 102). A rigorous proof of the extent of biotic and abiotic degradation of dictyotene by, for example, chiral analysis of the resulting alcohols is still lacking.
To evaluate the potential of the C11 hydrocarbons and their oxidation products to function as chemical defenses against herbivores, their effect on feeding habits of the generalist sea urchin Arbacia punctulata and the generalist amphipod Ampithoe longimana was investigated at naturally relevant concentrations (0.2 to 0.05% by weight in artificial food).112 While most of the compounds of Scheme 17 did not reduce sea urchin feeding, food consumption by amphipods was strongly and surprisingly affected by the parent hydrocarbon dictyotene 3, and by a mixture of crude hydroperoxides 85, 90, and by the cross-conjugated ketone 82 known from D. australis and D. plagiogramma.113 The two linearly conjugated cycloheptadienones 87 and 92 were less effective. The dienone 82 proved to be the only compound that more strongly deterred the sea urchin than the amphipod. The tropilidenes 84, 91, butylbenzene 101 and the isomeric benzaldehydes 96, 98, and 99 were not deterrent. As a matter of fact, dictyotene 3 and its degradation products along with certain C11 sulfur compounds turned out to be the first algal metabolites that were strongly active against sedentary mezo-grazers.114 Previous studies demonstrated that diterpene alcohols produced by the brown seaweed Dictyota significantly deterred feeding by temperate and tropical fishes and sea urchins, but had no effect on the amphipod A. longimana or the polychaete Platynereis dumerilii.115
5.2 C11-sulfur compounds from Dictyopteris; chemical defense against mezo-grazers
Certain seaweeds in the genus Dictyopteris produce significant amounts of C11-sulfur compounds that appear biosynthetically related to the C11 pheromones.113,114 Like the pheromones, the sulfur compounds compiled in Scheme 19 may originate from oxidative degradation of highly unsaturated eicosanoids via oxygenated intermediates as outlined in Scheme 20. The natural concentration of the C11-sulfur compounds isolated from thalli of the Mediterranean seaweed D. membranacea was ca. 0.1% of plant dry mass.114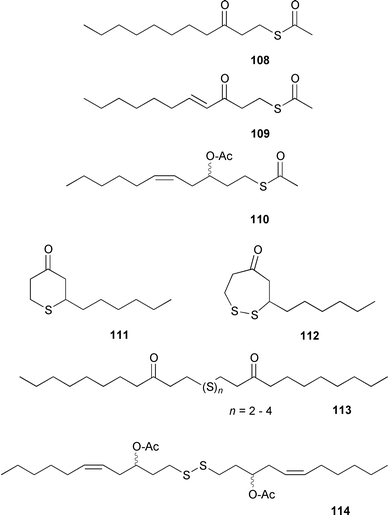 | ||
| Scheme 19 Sulfur containing compounds from Dictyopteris sp. | ||
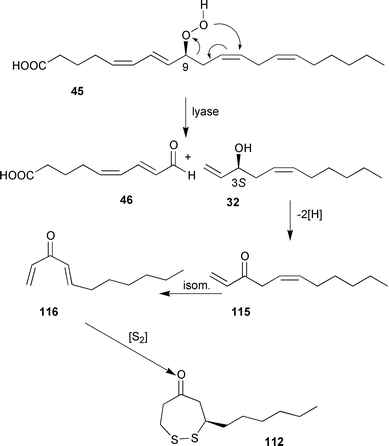 | ||
| Scheme 20 Proposed biosynthesis for 112.116 | ||
By analogy to the biosynthesis of the fungal metabolite oct-1-en-3-ol from 13-hydroperoxylinolenic acid, C11-sulfur compounds such as 111 and 112 may result from an oxidative cleavage of 9-hydroperoxyarachidonic acid 45 to yield undeca-1,5-dien-3-ol 32 along with the 9-oxo acid 46 as the second fragment.116 Undec-1-en-3-ol, dienol 32, trienol 34 and the corresponding acetates have been isolated from several brown algae of the genus Dictyopteris (see also Scheme 7).117
Further oxidation and isomerisation of the dienol 32 to the conjugated, unsaturated ketone 116 followed by introduction of sulfur via still unknown mechanisms could account for the simultaneous occurrence of C11 hydrocarbons and C11 sulfur compounds in the same Dictyopteris species. The postulated sequence may have several branching points. Addition of a “disulfide unit” to two molecules of the ketone 115 followed by reduction and acetylation accounts for the biosynthesis of the diacetate 114 and related compounds.114 The assumption of the diunsaturated ketone intermediate 115 is supported by the isolation of 109, which represents a mono-Michael-addition product of 116 and thioacetic acid.113 Oxidative degradation of other eicosanoids with either more or fewer double bonds could provide all the intermediates for the compounds in Scheme 19. When added to artificial diets at concentrations of 0.1% of dry weight, which corresponds to the natural concentration of the C11-sulfur compound, the dithiepan-5-one 112 significantly reduced feeding by the herbivorous amphipod A. longimana. The thiopyran-4-one 111 reduced feeding by 0.2% of algal dry mass.114 Since the ability of the dithiepanone 112 to suppress amphipod feeding was roughly comparable to that of extracts prepared from freshly collected D. membranacea, it appears likely that these compounds represent a novel class of seaweed secondary metabolites which target Ampithoe-like mezo-grazers that are often resistant to other defensive metabolites.114 Although the molecular target of the dithiepanone 112 is not known, it is interesting to note that this compound was partly able to inhibit phospholipase A2 activity.118
5.3 Lipoxygenase mediated chemical defense of diatoms
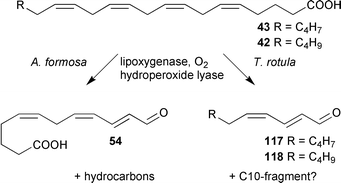
While the ecological role of C11-hydrocarbons released by calling female algal gametes, and brown algal thalli, is well understood, the benefit of hydrocarbon-production for diatoms has for a long time gone unrecognized. Diatoms are unicellular algae occurring in ocean and freshwater phytoplankton, as well as in biofilms on solid substrates. They are among the most important primary sources sustaining the marine food chain and suffer intense herbivore pressure. These algae propagate mainly by cell division; sexual reproduction occurs rarely and no involved pheromones have been reported so far. The significant release of reactive aldehydes co-occurring with C11-hydrocarbons (Schemes 5 and 6) suggests that lipoxygenase mediated fatty acid transformations might play a role in the chemical defense of these algae.Recent reports have clearly demonstrated the existence of a chemical defense mechanism relying on fatty acid degradation products. Miralto et al. detected the unsaturated aldehydes (2E,4Z)-deca-2,4-dienal 118 and (2E,4Z,7Z)-deca-2,4,7-trienal 117 in the marine planktonic diatom Thalassiosira rotula.119 Although these aldehydes neither reduced the food uptake of copepods (zooplankton grazers) nor resulted in intoxication of the herbivores, they play a role in the regulation of population dynamics in phytoplankton by reducing the hatching success from eggs of copepods.119–121 This observed activity explains the paradox that herbivorous copepods feeding on diatoms are less successful although these algae are considered a high-quality food source. Nevertheless, the reported concentration of ca. 1.5 mg l−1 decadienal 118 required to efficiently inhibit cell-division of the eggs seems quite high for a defensive compound released by unicellular planktonic algae. Investigation of the biosynthesis of the aldehydes provided insight into a defensive mechanism that might account for high local concentrations of 118 and 119 without requiring costly production of constitutive defensive metabolites. Stable isotope-labelled fatty acid incorporation showed that the volatile aldehydes derive from an oxygen-dependent reaction from highly unsaturated eicosanoids.92 These aldehydes are thus biosynthetically closely related to the 9- and 12-oxo acids 46 and 54 formed during the biosynthesis of C11- and C8-hydrocarbons in other diatoms (Schemes 5 and 6). Furthermore, the volatile aldehydes 117 and 118 compose the same reactive α,β,γ,δ-unsaturated aldehyde element as the oxo-acids which suggests a common role in the defense of these microalgae (Scheme 21).
 | ||
| Scheme 21 Biosynthesis of reactive aldehydes from diatoms. | ||
Although both unsaturated aldehydes and hydrocarbons are reported from numerous diatoms,85–87 quantification experiments with carefully isolated intact algae did not result in any detectable oxygenated fatty acid degradation products. This situation changes dramatically seconds after mechanical damage of the algae, and saturation levels of 45 fmol cell−156 and 4 fmol cell−1117 are reached after only 2–3 min.92 The rapid onset of enzymatic oxidative fatty acid degradation thus represents a wound-activated chemical defence of unicellular algae. This strategy—building up a high local concentration of defensive metabolites only in response to damage—allows to target secondary metabolites very efficiently. Exploitation of this rare mechanism could account even for the high concentrations of ca. 1.5 mg l−1 required to reduce copepod hatching success. During normal growth, cellular resources are invested in the production of eicosanoic fatty acids that are activated on demand for chemical defense, thus avoiding costly production of constitutive defensive secondary metabolites and minimising the risk of self-toxicity. Even if this mechanism does not increase the fitness of single algal cells that are consumed by the herbivore while their defense is activated, the overall success of the genetically very closely related individuals is increased by reducing herbivore pressure. During algal blooms—seasons with increased cell counts of certain algal species—this mechanism is especially successful, because most diatoms in the water column result from the fast division of parent cells.
References
- P. Karlson and M. Lüscher, Nature, 1959, 183, 55 CAS.
- G. Kochert, Annu. Rev. Plant Physiol. Plant Mol. Biol., 1978, 29, 461 Search PubMed.
- L. Jaenicke and W. Boland, Angew. Chem., Int. Ed. Engl., 1982, 21, 643 CrossRef.
- W. Boland, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 37 CAS.
- I. Maier, Progr. Phycolog. Res., 1995, 11, 51 Search PubMed.
- I. Maier and D. G. Müller, Biol. Bull., 1986, 170, 145 Search PubMed.
- M. G. Thuret, Ann. Sci. Nat. Bot., 1854, 4, 197 Search PubMed.
- I. Maier, A. Wenden and M. N. Clayton, J. Exp. Bot., 1992, 43, 1651 Search PubMed.
- A. Geller and D. G. Müller, J. Exp. Biol., 1981, 92, 53 Search PubMed.
- D. G. Müller, G. Gassmann and K. Lüning, Nature, 1979, 279, 430.
- I. Maier and D. G. Müller, Protoplasma, 1982, 113, 137 Search PubMed.
- I. Maier, D. G. Müller, C. Schmid, W. Boland and L. Jaenicke, Naturwissenschaften, 1988, 75, 260 CrossRef CAS.
- A. H. Cook, J. A. Elvidge and R. Bentley, Proc. R. Soc. (London) B, 1951, 138, 97 Search PubMed.
- D. G. Müller, L. Jaenicke, M. Donike and T. Akintobi, Science, 1971, 171, 815.
- I. Maier and M. N. Clayton, Bot. Acta, 1993, 106, 344 Search PubMed.
- K. Grob and F. Zürcher, J. Chromatogr., 1976, 117, 285 CrossRef CAS.
- I. Maier, G. Pohnert, S. Pantke-Böcker and W. Boland, Naturwissenschaften, 1996, 83, 378 CrossRef CAS.
- H. Kataoka, H. L. Lord and J. Pawliszyn, J. Chromatogr. A, 2000, 880, 35 CrossRef CAS.
- D. G. Müller, I. Maier and H. Müller, Photochem. Photobiol., 1987, 46, 1003 Search PubMed.
- H. Kawai, D. G. Müller, E. Fölster and D.-P. Häder, Planta, 1990, 182 Search PubMed.
- I. Maier, D. G. Müller and W. Boland, Z. Naturforsch., Teil C, 1994, 49, 601 Search PubMed.
- W. Boland, F. J. Marner, L. Jaenicke, D. G. Müller and E. Folster, Eur. J. Biochem., 1983, 134, 97 CAS.
- D. G. Müller, in Experimental Phycology A Laboratory Manual, 1988, Cambridge University Press, Cambridge Search PubMed.
- K. Kodama, K. Matsui, A. Hatanaka and T. Kajiwara, Phytochemistry, 1993, 32, 817 CrossRef CAS.
- D. G. Müller and G. Gassmann, Naturwissenschaften, 1980, 67, 462 CrossRef.
- D. G. Müller, H. Kawai, B. Stache, E. Folster and W. Boland, Experientia, 1990, 46, 534 Search PubMed.
- T. Kajiwara, A. Hatanaka, K. Kodama, S. Ochi and T. Fujimura, Phytochemistry, 1991, 30, 1805 CrossRef CAS.
- D. G. Müller, W. Boland, L. Jaenicke and G. Gassmann, Z. Naturforsch., Teil C, 1985, 40, 457 Search PubMed.
- S. Pantke-Böcker, G. Pohnert, I. Fischer-Lui, W. Boland and A. F. Peters, Tetrahedron, 1995, 51, 7927 CrossRef.
- D. G. Müller, A. Peters, G. Gassmann, W. Boland, F. J. Marner and L. Jaenicke, Naturwissenschaften, 1982, 69, 290 CrossRef.
- D. G. Müller, M. N. Clayton, M. Meinderts, W. Boland and L. Jaenicke, Naturwissenschaften, 1986, 73, 99 CrossRef.
- D. G. Müller, G. Gassmann, W. Boland, F. J. Marner and L. Jaenicke, Science, 1981, 212, 1040.
- J. A. Phillips, M. N. Clayton, I. Maier, W. Boland and D. G. Müller, Phycologia, 1990, 29, 367 Search PubMed.
- T. Schotten, W. Boland and L. Jaenicke, Tetrahedron Lett., 1986, 27, 2349 CrossRef CAS.
- D. G. Müller, I. Maier and G. Gassmann, Phycologia, 1985, 24, 475 Search PubMed.
- I. Maier, C. Hertweck and W. Boland, Biol. Bull., 2001, 201, 121 Search PubMed.
- D. G. Müller, M. N. Clayton, G. Gassmann, W. Boland, F. J. Marner, T. Schotten and L. Jaenicke, Naturwissenschaften, 1985, 72, 97 CrossRef.
- D. G. Müller and K. Seferiadis, Z. Pflanzenphys., 1977, 84, 166 Search PubMed.
- D. G. Müller and G. Gassmann, J. Plant Physiol., 1985, 118, 401 Search PubMed.
- W. Boland, G. Pohnert and I. Maier, Angew. Chem., Int. Ed. Engl., 1995, 34, 1602 CrossRef CAS.
- D. G. Müller, M. Clayton, G. Gassmann, W. Boland, F. J. Marner and L. Jaenicke, Experientia, 1984, 40, 211 Search PubMed.
- W. Boland, U. Flegel, G. Jordt and D. G. Müller, Naturwissenschaften, 1987, 74, 448 CrossRef CAS.
- D. G. Müller, R. Westermeier, A. Peters and W. Boland, Bot. Mar., 1990, 33, 251 Search PubMed.
- H. Kawai, W. Boland and D. G. Müller, Jpn. J. Phycol. (Sorui), 1994, 42, 227 Search PubMed.
- D. G. Müller, Biochem. Physiol. Pflanzen, 1974, 165, 212 Search PubMed.
- W. Boland, W. A. Konig, R. Krebber and D. G. Müller, Helv. Chim. Acta, 1989, 72, 1288 CrossRef CAS.
- W. Boland, K. Mertes, L. Jaenicke, D. G. Müller and E. Folster, Helv. Chim. Acta, 1983, 66, 1905 CrossRef CAS.
- J. A. Phillips, M. N. Clayton, I. Maier, W. Boland and D. G. Müller, Br. Phycological J., 1990, 25, 295 Search PubMed.
- I. Maier, D. G. Müller, G. Gassmann, W. Boland, F. J. Marner and L. Jaenicke, Naturwissenschaften, 1984, 71, 48 CrossRef CAS.
- W. Boland, L. Jaenicke and D. G. Müller, Naturwissenschaften, 1985, 72, 147 CrossRef CAS.
- D. G. Müller, W. Boland, F. J. Marner and G. Gassmann, Naturwissenschaften, 1982, 69, 501 CrossRef.
- D. G. Müller, W. Boland, U. Becker and T. Wahl, Biol. Chem. Hoppe-Seyler, 1988, 369, 655 Search PubMed.
- D. Wirth and W. Boland, Helv. Chim. Acta, 1990, 73, 916 CrossRef CAS.
- A. Peters and D. G. Müller, Helgol. Meeresunters., 1985, 39, 441 Search PubMed.
- D. G. Müller, G. Gassmann, F. J. Marner, W. Boland and L. Jaenicke, Science, 1982, 218, 1119.
- D. Wirth, W. Boland and D. G. Müller, Helv. Chim. Acta, 1992, 75, 751 CrossRef CAS.
- T. Kajiwara, K. Kodama and A. Hatanaka, in Volatile Attractants from Plants, ACS Symposium Series, American Chemical Society, Washington, DC, 1993, vol. 525, p. 103 Search PubMed.
- M. Hombeck, G. Pohnert and W. Boland, Chem. Commun., 1999, 243 RSC.
- W. Boland, U. Niedermeyer and L. Jaenicke, Helv. Chim. Acta, 1985, 68, 2062 CrossRef CAS.
- J. Keitel, I. Fischer-Lui, W. Boland and D. G. Müller, Helv. Chim. Acta, 1990, 73, 2101 CrossRef CAS.
- F. J. Marner, D. G. Müller and L. Jaenicke, Z. Naturforsch., Teil C, 1984, 39, 689 Search PubMed.
- W. Boland, L. Jaenicke, D. G. Müller and G. Gassmann, Experientia, 1987, 43, 466 Search PubMed.
- W. Boland and D. G. Müller, Tetrahedron Lett., 1987, 28, 307 CrossRef CAS.
- D. Wirth, I. Fischer-Lui, W. Boland, D. Icheln, T. Runge, W. A. Konig, J. Phillips and M. Clayton, Helv. Chim. Acta, 1992, 75, 734 CrossRef CAS.
- R. E. Moore, J. A. Pettus and J. Mistysyn, J. Org. Chem., 1974, 39, 2201 CrossRef CAS.
- D. G. Müller and C. E. Schmid, Biol. Chem. Hoppe-Seyler, 1988, 369, 647 Search PubMed.
- W. Boland, K. Jakoby, L. Jaenicke, D. G. Müller and E. Folster, Z. Naturforsch., Teil C, 1981, 36, 262 Search PubMed.
- W. Boland, F. P. Hoever and B. W. Kruger, Z. Naturforsch., Teil C, 1989, 44, 829 Search PubMed.
- G. P. Bowell, J. A. Callow, M. E. Callow and L. V. Evans, J. Cell Sci., 1979, 36, 19 Search PubMed.
- C. Brownlee, New Phytol., 1994, 127, 399 Search PubMed.
- D. G. Müller, Naturwissenschaften, 1972, 59, 166 CrossRef.
- C. E. Schmid, Hydrobiologia, 1993, 261, 437 Search PubMed.
- R. G. Berger, F. Drawert, H. Kollmannsberger, S. Nitz and B. Schraufstetter, J. Agric. Food Chem., 1985, 33, 232 CrossRef CAS.
- P. Schreier and H. Idstein, Dtsch. Lebensm.-Rundsch., 1984, 80, 335 Search PubMed.
- F. Bohlmann, C. Zdero, D. Berger, A. Suwita, P. Mahanta and C. Jeffrey, Phytochemistry, 1979, 18, 79 CrossRef CAS.
- W. Boland and K. Mertes, Eur. J. Biochem., 1985, 147, 83 CAS.
- C. Neumann and W. Boland, Biol. Chem. Hoppe-Seyler, 1989, 370, 792 Search PubMed.
- C. Neumann and W. Boland, Eur. J. Biochem., 1990, 191, 453 CAS.
- V. E. Vaskovsky, S. V. Khotimchenko, B. M. Xia and H. F. Li, Phytochemistry, 1996, 42, 1347 CrossRef CAS.
- M. Honya, T. Kinoshita, M. Ishikawa, H. Mori and K. Nisizawa, J. Appl. Phycol., 1994, 6, 25 Search PubMed.
- A. L. Jones and J. L. Harwood, Phytochemistry, 1992, 31, 3397 CrossRef CAS.
- M. Shameel, W. Shaikh and R. Khan, Bot. Mar., 1991, 34, 425 Search PubMed.
- K. Stratmann, W. Boland and D. G. Müller, Angew. Chem., Int. Ed. Engl., 1992, 31, 1246 CrossRef.
- K. Stratmann, W. Boland and D. G. Müller, Tetrahedron, 1993, 49, 3755 CrossRef CAS.
- F. Jüttner and U. Durst, Arch. Hydrobiol., 1997, 138, 451 Search PubMed.
- T. Wendel and F. Jüttner, Phytochemistry, 1996, 41, 1445 CrossRef CAS.
- F. Jüttner and H. Müller, Naturwissenschaften, 1979, 66, 363 CrossRef.
- J. B. Derenbach and D. Pesando, Mar. Chem., 1986, 19, 337 CrossRef CAS.
- G. Pohnert and W. Boland, Tetrahedron, 1996, 52, 10073 CrossRef CAS.
- M. Hamberg, Adv. Prostag. Thrombox. Leukot. Res., 1991, 21, 117 Search PubMed.
- M. Hombeck and W. Boland, Tetrahedron, 1998, 54, 11033 CrossRef CAS.
- G. Pohnert, Angew. Chem., Int. Ed., 2000, 39, 4352 CrossRef CAS.
- Y. Yamamoto, A. Yoshihiko, K. Matsui, H. Shimizu and T. Kajiwara, Z. Naturforsch., Teil C, 2001, 56, 6 Search PubMed.
- J. M. Brown, B. T. Golding and J. J. Stofjo, J. Chem. Soc., Perkin Trans. 2, 1978, 436 RSC.
- W. Pickenhagen, F. Näf, G. Ohloff, P. Müller and J.-C. Perlberger, Helv. Chim. Acta, 1973, 56, 1868 CrossRef CAS.
- U. Ader, P. Breitgoff, P. Klein and M. P. Schneider, Tetrahedron Lett., 1989, 30, 1793 CrossRef CAS.
- K. Takai, K. Nitta and K. Utimoto, J. Am. Chem. Soc., 1986, 108, 7408 CrossRef CAS.
- G. Pohnert and W. Boland, Tetrahedron, 1997, 53, 13681 CrossRef CAS.
- W. Boland, L. Jaenicke, D. G. Müller and A. Peters, Eur. J. Biochem., 1984, 144, 169 CAS.
- G. Pohnert and W. Boland, Tetrahedron, 1994, 50, 10235 CrossRef CAS.
- J. E. Baldwin and V. P. Reddy, J. Am. Chem. Soc., 1987, 109, 8051 CrossRef CAS.
- J. E. Baldwin and V. P. Reddy, J. Org. Chem., 1988, 53, 1129 CrossRef CAS.
- E. Havinga and J. Schlatmann, Tetrahedron, 1961, 16, 146 CrossRef.
- J. Schlatmann and E. Havinga, Recl. Trav. Chim. Pays-Bas-J. Roy. Neth. Chem. Soc., 1961, 80, 1101 Search PubMed.
- J. Piel, Diploma Thesis, Universität Bonn, 1994.
- P. Erbes, PhD Thesis, Universität Karlsruhe, 1992.
- I. Schnitzler, PhD Thesis, Universität Bonn, 1999.
- W. Adam and M. Balci, J. Am. Chem. Soc., 1979, 101, 7542 CrossRef CAS.
- J. A. Turner and W. Herz, J. Org. Chem., 1977, 42, 1900 CrossRef CAS.
- A. E. Asato and R. E. Moore, Tetrahedron Lett., 1973, 49, 4941 CrossRef.
- T. J. McMurry and J. T. Groves, Cytochrome P-450, Structure, Mechanism, and Biochemistry, Plenum, New York, 1986 Search PubMed.
- M. E. Hay, J. Piel, W. Boland and I. Schnitzler, Chemoecology, 1998, 8, 91 CAS.
- R. E. Moore, J. Mistysyn and J. A. Pettus, J. Chem. Soc., Chem. Commun., 1972, 326 RSC.
- I. Schnitzler, W. Boland and M. E. Hay, J. Chem. Ecol., 1998, 24, 1715 Search PubMed.
- M. E. Hay and W. Fenical, Oceanography, 1996, 9, 10 Search PubMed.
- I. Schnitzler, G. Pohnert, M. E. Hay and W. Boland, Oecologia, 2001, 126, 515 Search PubMed and references cited therein.
- T. Kajiwara, K. Kodama and A. Hatanaka, Bull. Jpn. Soc. Sci. Fish., 1982, 48, 211 Search PubMed.
- A. M. S. Mayer, V. J. Paul, W. Fenical, J. N. Norris, M. S. de Carvallo and R. S. Jacobs, Hydrobiologia, 1983, 260, 521 Search PubMed.
- A. Miralto, A. Ianora, S. A. Poulet, G. Romano, I. Buttino and S. Scala, Mar. Biotechnol., 1999, 1, 401 Search PubMed.
- A. Miralto, G. Barone, G. Romano, S. A. Poulet, A. Ianora, G. L. Russo, I. Buttino, G. Mazzarella, M. Laabir, M. Cabrini and M. G. Giacobbe, Nature, 1999, 402, 173 CrossRef CAS.
- I. Buttino, A. Miralto, A. Ianora, G. Romano and S. A. Poulet, Mar. Biol., 1999, 134, 147 CrossRef.
| This journal is © The Royal Society of Chemistry 2002 |