
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nickel phosphide: the effect of phosphorus content on hydrogen evolution activity and corrosion resistance in acidic medium†
Anthony R. J.
Kucernak
* and
Venkata N.
Naranammalpuram Sundaram‡
Department of Chemistry, Imperial College London, London SW7 2AZ, UK. E-mail: anthony@imperial.ac.uk; Fax: +44 (0)20 75945804; Tel: +44 (0)20 75945831
First published on 20th August 2014
Abstract
Transition metal phosphides possess novel, structural, physical and chemical properties and are an emerging new class of materials for various catalytic applications. Electroplated or electrolessly plated nickel phosphide alloy materials with achievable phosphorus contents <15 at% P are known to be more corrosion resistant than nickel alone, and have been investigated as hydrogen evolution catalysts in alkaline environments. However, there is significant interest in developing new inexpensive catalysts for solid polymer electrolyte electrolysers which require acid stable catalysts. In this paper, we show that by increasing the phosphorus content beyond the limit available using electroplating techniques (∼12 at% P), the nickel based phosphides Ni12P5 and Ni2P with higher levels of phosphorus (29 and 33 at% P) may be utilised for the hydrogen evolution reaction (HER) in acidic medium. Corrosion resistance in acid is directly correlated with phosphorus content – those materials with higher phosphorus content are more corrosion resistant. Hydrogen evolution activity in acid is also correlated with phosphorus content – Ni2P based catalysts appear to be more active for the hydrogen evolution reaction than Ni12P5. Electrochemical kinetic studies of the HER reveal high exchange current densities and little deviation in the Tafel slope especially in the lower overpotential regime for these nickel phosphide catalysts. The electrochemical impedance spectroscopy response of the respective system in acidic medium reveals the presence of two time constants associated with the HER.
Introduction
Rapid depletion of fossil fuels coupled with the adverse effect of greenhouse gases on the environment by burning conventional fossil fuels leads us to search for renewable and eco-friendly energy resources.1 Hydrogen has been suggested as one of the principle eco-friendly renewable energy vectors.2 However, the majority of industrial production of hydrogen involves the steam reformation process which again depends on the use of conventional fossil fuels. Water electrolysis powered by renewable electricity resources offers one of the best current methods available for producing renewable and environmentally friendly pure hydrogen without the additional introduction of greenhouse gases like carbon dioxide into the atmosphere. The industrial water splitting reaction has historically involved the use of alkaline based electrolytes and electro-catalysts such as Raney nickel or Raney cobalt.3,4 Even though the cost of industrial electrolysis is higher than other processes of producing hydrogen through gas reformation of fossil fuels,5 electrolysis methods offer one of the best environmentally friendly routes available at our disposals. High maintenance cost, higher overvoltage required to initiate electrolysis at appropriate rates, low power efficiency, and safety issues arising out of the use of alkaline electrolytes are some of the problems facing industrial alkaline based water electrolysis. Polymer membrane electrolyte based technology using acid based solid electrolytes6 could be one of the potential future techniques to replace the widely used industrial alkaline based process because of the flexibility in design & usage, high power efficiency, low overvoltage relative to that of alkaline systems, high current densities that could be achieved for the water splitting reaction, and lower maintenance & relatively lower safety issues. However the acid based polymeric membrane based system suffers from the use of noble metal electro-catalysts such as Pt,7 Pd,8 Ru,9 and Ir,10 for the water splitting reaction which increases the production cost of such systems. Additionally conventional non-noble metal electrocatalysts suffer from the poor stability in the acidic environment.11 Therefore there is significant interest in finding out a suitable alternative non-noble electrocatalyst for the hydrogen evolution reaction (HER) in acidic medium especially with improved stability. In the literature, several approaches have been utilised to address the issues of using non-noble metal catalysts for the HER in acidic medium. Such approaches involve, for instance, a study of the nature of the HER itself,12 electrocatalyst morphology, structure, chemical & electronic properties of the electrode materials,13–17 activation of the catalyst by different routes namely chemical & electrochemical treatments, etc.The aim of our present investigation is to study the electrochemical characteristics of the non-noble metal catalyst based on bulk nickel phosphides towards the hydrogen evolution reaction in acidic medium. Apart from noble metal electrocatalysts, metals like nickel, cobalt, molybdenum, and tungsten are some of the well-known transition metals found to have high intrinsic electrocatalytic activity18 for the water splitting reaction. However these metals suffer from the lack of long term stability in the acidic environment which compromises the use of these catalysts in the acidic environment. Different approaches in the literature have been utilised to improve the overall stability of the metals in the harsh acidic environment. Alloying with secondary metals and non-metals, and doping with secondary elements such as phosphorous, carbon, nitrogen, boron, etc.19–23 are some of the best available methods.
Transition metal phosphides are an interesting class of materials and offer many interesting features such as being low band gap semiconductors (therefore being electronically conductive), good stability in acidic & basic media compared to that of pure metal, in-expensive and availability of facile chemical routes for their synthesis24–26 and easy access to a number of different phases. Although many transition metals are unstable in even weak acids, metal rich phosphides of the form M3P (e.g. Ni3P, Cr3P, Fe3P, Ti3P) are only dissolved in hot oxidising acids such as HNO3. As the phosphorus content is increased, corrosion resistance further improves and MP phosphides are only dissolved in hot aqua regia (e.g., CrP, VP, TiP, TaP).27 Protective electroless nickel coatings and autocatalytic nickel–phosphorus alloys (typically less than 10 at% P) are used in a wide variety of objects due to their high corrosion resistance. Three aspects contribute to their corrosion resistance:
• Metal dissolution is thermodynamically less favoured when alloyed with phosphorus;
• Metal phosphides often form amorphous films which are immune to attack at grain boundaries;
• On oxidation, phosphorus may be oxidised to phosphate, which is much less soluble than the other ionic species typically formed during metal dissolution.
Owing to their novel & superior structural, physical and chemical properties,24–26 they have been considered for a variety of different applications. For example, transition metal phosphides could be potential insertion anodes for lithium ion batteries,28 homogenous catalysts for hydrogenation of alkenes,24etc. Recently there was a communication highlighting the use of nanostructure nickel phosphide as the electrocatalyst for the hydrogen evolution reaction (HER).29 Nickel electro and electroless plating in the presence of a phosphorus source such as sodium hypophosphite22,30–33 often has been used to introduce phosphorus content into the electroless and/or electrodeposited nickel to improve the corrosion resistant properties of the metal. However, nickel plating typically achieves a maximum phosphorus loading of about 12 to 15 at% (close to the eutectic composition of the alloy at 20 at%) which ultimately is not good enough to sustain the metallic stability in the acidic environment for extended periods of time. On the other hand, true metal phosphides offer an attractive alternative to phosphorus doping. Additionally some of the well-known phosphides are increasingly available commercially. Recently there has been interest in the use of metal phosphides and sulphides as highly active electrocatalysts for the hydrogen evolution reaction.29,34–36
In this paper, we have used nickel phosphides as electrocatalysts for the hydrogen evolution reaction in acidic medium. We have utilised both commercially available nickel phosphide powders (reduced in size by different ball milling treatments) and nickel phosphides synthesized by a hydrothermal route. We compare electrocatalytic hydrogen evolution activity in acidic medium using electrochemical techniques for the different nickel phosphide phases. Our attempt is to address the issue of electrocatalytic activity of bulk nickel phosphides as many of the active HER catalysts such as hexagonal MoS2 (ref. 37) are only active in the nanostructure form and are inactive for the HER in bulk form. Additionally we have looked at the electrochemical kinetics & electrochemical ac impedance analysis of the materials along and the effect of size reduction on the subsequent electrocatalytic activity toward the HER.
Experimental
Preparation of nickel phosphide samples
Nickel chloride hexahydrate (AR Grade >98%), sodium hypophosphite monohydrate (>99% purity), red phosphorus (99.99%), sulphuric acid (>98%), potassium permanganate, and hydrogen peroxide (30 wt%) were used. Conductivity water from a Millipore MilliQ water system with a resistivity of >18 MΩ cm was used to make up all the required solutions. All glassware was cleaned by immersing in acidified potassium permanganate overnight followed by thorough washing with conductivity water followed by washing with freshly prepared and diluted piranha solution. The glassware was further rinsed several times with conductivity water and dried in an air oven before use (caution: piranha solution is a highly corrosive mixture of hydrogen peroxide and sulphuric acid and extreme care must be taken during its handling and use).Ni2P (100 mesh) was purchased from Sigma Aldrich and used without any further purification. Size reduction of Ni2P was achieved by planetary ball milling methods. A known quantity of as-received Ni2P powder was weighed (2 g) and transferred to a ball mill jar of hardened steel of 50 or 80 mL volume using 15 zirconia balls (10 mm diameter). Ball milling was carried out using a Planetary Ball Mill PM 100 model from Retsch (Germany). Different samples of Ni2P were made by varying the time interval of ball-milling ranging from 2 hours to 9 hours at a constant rotation rate of 500 rpm. After each one hour duration of milling, there was a pause of 15 min. Rotation reversal was also employed after each cycle of ball milling to achieve uniform grinding of the powder. After ball milling, the samples were washed with water, ethanol and vacuum dried before further use. Ni12P5 was prepared hydrothermally according to reported literature38 work with modification using sodium hypophosphite and nickel chloride as phosphorus and nickel sources, respectively. For comparison, a sample of the electrodeposited nickel–phosphorus alloy (8 at% P), as has been previously used as a hydrogen evolution catalyst under alkaline conditions was also tested.33 This material was prepared by nickel electroplating consisting of deposition from a bath containing 0.1 M NiSO4, 0.3 M NaH2PO2, 0.15 M H3BO3, and 0.1 M NH4Cl at a current density of 10 mA cm−2 and electrodepositing a layer onto a gold foil electrode. The specific surface area and pore size of the catalyst materials were studied using a Micromeritics TriStar 3000 analyzer (Micromeritics UK Ltd.) with pure N2, based on the Brunauer, Emmet, Teller (BET) and the Barrett, Joyner and Halenda (BJH) methods.
Electrochemical characterization
Electrochemical measurements were performed in a standard three-electrode electrochemical glass cell (Ace Glass).A very thin film of nickel phosphide on a glassy carbon electrode (loading of 150 μg cm−2, geometric area of glassy carbon 0.192 cm2; Pine Instruments) was used as the working electrode. Utilising the BET surface areas of these materials the roughness factor of the electrodes (real electrocatalyst surface area ratioed to geometric area) corresponds to 1.5 (as received Ni2P), 1.8 (6 h ball-milled Ni2P), 2.1 (9 h ball-milled Ni2P) and 24.2 (Ni12P5) assuming 100% utilisation of the catalyst. This loading is 10–100 fold lower than that might be used in a real electrolyser, but allows us to accurately follow the performance of the catalyst under ideal conditions. Deposition was achieved by casting thin films using 0.02 wt% Nafion (prepared from 5 wt% stock solution; Sigma Aldrich) in a mixture of water & low carbon chain alcohols. A platinum coil was used as the counter electrode and a reversible hydrogen electrode (Hydroflex from Gaskatel GmbH) as the reference electrode. Care was taken to avoid excessive polarisation of the counter electrode to limit platinum dissolution and subsequent redeposition on the working electrode. Prior to casting the thin film of nickel phosphide, the glassy carbon working electrode was polished to a mirror like finish using alumina slurry (Buehler) of decreased pore size on a Buehler polish pad and washed repeatedly with conductivity water, sonicated for 10 minutes (Powersonic P230D), and washed again before use.
All the measurements were performed in an oxygen free environment by purging the solution with ultra-high pure nitrogen (>99.999%, Air products BIPA) at least for 30 minutes and subsequently maintaining a nitrogen atmosphere over the entire duration of the electrochemical experiments. Electrochemical measurements were performed using a Gamry Reference 600 model potentiostat–galvanostat equipped with Framework/Echem analyst software. Activation of the electrocatalyst could be achieved by cycling the electrode in the negative direction from 0 to −0.4 V vs. RHE for 5 cycles until a stable voltammogram and OCV were achieved. Electrochemical impedance spectroscopy was performed by scanning the frequency from 100 kHz to 200 mHz at 10 points per decade. A sinusoidal perturbation of 10 mVpp was used.
X-Ray diffraction measurements were performed on a PANalytical X'Pert Pro XRD system with a Cu anode (Kα = 1.54 Å) at 40 kV and 120 mA. Field-emission scanning electron microscopy (FE-SEM) analysis was performed using a LEO Gemini 1525 model FEGSEM coupled with an energy dispersive spectrometer (EDS).
Results and discussion
Characterisation of nickel phosphides
X-Ray diffraction was utilised to characterise the as received Ni2P, ball milled Ni2P for different time intervals and hydrothermally synthesised Ni5P12. Fig. 1(a) represents the resultant X-ray diffraction pattern of bulk and ball milled nickel phosphide powders. The patterns are consistent with that reported for nickel phosphide (XRD file number 01-074-1385) with a hexagonal structure (similar to the Fe2P structure) and the space group P![[6 with combining macron]](https://www.rsc.org/images/entities/char_0036_0304.gif) 2m with the following lattice parameters a = b = 5.859 Å and c = 3.382 Å. As the ball milling time interval increases from 6 to 9 hours, there is some broadening of (111), (201), (210), (211), (102) reflections corresponding to the change in the average crystalline size of Ni2P powders.
2m with the following lattice parameters a = b = 5.859 Å and c = 3.382 Å. As the ball milling time interval increases from 6 to 9 hours, there is some broadening of (111), (201), (210), (211), (102) reflections corresponding to the change in the average crystalline size of Ni2P powders.
 | ||
| Fig. 1 (a) X-Ray diffraction pattern of bulk hexagonal Ni2P and those ball milled for different time intervals (6 & 9 h) (b). X-Ray diffraction pattern of tetragonal Ni12P5. Stick models for standard Ni2P (reference number 01-074-1385) and Ni12P5 (reference number 00-022-1190) are given at the bottom of the figure for comparison. | ||
On the other hand hydrothermally synthesised nickel phosphides show major reflections (400), (240), and (312) corresponding to the metal rich tetragonal Ni12P5 (XRD-reference code 00-022-1190) with the following lattice parameters a = b = 8.646 Å and c = 5.07 Å, Fig. 1(b). A small amount of a secondary phase of unknown composition is evident.
Fig. 2 represents the scanning electron microscopy images of as-received, ball milled and hydrothermally synthesised nickel phosphide powder. As received Ni2P exhibits varied shapes with the average crystalline size in the order of several microns (10–15 μm) having a distinctive rough surface morphology. On the other hand, 6 hour ball milled Ni2P catalysts show a comparatively smoother surface with the average crystalline size in the range of only a few microns (2–5 μm). As the ball milling time increases to 9 hours, the average size of nickel phosphide reduces even further (1–2 μm) with again varied shapes of Ni2P powders. 9 hour ball milled Ni2P powder shows a very smooth surface morphology compared to that of either as-received or 6 h ball milled powders. The origin of the emergent homogeneity in ball milled Ni2P powders could be due to multiple reasons.39,40 During the course of the ball milling process, the Ni2P powders undergo high energy collision with the milling balls and the subsequent compressive force of the ball milling process results in smoother particles. Additionally, surface Ni and P atoms in nickel phosphide might undergo reorientation during the course of ball milling due to the local heat40 generated by colliding balls and subsequent annealing resulting in a homogenous and smoother Ni2P electrocatalyst as opposed to the starting Ni2P compounds. Moreover, any structural transformation of the ball milled Ni2P is ruled out as the corresponding X-Ray diffraction shows a near identical diffraction pattern to that of the as-received Ni2P powders. Hydrothermally synthesised Ni12P5, on the other hand, shows continuous chain like structures with the average crystalline size in the range of 200–500 nm.
 | ||
| Fig. 2 FE-SEM micrograph of (a) as received Ni2P; (b) 6 hour and (c) 9 hour ball milled Ni2P; and (d) hydrothermally synthesised Ni12P5. Listed on the diagrams are also the measured BET surface areas of the catalysts. | ||
Electrochemistry of nickel phosphide electrodes
The corrosion behaviour of the active anode material in an electrolyser is an important parameter to understand. Although during operation, the hydrogen evolving cathode will be polarised at reducing potentials, during the period of inoperation the potential may rise quite high due to the interaction between the catalyst material and oxygen which diffuses into the cathode compartment. Likewise, improper operation of the electrolyser may transiently lead to the application of oxidising potentials to the cathode. Usually, tests for new hydrogen evolution catalysts do not consider corrosion of the material at higher potentials, but instead look at the stability of the catalyst at operating potentials. It should be noted that under hydrogen evolution conditions, the likelihood of corrosion is much reduced due to the low electrochemical potential. Electrodeposited or electroless deposited Ni–P alloys having a maximum of 12–15 at% of phosphorus (close to the eutectic composition of the alloy at 20 at%) display reduced active anodic metal dissolution in the acidic environment in the potential regime where the pure metal undergoes active dissolution and hence Ni–P alloys are classically known41–43 to have superior anticorrosion properties to pure nickel. Various mechanisms and experimental evidence41–43 have been demonstrated in the literature for this superior anticorrosion property. However the actual reason is still not very well understood and there is a great deal of interest in finding out the actual mechanism. Prospective reasons for enhanced passivity include:• Formation of a protective phosphate like thin film on the alloy surface which subsequently acts as a diffusion barrier for active dissolution;
• Adsorption of hypophosphite (used as a phosphorous source during metal deposition) at the electrode/electrolyte interface thus forming a diffusion barrier;
• Formation of a surface phosphorus-rich film due to the preferential leaching of the metal;
• Partial covalent bond formation between nickel and phosphorus leading to a partial negative charge on the phosphorus atoms similar to that of metal phosphides and thus altering nickel's structural and electronic properties.
The steady state polarisation plots (potentiodynamic) for the electrodeposited nickel–phosphorus alloy and the four high phosphorus content nickel phosphide based catalysts in 0.5 M H2SO4 are given in Fig. 3. In the case of the electrodeposited Ni–P alloy, once the potential is scanned beyond the corrosion potential, further anodic polarisation leads to a current plateau between 0 and +0.4 V vs. RHE beyond which there is a rapid increase in the observed current due to bulk metal dissolution in the acidic environment. Above 0.6 V, the current rapidly falls – this is not due to passivation of the material, but rather because all of the Ni–P alloy has been dissolved from the electrode! For comparison, the current for the gold foil electrode on which the Ni–P alloy was electrodeposited is also shown over the potential range of 0.15–1.2 V (these results below the lower limit are not shown to avoid cluttering the low potential region). It can be seen that the Ni–P alloy electrode current rapidly approaches that of the gold foil at higher potentials. Evidently, the electrodeposited Ni–P alloy material is unstable in acid at moderate potentials, and dissolves, leaving just the gold foil. This work is confirmed by the work of others on the corrosion of electrodeposited Ni–P alloys in 0.1 M H2SO4.41–43 In contrast, the high-phosphorus nickel phosphide based catalysts show markedly different behaviour. Upon reaching the corrosion potential, further anodic polarisation of all the four high-phosphorus nickel phosphide based catalysts displayed no sharp increase in current and displayed a continuous current plateau (with lower values than the electrodeposited Ni–P alloy) extending up to high potentials (1.2 V vs. RHE). Evidently these materials are much more corrosion resistant in acidic solution than the “low-phosphorus” content electrodeposited material. In the present scenario, all the four catalysts thus resist bulk dissolution of metallic nickel in acidic medium over a large potential range.
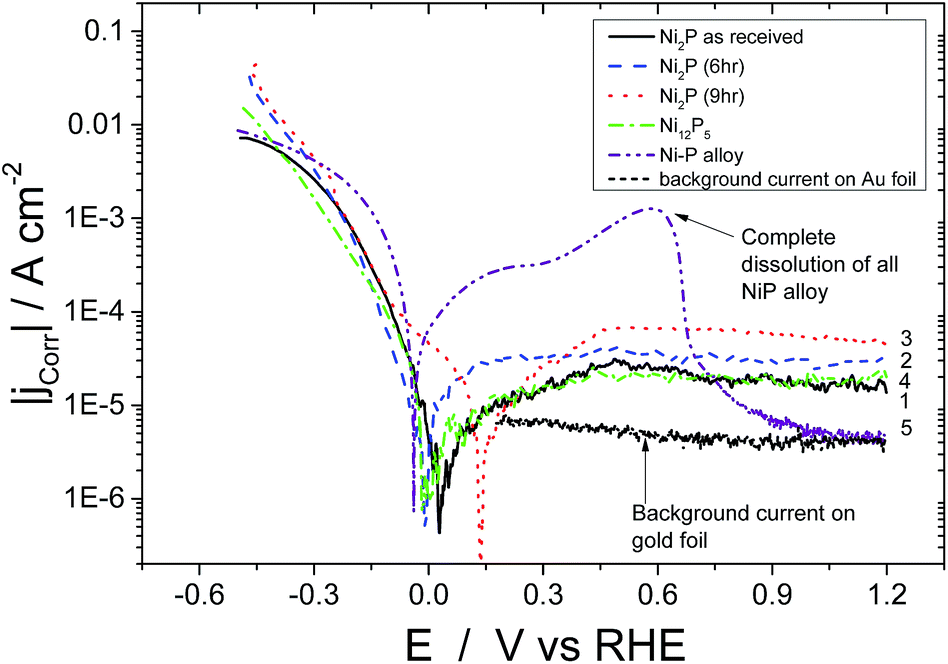 | ||
| Fig. 3 Potentiodynamic steady state scan for (1) as obtained Ni2P (2) 6 h, and (3) 9 h ball milled Ni2P, (4) Ni12P5 and (5) Ni–P alloy. 0.5 M H2SO4 N2 purged medium at 298 K and 1 mV s−1 scan rate. Current densities are displayed ratioed to the geometric area of the electrode with a catalyst loading of 150 μg cm−2. | ||
This would appear to be correlated with the greater than two-fold increase in the phosphorus content of our materials compared to the electrodeposited material. Ni2P and Ni12P5 have covalent bonding with a net negative charge on the phosphorus atom and hence this covalent bonding could consequently alter the electronic properties of metallic nickel resisting bulk metal dissolution in an acidic environment over the considerable potential range. Ni12P5 and as-received Ni2P show similar values of corrosion current in the plateau region. As-received Ni2P, Ni12P5 and 6 hour ball milled Ni2P show almost the same corrosion potentials (Ecorr of 0.020 V and 0.05 V vs. RHE, respectively). On the other hand, 9 hour ball milled Ni2P shows a shift in the corrosion potential of about 130 mV in the positive direction vs. RHE when compared to the other three nickel phosphide based electrocatalysts. The origin of such a shift in the corrosion potential could be attributed to multiple reasons. Surface Ni and P atoms in nickel phosphide might undergo reorientation during the course of the ball milling process due to the local heat generated by colliding balls and subsequent annealing resulting in a homogenous, more amorphous Ni2P surface as opposed to the starting compounds as discussed earlier. It has to be noted here that nickel phosphide does not undergo any bulk structural change over the course of ball milling as evidenced by the X-Ray diffraction pattern. For Ni12P5, the observed corrosion potential is the lowest among the high phosphorus nickel phosphide catalysts, although this corrosion potential is nonetheless more positive than the electrodeposited Ni–P alloy. This indicates that Ni12P5 might be inherently less stable in the acidic environment as opposed to Ni2P, although clearly still more stable than electrodeposited Ni–P alloys. Even though metal rich phosphides offer somewhat better electronic conductivity, they are less stable in acidic medium compared to that of phosphorus rich materials. The metal to phosphorus ratio for Ni2P is 2 whereas for Ni12P5 the ratio is 2.4. In comparison, for the electrodeposited nickel–phosphorus alloys with the highest achievable phosphorus content, the ratio is 4. Comparison between the catalysts is made somewhat difficult because of the difference in the particle size, although such changes should not affect the corrosion potential. However, it would be expected to affect the ordering of the passive current densities which are based on the geometric, not real surface areas. Below, we correct for surface area effects for the different catalysts (Fig. 5).
Hydrogen evolution reaction
The phosphorus content is also manifested in HER activity in that the phosphorus content appears correlated with the HER activity – higher phosphorus content produces higher HER activity.Discharge step – Volmer reaction
| H3O+ + e− ⇆ Hads + H2O, b = 2.303RT/αnF ≈ 120 mV |
Desorption – Heyrovsky reaction
| Hads + H3O+ + e− ⇆ H2 + H2O, b = 2.30RT/(1 + α) F ≈ 40 mV |
Recombination – Tafel reaction
| Hads + Hads ⇆ H2, b = 2.303RT/2F ≈ 30 mV |
An overview of the hydrogen evolution reaction in acidic medium is given above with three possible rate limiting steps. If the kinetics of the HER is limited by the discharge process (Volmer step) then one would expect a Tafel slope of 120 mV per decade. On the other hand, if the desorption process (Heyrovsky reaction) is the rate determining step then the expected slope would be 40 mV per decade. Finally, if the recombination reaction (Tafel reaction) is the rate limiting step, a slope of 30 mV per decade at 20 °C is expected. Another useful parameter in the HER for an electrocatalyst is the electron transfer barrier coefficient (symmetry factor), β, which is equal to the transfer coefficient (α) for the Volmer reaction step but for the Heyrovsky reaction β = α − 1.
On bulk metallic nickel electrodes in acidic medium it has been reported44 that the kinetics of the hydrogen evolution reaction is limited by the Volmer reaction step, i.e. the rate at which hydrogen adsorbs on the metal to form M − Hads. It is generally reported to be in the region of ∼150 mV per decade.45 The origin of such high Tafel slopes is reported45 to be indicative of thin surface oxides on the metallic nickel. It has been claimed that since Ni-oxides have lower electronic conductivity compared to that of the pure metal, the electron transfer rate through the oxide film is comparatively hindered to that of metallic nickel. Therefore, there is a deviation of the Tafel slope from the theoretically predicted 120 mV per decade value for the Volmer rate limiting step.
Results for the HER on aged nickel phosphide based electrocatalysts on a rotating electrode (to avoid bubble build up) in 0.5 M H2SO4 are plotted in Fig. 4. Results for the electrodeposited Ni–P alloy are not displayed, as this material was too unstable under acidic conditions to obtain reproducible results. Tafel analysis and the experimental parameters derived from these plots are given in Table 1.
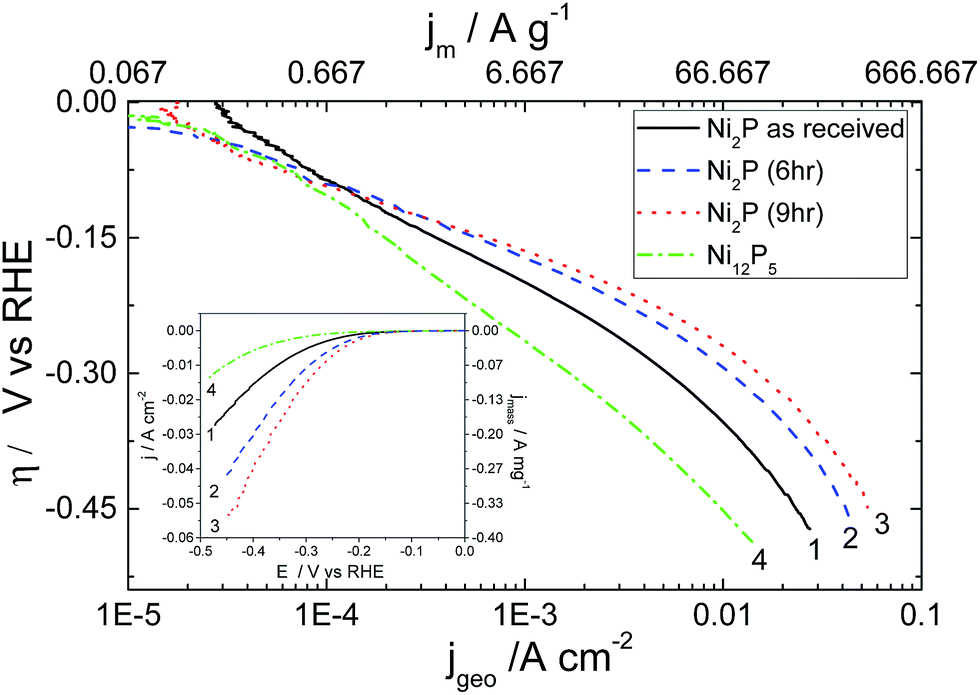 | ||
| Fig. 4 Steady state Tafel plot for the HER on (1) as obtained Ni2P (2) 6 h, and (3) 9 h ball milled Ni2P and (4) Ni12P5 coated GC electrode all with a metal phosphide loading of 150 μg cm−2. Current densities are displayed ratioed to the geometric area of the electrode, and the top axis gives the mass-specific current density. Equilibrium potential of the HER under the present condition of 0.5 M H2SO4 at pH = 0.25 (Erev = −0.015 V) is used for the over-potential calculation. 10 mV s−1 is the scan rate. The electrode rotated at 1600 rpm to achieve the steady state during the measurements. Inset: raw data for the Tafel plot. | ||
| Catalyst | Region 1 0 to −140 mV | Region 2 −140 to −450 mV | ||||
|---|---|---|---|---|---|---|
| B/mV dec−1 | j sp o/μA cm−2 | α | B/mV dec−1 | j sp o/μA cm−2 | α | |
| Ni2P as recv. | 128 | 0.13 | 0.46 | 167 | 37.3 | 0.35 |
| Ni2P (6 h) | 79 | 6.21 | 0.72 | 165 | 74.6 | 0.36 |
| Ni2P (9 h) | 84 | 2.90 | 0.70 | 159 | 72.5 | 0.37 |
| Ni12P5 | 108 | 0.37 | 0.55 | 177 | 1.37 | 0.33 |
The Tafel slopes observed for hydrothermally synthesized Ni12P5 at low overpotential (Region 1 between −5 mV and −140 mV vs. RHE) and as-received Ni2P are close to 120 mV per decade expected if the kinetics of the HER is limited by the discharge step (Volmer reaction). On the other hand, 6 h and 9 h ball milled Ni2P under identical condition shows deviation to a lower value with a concurrent increase in the transfer coefficients.
A recent report for the HER29 using nanostructured Ni2P based electrocatalysts shows very low Tafel slopes suggesting that the Heyrovsky step is the rate limiting step in this lower potential regime. This indicates the possibility for mixed kinetics in the low overpotential regime for 6 h and 9 h ball milled Ni2P electro-catalysts unlike as-received Ni2P or nanostructured Ni5P12. As the overpotential increases (−150 mV to −450 mV vs. RHE), there is a noticeable deviation from Tafel-like behaviour in the plots of all Ni2P electrocatalysts as inferred from the significant curvature in region 2. Large deviations from Tafel behaviour for the HER on these electrocatalysts may be due to uncompensated resistance (iR drop), mass transport limitation through narrow pores of the electrocatalyst, or formation of metal hydrides which subsequently acts as a barrier for the hydrogen evolution reaction. Additionally as discussed earlier, the electronic properties of nickel phosphides are quite different from that of metallic nickel or the electrodeposited Ni–P alloy catalyst due to the existence of covalent interactions between the Ni 3d and P 3p electronic bands.42,46 In our experiments, we have compensated our curves for iR effects utilising the measured solution resistance (see ESI†). Likewise, mass transport limitation through narrow pores is limited by employing very low catalyst loadings. Furthermore, shielding effects due to bubble formation is prevented by performing the measurements on a rotating disk electrode under forced convection. Hence, this curvature appears to be more associated with the electrokinetics of the reaction.
Careful examination of the exchange current density based on the real catalyst area determined from BET measurements (Table 1) reveals good electrocatalytic activity for all the three Ni2P based electrocatalysts. It would appear that ball-milling doubles the specific exchange current density in the large overpotential region compared to that achieved with the as-received Ni2P. Hence the effect of ball milling appears to be two fold – after a short period the ball milling activates the surface, and over longer periods it slightly increases the surface area of the material. Hydrothermally prepared Ni12P5 shows rather poor exchange current densities, although its performance is significantly improved by the higher intrinsic surface area of the material.
Fig. 5 shows the merit plot for the different catalysts tested. This plot compares the average corrosion current over the potential range of 0.15–1.2 V (0.15–0.4 for the Ni–P alloy) with the intrinsic exchange current density for hydrogen evolution in the large overpotential region (Region 2) based on the specific surface area of the electrode (i.e. real catalyst surface area determined by BET measurement and catalyst loading). As “good” hydrogen evolution catalysts should show low corrosion current densities but high exchange current densities, the upper left corner is the preferred operational area. It can be seen from this plot that the six-hour ball milled Ni2P catalysts are the materials which come closest to this position.
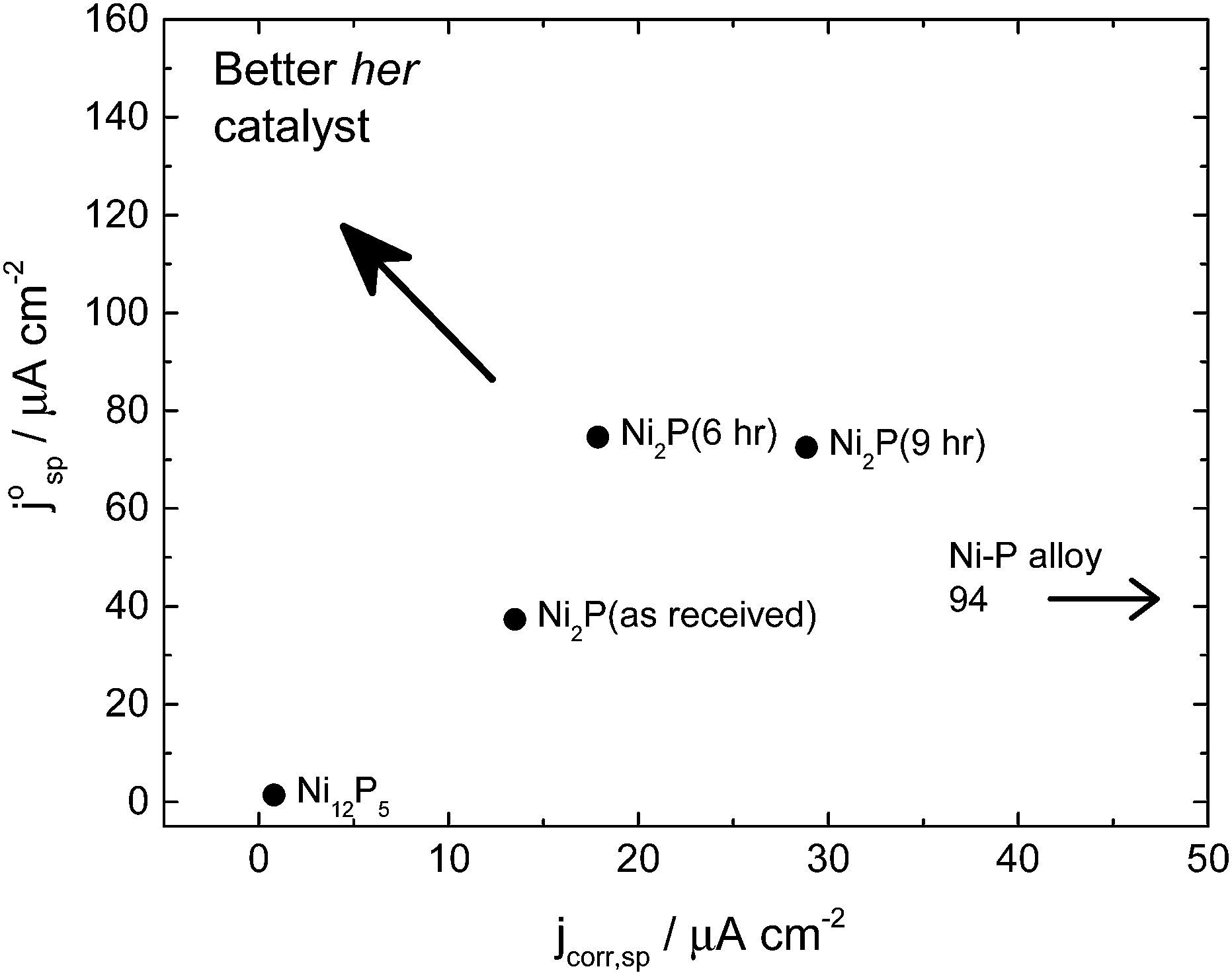 | ||
| Fig. 5 Plot of the HER specific exchange current density (−140 to −450 mV) versus limiting specific corrosion current density (0.15–1.2 V) for different nickel phosphide catalysts in 0.5 M H2SO4. Better catalysts operate in the upper left hand corner. The Ni–P alloy catalyst has a corrosion current density of 94 μA cm−2 and so is off to the far right of this graph. | ||
The performance of the catalysts tested in this paper is compared to a number of other materials in the literature which were tested under comparable conditions and for which appropriate experimental data are available, Table 2 (see also ESI†). Current densities are converted to specific current densities, and thence to turnover numbers at potentials of −0.1 and −0.2 V. We find that in comparison to a higher surface area Ni2P,29 the ball milled material shows activity which is 9-fold (−0.1 V) and 4-fold (−0.2 V) more active. Although size effects are well known in the catalyst literature, these typically only occur at a very small particle size, more likely is the effect that ball milling has on producing a clean and uncontaminated surface. The Ni2P catalyst is found to be more active than both CoP and MoS towards the HER.
| Material | Loading/mg cm−2 | Surface area/m2 g−1 | R f/cm2 cm−2 | j −0.10 V,sp/A cm−2 | j −0.20 V,sp/A cm−2 | TOF−0.1 V/s−1 | TOF-0.2 V/s−1 | Ref. |
|---|---|---|---|---|---|---|---|---|
| a Loading and specific surface area are not reported, but the roughness factor is reported. NR: not reported. | ||||||||
| Ni2P(as recv.) | 0.15 | 1.0 | 1.5 | −0.089 | −0.673 | 0.140 | 1.051 | This work |
| Ni2P(6 h) | 0.15 | 1.18 | 1.77 | −0.085 | −1.113 | 0.132 | 1.737 | This work |
| Ni2P(9 h) | 0.15 | 1.38 | 2.07 | −0.062 | −1.266 | 0.096 | 1.975 | This work |
| Ni12P5 | 0.15 | 16.1 | 24.15 | −0.004 | −0.016 | 0.019 | 0.081 | This work |
| Ni2P | 1 | 32.8 | 328 | −0.010 | −0.320 | 0.015 | 0.5 | 29 |
| CoP | 0.9 | 59.1 | 531.9 | −0.048 | NR | 0.06 | NR | 35 |
| MoSa | NR | NR | 90 | NR | −0.111 | NR | 0.3 | 36 |
The high value of the exchange current density is a measure of high HER catalytic activity of any catalyst, however the true measure of the HER electrocatalytic activity could be ascertained from the linear polarisation data as well as using current versus time transients recorded by fixing the overpotential required for the HER and measuring the resultant current density. Combined with corrosion plots, these are liable to give a better understanding of how a catalyst is liable to function within an electrolyser.
Current versus time transients for as-received Ni2P, 6 h and 9 h ball milled Ni2P and hydrothermally synthesized Ni12P5 in acidic medium are depicted in Fig. 6. It is important to remember that these electrodes have very low loading and in a real electrolyser the loading would be 10–100 fold greater (and hence current densities would likewise be increased). From the graph, it is clear that the 9 h and 6 h ball milled Ni2P electro-catalysts show the best activity with similar performance considering the slightly larger surface area of the 9 h ball-milled catalyst. These performances are over double that of the as-received Ni2P. Such a response is expected from the exchange current densities and specific surface areas of these catalysts. All Ni2P catalysts show a decay in current as a function of time. In contrast, the Ni12P5 electrocatalyst gives a lower electrocatalytic activity although the current is very stable. It would appear that the metal-rich Ni12P5 phase is less active than Ni2P, but more stable in terms of hydrogen production and with a lower corrosion current.
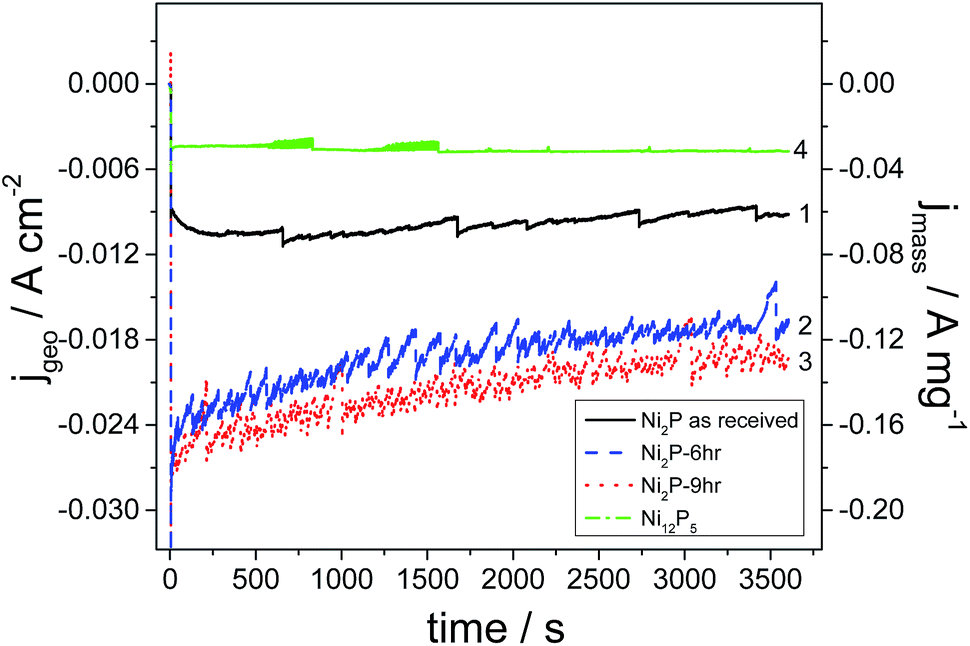 | ||
| Fig. 6 Chronoamperometric HER evolution curves in 0.5 M H2SO4 at −0.4 V vs. RHE for 1 hour reaction on glassy carbon electrodes coated with 150 μg cm−2 (1) as-received Ni2P (2) 6 h and (3) 9 h ball milled Ni2P, and (4) Ni12P5. | ||
Electrochemical impedance analysis
The performance of the electrocatalysts may be affected by a blocking film which contributes to their passivity. The interfacial properties and kinetics of nickel phosphide based catalysts towards the HER could be best understood further by electrochemical impedance spectroscopy (EIS). Electrochemical impedance spectroscopy being a non-invasive yet powerful technique has been utilised extensively to explain the electrochemical kinetics of the hydrogen evolution reaction on a number of different electrodes.44Equivalent circuit models utilising an electrochemical reaction as the circuit elements have been proposed to explore the electrode/electrolyte interphase and understand the physical meaning of various processes associated with the HER in metal/metal oxide/semiconductor based materials. Various models have been proposed based on:
• a single process – i.e. the kinetics of the HER, producing a single time constant (1T).
• Two processes operating in parallel (2TP).
• The porous model which appears as two processes operating in series (2TS).
The latter two models involving more than one characteristic process associated with the HER.
In the equivalent circuit analysis, the capacitance associated with any process is replaced with a frequency dependent constant phase element owing to porosity,47 and surface inhomogeneity48 at the atomic level and uneven charge distribution on the electrode surface. The 1T model has been largely used to describe the HER on Pt based electrodes49 as well as the HER on smooth electrode surfaces17 where the associated time constant is related to the kinetics of the hydrogen evolution reaction. On the other hand, the double time constant models have been used to describe the HER behaviour of smooth as well as porous electrodes.50 In the 2 TP model, (see Fig. 7(a) inset) the first time constant has been attributed to the kinetics of the charge transfer process in the HER whereas the second time constant is associated with the adsorption of hydrogen on the electrode surfaces. Molecular or atomic hydrogen generated by the application of overpotential forms hydride type compounds by strong adsorption on electrodes like nickel. This is also compounded by the strong affinity of transition metals like Ni and Co for hydrogen adsorption. This hydride type compound resists further evolution of hydrogen and thus acts as a barrier for the HER. Hence both the time constants in this particular model (2TP, Fig. 7(a) inset) are related to the kinetics of the HER and both of them thus change with applied overpotential. On the other hand, in the 2TS model only one time constant is related to the kinetics of the HER whereas the second time constant is related to the porosity of the electrode. The first time constant is associated with the charge transfer process and changes with overpotential whereas the second time constant due to the porosity of the electrode is independent of the applied over-potential. Therefore experimentally observed EIS data could be fitted with an equivalent circuit and after verifying the suitability of the model (either 1T or 2TP or 2TS), the resultant equivalent circuit parameters could be used to explain the specific process such as charge transfer kinetics, porosity or adsorption of hydrogen associated with the specific electrode under the investigated experimental conditions.
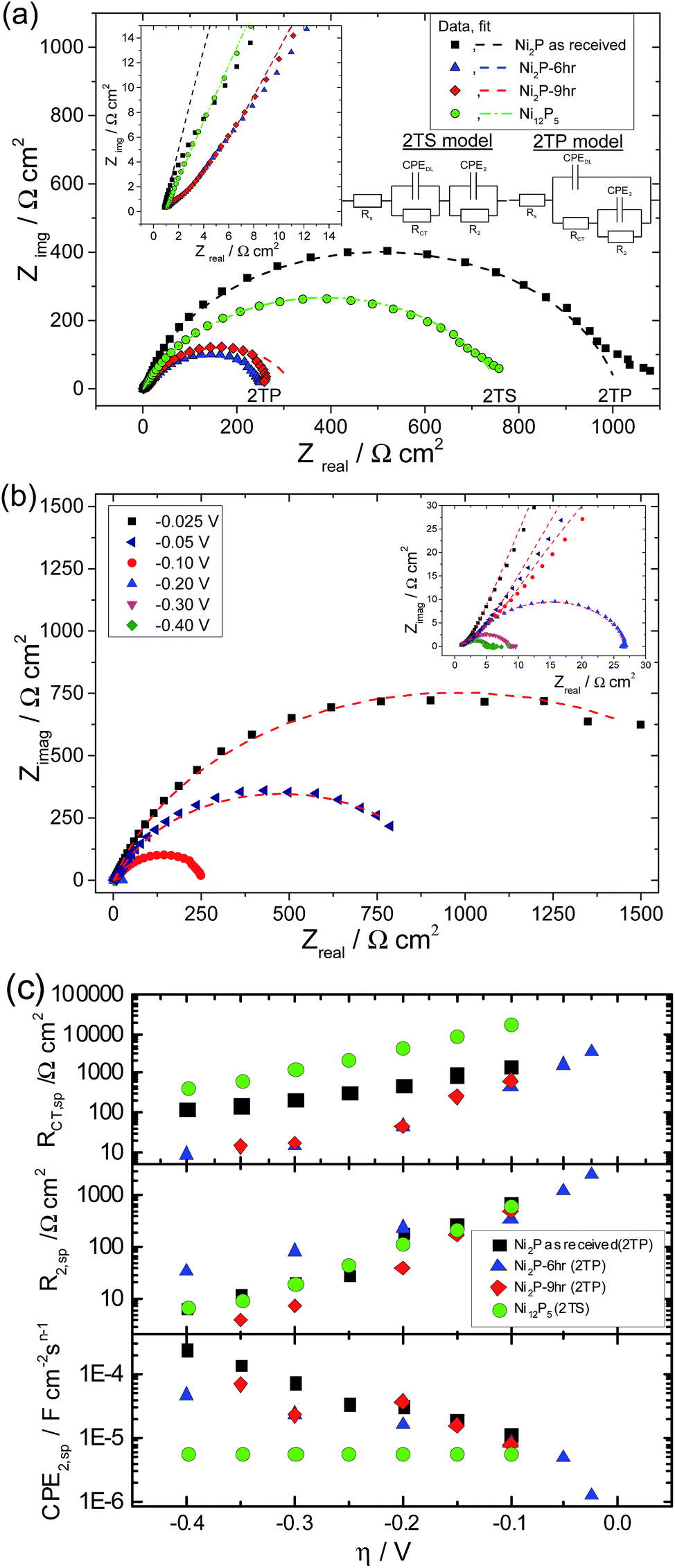 | ||
| Fig. 7 (a) Nyquist plots for as received Ni2P; 6 h and 9 h ball milled Ni2P and Ni12P5 electrodes at an applied potential of −0.100 V vs. RHE in 0.5 M H2SO4. An expanded high frequency region showing the high frequency semicircle is given as an inset. Data points represent the experimental data and the solid line represents the non-linear least squares fit using the 2TP model for the Ni2P based catalysts and the 2TS model for Ni12P5. (b) Nyquist plot of the 6 h ball milled Ni2P material as a function of applied potential. The inset is an expansion of the high frequency region. (c) Potential dependence of parameters (corrected to the real surface area) obtained from fits of the EIS spectra. Frequency range: 100 kHz to 0.2 Hz at 10 points per decade. A sinusoidal perturbation of 10 mVpp was used. | ||
Nyquist plots of glassy carbon electrodes coated in either as-received Ni2P, 6 & 9 h ball milled Ni2P or Ni12P5 in 0.5 M H2SO4 at a fixed applied electrode potential of −0.100 V vs. RHE are given in Fig. 7(a). For one of these samples (6 h ball milled Ni2P), the Nyquist plot as a function of applied dc bias is also provided, Fig. 7(b). Nyquist plots for the electrocatalysts clearly depict two time constants one appearing as a large semicircle in the low frequency region and the other appearing in the form of a small semicircle in the high frequency region for the entire overpotential range of the HER. The presence of two characteristic processes associated with the HER with two different time-constants could also be inferred clearly from the high frequency region displayed in the corresponding Bode plot (see ESI†). Therefore the EIS response of nickel phosphide based electrodes is fitted with both the two time constant parallel (2TP) and series models (2TS). Non-linear least square fitting of the experimental data using the 2TP model for as-received and ball-milled (6 h & 9 h) Ni2P yields excellent results. In comparison, attempts to fit the Ni2P data to the 2TS model the electrocatalysts lead to poor fitting of the experimentally observed data.
On the other hand, the Ni12P5 experimental EIS spectrum matches well with the 2TS porous equivalent circuit. Equivalent circuit analysis with corresponding derived parameters could give a better indication of the physical process associated with the HER on nickel phosphide based electrodes. Extracted parameters (corrected to real surface area) are plotted as a function of overpotential in Fig. 7(c). The extracted parameters for the fits are given in the tabular form in the ESI.†
It has been demonstrated previously for solid electrodes that if the radius of the high frequency semicircle is independent of applied overpotential then it has been assigned to the porosity of the electrode whereas the low frequency semicircle which varies with overpotential is associated with HER kinetics.44,51 However when both the high and low frequency semicircles vary with applied overpotential, then one time constant has been associated with adsorption of hydrogen on the electrodes forming hydride type compounds while the other time constant is associated with the kinetics of charge transfer in the HER. Additionally it has been demonstrated that the time constant associated with the adsorption of hydrogen is usually very large compared to that of the charge transfer kinetics and hence the low frequency semicircle is typically associated with the adsorption process whereas the high frequency semicircle is associated with the HER kinetics. For Ni2P based electrodes, CPEDL remains almost constant with a very small standard deviation and the corresponding resistance RCT decreases with overpotential in a semilogarithmic fashion, Fig. 7(c). On the basis of the above behaviour it could be concluded that RCT and CPEDL are associated with the kinetics of the HER. The effect of ball-milling is to produce a 10-fold reduction in RCT. Further ball milling, although leading to an increase in total surface area does not improve RCT further (i.e. 6- and 9 hour RCT values are coincident). On the other hand, CPE2 increases as the overpotential is increased with concurrent decreases in R2. This is not the characteristic behaviour of a porous electrode wherein the time constant remains invariant with changes in applied overpotential. Additionally the second time constant values are much higher than the time constant associated with the kinetics of the charge transfer process (CPEDL & RCT) which suggests that CPE2 & R2 are associated with the adsorption behaviour of hydrogen on Ni2P based electrodes. On the other hand, for Ni12P5, the 2TS model gives the best fit whilst the 2TP model fit of the experimental data leads to large errors. CPEDL derived using the 2TP model is almost constant with little deviation whereas RCT is significantly worse than that for the Ni2P samples. Additionally, R2 and CPE2 also decrease with applied overpotential unlike the Ni2P based electrodes. The calculated second time constant (CPE2 & R2) is smaller than the first time constant values. The physical meaning of the second charge (or mass) transfer process which changes with applied overpotential is not very well understood yet at Ni12P5 electrodes. Similar observations have also been reported in the literature for the 2TS model equivalent circuit analysis of metallic electrodes and porous electrodes.17,44,47–50 Of the investigated nickel phosphide based catalysts, Ni12P5 has the lowest mass-activity (see Fig. 6) as well as higher Tafel slopes. One possibility for this is the presence of a native thin oxide or phosphate coating on the Ni12P5 surfaces and the subsequent need to overcome this barrier for the HER. It is worth mentioning that Ni12P5 has the smallest average crystalline size and the highest stability in acidic medium as inferred from the previously discussed steady state voltammetry experiments, Fig. 3.
Conclusions
We have examined three different nickel phosphides of different compositions for both their passivity in acid and their activity in the hydrogen evolution reaction. In analysing these catalysts, it is important to understand their passivity across the entire potential range, as corrosion of electrocatalysts during periods of electrolyser inactivity, or during cell reversal will lead to oxidising potentials being applied to the hydrogen evolution catalyst.We have shown that by increasing the phosphorus content of nickel phosphides beyond the amount normally achieved in electrodeposited nickel phosphide films it is possible to extend the passivity of these films across the entire potential region (−0.45 to 1.1 V vs. RHE) expected in a solid polymer based electrolyser.
Ball milled Ni2P could be an excellent electrocatalyst for the hydrogen evolution reaction in acidic medium as inferred from the high exchange current density for the latter electrode and very small deviation in the Tafel slope with high transfer coefficients especially in the low overpotential region. Even though the Ni12P5 phase demonstrates very good passivity, it would appear that large amounts of the surface are inaccessible to the electrochemical reaction due to the formation of a blocking or passive layer. This is confirmed by the Tafel slope especially in the lower overpotential region, and the instability of Ni12P5 in acidic medium. This suggests that Ni12P5 is most likely inferior for hydrogen evolution compared to Ni2P based electrodes. An equivalent circuit model is proposed for both Ni2P and Ni12P5 based electrodes in acidic medium respectively based on either the two time constant parallel or serial model. The kinetics of the HER and probably the adsorption of hydrogen on the Ni2P based electrodes at large overpotentials contribute to the two time constants which change with the applied overpotential. On the other hand, even though Ni12P5 shows two charge transfer processes in the electrochemical impedance analysis, only one of those varies with potential and hence is associated with the kinetics of the HER.
Acknowledgements
The authors wish to acknowledge the UK Engineering and Physical Sciences Research Council for funding this work under grant EP/I013032/1.Notes and references
- M. Höök and X. Tang, Energy Policy, 2013, 52, 797–809 CrossRef PubMed.
- M. S. Dresselhaus and I. L. Thomas, Nature, 2001, 414, 332–337 CrossRef CAS PubMed.
- Y. Choquette, H. Menard and L. Brossard, Int. J. Hydrogen Energy, 1990, 15, 21–26 CrossRef CAS.
- S. Marini, P. Salvi, P. Nelli, R. Pesenti, M. Villa, M. Berrettoni, G. Zangari and Y. Kiros, Electrochim. Acta, 2012, 82, 384–391 CrossRef CAS PubMed.
- S. Dunn, Int. J. Hydrogen Energy, 2002, 27, 235–264 CrossRef CAS.
- M. Carmo, D. L. Fritz, J. Mergel and D. Stolten, Int. J. Hydrogen Energy, 2013, 38, 4901–4934 CrossRef CAS PubMed.
- S. A. Grigoriev, P. Millet and V. N. Fateev, J. Power Sources, 2008, 177, 281–285 CrossRef CAS PubMed.
- A. Döner, F. Tezcan and G. Kardaş, Int. J. Hydrogen Energy, 2013, 38, 3881–3888 CrossRef PubMed.
- S. Ardizzone, G. Fregonara and S. Trasatti, Electrochim. Acta, 1990, 35, 263–267 CrossRef CAS.
- I. Kodintsev and S. Trasatti, Electrochim. Acta, 1994, 39, 1803–1808 CrossRef CAS.
- J. Divisek, H. Schmitz and B. Steffen, Electrochim. Acta, 1994, 39, 1723–1731 CrossRef CAS.
- B. E. Conway and G. Jerkiewicz, Electrochim. Acta, 2000, 45, 4075–4083 CrossRef CAS.
- H. Ezaki, M. Morinaga and S. Watanabe, Electrochim. Acta, 1993, 38, 557–564 CrossRef CAS.
- J. G. Highfield, E. Claude and K. Oguro, Electrochim. Acta, 1999, 44, 2805–2814 CrossRef CAS.
- C. Hitz and A. Lasia, J. Electroanal. Chem., 2001, 500, 213–222 CrossRef CAS.
- W. Hu, Int. J. Hydrogen Energy, 2000, 25, 111–118 CrossRef CAS.
- M. Metikoš-Huković and A. Jukić, Electrochim. Acta, 2000, 45, 4159–4170 CrossRef.
- S. Trasatti, J. Electroanal. Chem., 1972, 39, 163–184 CrossRef CAS.
- E. Navarro-Flores, Z. Chong and S. Omanovic, J. Mol. Catal. A: Chem., 2005, 226, 179–197 CrossRef CAS PubMed.
- R. Karimishervedani and A. Rezamadram, Int. J. Hydrogen Energy, 2008, 33, 2468–2476 CrossRef CAS PubMed.
- C. C. Hu and A. Bai, J. Appl. Electrochem., 2001, 31, 565–572 CrossRef CAS.
- T. Burchardt, Int. J. Hydrogen Energy, 2000, 25, 627–634 CrossRef CAS.
- L. Vračar and B. E. Conway, Int. J. Hydrogen Energy, 1990, 15, 701–713 CrossRef.
- A.-M. Alexander and J. S. J. Hargreaves, Chem. Soc. Rev., 2010, 39, 4388 RSC.
- B. M. Barry and E. G. Gillan, Chem. Mater., 2008, 20, 2618–2620 CrossRef CAS.
- A. E. Henkes, Y. Vasquez and R. E. Schaak, J. Am. Chem. Soc., 2007, 129, 1896–1897 CrossRef CAS PubMed.
- M. V. Gerasimov and Y. N. Simirskii, Metallurgist, 2008, 52, 477–481 CrossRef CAS.
- D. C. C. S. Gosselink, PhD, University of Waterloo, 2006.
- E. J. Popczun, J. R. McKone, C. G. Read, A. J. Biacchi, A. M. Wiltrout, N. S. Lewis and R. E. Schaak, J. Am. Chem. Soc., 2013, 135, 9267–9270 CrossRef CAS PubMed.
- M. Popczyk, A. Budniok and A. Lasia, Int. J. Hydrogen Energy, 2005, 30, 265–271 CrossRef CAS PubMed.
- Y. You, C. Gu, X. Wang and J. Tu, J. Electrochem. Soc., 2012, 159, D642–D648 CrossRef CAS PubMed.
- M. Palaniappa, G. V. Babu and K. Balasubramanian, Mater. Sci. Eng., 2007, 471, 165–168 CrossRef PubMed.
- R. K. Shervedani and A. Lasia, J. Electrochem. Soc., 1997, 144, 2652–2657 CrossRef CAS PubMed.
- P. Xiao, M. A. Sk, L. Thia, X. Ge, R. J. Lim, J.-Y. Wang, K. H. Lim and X. Wang, Energy Environ. Sci., 2014, 7, 2624–2629 CAS.
- E. J. Popczun, C. G. Read, C. W. Roske, N. S. Lewis and R. E. Schaak, Angew. Chem., Int. Ed., 2014, 53, 5427–5430 CrossRef CAS PubMed.
- J. D. Benck, Z. B. Chen, L. Y. Kuritzky, A. J. Forman and T. F. Jaramillo, ACS Catal., 2012, 2, 1916–1923 CrossRef CAS.
- T. F. Jaramillo, K. P. Jorgensen, J. Bonde, J. H. Nielsen, S. Horch and I. Chorkendorff, Science, 2007, 317, 100–102 CrossRef CAS PubMed.
- Y. Ni, K. Liao and J. Li, CrystEngComm, 2010, 12, 1568 RSC.
- M. Sopicka-Lizer, High-energy Ball Milling: Mechanochemical Processing of Nanopowders, Woodhead Publishing Limited, 2010 Search PubMed.
- K. S. Venkataraman and K. S. Narayanan, Powder Technol., 1998, 96, 190–201 CrossRef CAS.
- J. L. Carbajal and R. E. White, J. Electrochem. Soc., 1988, 135, 2952–2957 CrossRef CAS PubMed.
- B. Elsener, D. Atzei, A. Krolikowski, V. Rossi Albertini, C. Sadun, R. Caminiti and A. Rossi, Chem. Mater., 2004, 16, 4216–4225 CrossRef CAS.
- B. Elsener, M. Crobu, M. A. Scorciapino and A. Rossi, J. Appl. Electrochem., 2008, 38, 1053–1060 CrossRef CAS.
- B. Losiewicz, Int. J. Hydrogen Energy, 2004, 29, 145–157 CrossRef CAS.
- A. Krolikowski and A. Wiecko, Electrochim. Acta, 2002, 47, 2065–2069 CrossRef CAS.
- C. Hausleitner and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 5689–5709 CrossRef CAS.
- M. Kramer and M. Tomkiewicz, J. Electrochem. Soc., 1984, 131, 1283–1288 CrossRef CAS PubMed.
- S. Omanovic and S. G. Roscoe, J. Colloid Interface Sci., 2000, 227, 452–460 CrossRef CAS PubMed.
- O. Savadogo and E. Ndzebet, Int. J. Hydrogen Energy, 2001, 26, 213–218 CrossRef CAS.
- R. Simpraga, G. Tremiliosi, S. Y. Qian and B. E. Conway, J. Electroanal. Chem., 1997, 424, 141–151 CrossRef CAS.
- B. Børresen, G. Hagen and R. Tunold, Electrochim. Acta, 2002, 47, 1819–1827 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Bode plot for data in Fig. 7(b) and the table of fitting parameters for EIS data in Fig. 7(a) and (b). See DOI: 10.1039/c4ta03468f |
| ‡ Current address: Department of Chemistry, Central University of Karnataka, Gulbarga, Karnataka, India |
| This journal is © The Royal Society of Chemistry 2014 |