New silica based polymeric systems designed for the solid–liquid extraction of uranyl ions
F.
Caprasse
a,
D.
Leroy
*a,
L.
Martinot
a,
S.
Lambert
b,
J. P.
Pirard
b,
J.
Guillaume
c,
C.
Jérôme
d and
R.
Jérôme
d
aCoordination and Radiochemistry, University of Liège, Bat B 16, Sart Tilman, 4000, Liège, Belgium
bApplied Physical Chemistry, University of Liège, Bat B 6, Sart Tilman, 4000, Liège, Belgium
cEngineering High School of Brussels, Brussels, Belgium
dCenter for Education and Research on Macromolecules (CERM), University of Liège, Bat B 6, Sart Tilman, 4000, Liège, Belgium
First published on 19th November 2001
Abstract
3-(Triethoxysilyl)propylacrylamide monomer was synthesized for the first time and then copolymerized with acrylamide (Aam), allyltriethoxysilane (TEA) or 3-(triethoxysilyl)propylacrylate (TMPAac). These three silylated copolymers were investigated as uranyl complexing agents. In another experiment, the copolymers were processed with tetraethylorthosilicate (TEOS) following a sol–gel process to prepare new microporous gels suited for solid–liquid uranium extraction from liquid wastes. The gels were prepared with uranyl as imprinted gels and without uranyl ions in solution to obtain non-imprinted gels. The effect on the uranyl binding capacities of the gels was studied. The imprinted gels were also dipped in ternary solutions of thorium, lanthanum and uranium. Selectivity toward uranyl was observed for uranyl imprinted gels. The stability of the different matrices against dynamic leaching and gamma irradiation was also studied.
Introduction
The use of polymers for the treatment of low level activity liquid wastes (LLW) appears to be a promising pathway.1In these processes, water soluble polymers are involved and they are selected depending on their capacity to complex UO22+ ions in aqueous acidic (HNO3) media.
In our laboratory, we measured the stability constant (K) of various complexes (polymer/UO22+ or monomer/UO22+) and we found that both poly(acrylamidomethylpropanesulfonic acid) (PAMPS) and poly(acrylamidoglycolic acid) (PAGA) are very promising materials.2 The insolubilisation of the complexes is achieved either by addition of a polyanion such as polystyrenesulfonate (PSSO3−Na+) or by doping a conducting polymer (PPy), which is insoluble in water, with the two quoted polymers.3 The potential of the final compound to fix uranium is ascertained by leaching tests (static or dynamic).
Another method for the treatment of liquid radioactive wastes has been patented4 recently by the French Commissariat à l′Energie Atomique (CEA). This process involves silica containing organic ligands such as amino, ether, hydroxy, amido or pyridino groups which are able to complex americium and plutonium. In these compounds the presence of the Si–O–Si backbone imparts good stability towards acidic solutions and γ irradiation. The preparation of these materials, by the sol–gel process, starting from R′(RO)2Si–O–Si(OR)3 (where R′ is a ligand function and R is ethyl or methyl) leads to a porous material suitable for the treatment of liquids. However, the total content of fixed metal ions (Am and Pu) remains weak (1–5 wt%)4 as compared to conducting polymer composites we had prepared (5–40 wt%).3 The slow hardening times of the gels, one month in some cases, also hinder their practical use.
With the aim of retaining the good stability of the silica matrix and to increase the capacity of uranyl fixation, we decided to replace the R′ complexing group by a macromolecule containing both complexing sites (amide functions) and silylated functions which will react in the sol–gel process. Results of uranyl complexation by three silylated comonomers (Fig 1) copolymerized with acrylamide (Aam): 3-(triethoxysilyl)propylacrylamide (TEPAam), Aam/TEPAam, 3-(trimethoxysilyl)propylacrylate (TMPAac), Aam/TEPAac and 3-(triethoxysilyl)allyl(allyltriethoxysilane) (TEA), Aam/TEA, are reported. In a second step, the most efficient copolymer (Aam/TEPAam) is incorporated in to a silica matrix by the sol–gel process and used for uranium extraction.
 | ||
| Fig. 1 Structures of the silylated (co)monomers TEPAam, TEA and TMPAac. | ||
The synthesis, characterization and testing of stability against leaching and gamma irradiation of this new material is then investigated (approach I).
In addition, with a view to the treatment of aqueous uranium solutions, Bristish Nuclear Fuel Ltd. (BNFL) patented a different process based on the polymer imprinting technique.5 In this process, chloroacrylic acid (a good ligand for UO22+) is reacted with a cross-linking agent (ethyleneglycoldimethacrylate: EGDMA) in the presence of UO22+ to produce an insoluble uranyl complex. Uranium is then removed by ultrasonification in concentrated HNO3. In this manner, calibrated pores corresponding to the UO22+ ion size are produced, giving to the material a selectivity towards uranyl which can be exploited for separation processes.
In the second part of this paper (approach II), the preparation of silica gels with the above quoted copolymers, in the presence of UO22+ ions, by the sol–gel technique is carried out in order to evaluate the possibility to create uranyl calibrated pores inside the material. The removal of UO22+ from the so-prepared material is carried out as in the BNFL process and the uranium enrichment of such gels from a UO22+–Th4+–La3+ ternary equimolar solution evaluated.
Our aim is thus the preparation of new materials by combining interesting features of both the CEA and BNFL patents. The capacity of these products to be suitable for waste disposals is checked by leaching tests and by measurements of stability to high dose gamma irradiation.
Experimental
Preparation of silylated monomers (TEPAam, TMPAac and TEA)
3-(Trimethoxysilyl)propylacrylate (TMPAac) (Aldrich: 47,514-9) and 3-(triethoxysilyl)allyl(allyltriethoxysilane) (TEA) (Aldrich: A3,630-1) are commercially available. 3-(Triethoxysilyl)propylacrylamide (TEPAam) was prepared by a condensation reaction between acryloyl chloride and 3-aminopropyltriethoxysilane in an ice bath. THF was dried from Na/benzophenone and distilled before use. Triethylamine (Aldrich: 47,128-3) was also distilled before use. The reaction between acryloyl chloride (Aldrich: A2-410-9) and 3-aminopropyltriethoxysilane (Aldrich: 28,177-8) was carried out with a tenfold excess of acryloyl chloride with respect to the amine, under dry atmosphere due to the sensitivity of the reagents and products toward hydrolysis. After 10 h of reaction, TEPAam was obtained with a reaction yield of 90–95% by evaporation of the solvent and of the excess of acryloyl chloride. Its structure was confirmed by 13C (Fig 2) and 1H NMR (Fig 3) and by mass spectrometry (molecular ion: m/z = 275).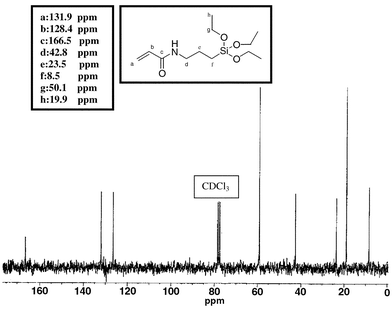 | ||
| Fig. 2 13C NMR spectrum of TEPAam. | ||
 | ||
| Fig. 3 1H NMR spectrum of TEPAam. | ||
Preparation of silylated copolymers
The three silylated comonomers (TEA, TEPAam and TMPAac) were copolymerized with acrylamide by classical radical polymerization in dry THF (nitrogen atmosphere, T = 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) °C, reflux condensor), with azobis(isobutyronitrile) (AIBN) as initiator (2 wt% based on total monomer concentration: 0.1 M). Three different comonomer concentration ratios were used for each copolymers preparation (50∶50, 80∶20 and 20∶80 mol%) so as to screen a wide coverage of feed compositions. The polymerization was stopped after 80% conversion by precipitation in heptane. The compositions of the three copolymers are listed in Table 1. The Aam/TEPAam and Aam/TMPAac copolymers were characterized by a high silylated comonomer content. As a consequence, these two copolymers were very sensitive to cross-linking and precipitation following
the hydrolysis of Si–OR functions. It was nevertheless possible to solubilize them again by an appropriate NaOH treatment.6 The copolymer Aam/TEA, in contrast to the two others, contains only 3 mol% of TEA and was less prone to spontaneous precipitation.
°C, reflux condensor), with azobis(isobutyronitrile) (AIBN) as initiator (2 wt% based on total monomer concentration: 0.1 M). Three different comonomer concentration ratios were used for each copolymers preparation (50∶50, 80∶20 and 20∶80 mol%) so as to screen a wide coverage of feed compositions. The polymerization was stopped after 80% conversion by precipitation in heptane. The compositions of the three copolymers are listed in Table 1. The Aam/TEPAam and Aam/TMPAac copolymers were characterized by a high silylated comonomer content. As a consequence, these two copolymers were very sensitive to cross-linking and precipitation following
the hydrolysis of Si–OR functions. It was nevertheless possible to solubilize them again by an appropriate NaOH treatment.6 The copolymer Aam/TEA, in contrast to the two others, contains only 3 mol% of TEA and was less prone to spontaneous precipitation.
The silylated monomer content in the copolymers was determined by thermogravimetric analysis (TGA) by considering the remaining SiO2 plateau (600–1000°C) at the end of the temperature scan.
Preparation of copolymer–silica gels (approach I)
The copolymer–silica gels were prepared in a PVC mold by addition of tetraethylorthosilicate (TEOS) to an aqueous solution of the copolymers (0.1 M) in order to achieve different polymer to silica ratios; various amounts of TEOS were added so that the silylated unit/TEOS ratio ranged from 0.01 to 1. A catalyst (NH4F or HCl: 0.05 M) was added in the second step (Table 2). Vigorous stirring was applied for 10 min, and the solutions allowed to settle until gelification occurred. The gel was allowed to stand for 24 h before being vacuum heated at 100°C for 3 h and was recovered as an insoluble powdery material.
It should be noted that since the copolymer solution is alkaline, resulting from the required NaOH treatment, the pH was adjusted to 1 with nitric acid before adding the TEOS . The mechanism of formation of the gel starting from the silylated copolymers and TEOS is shown in Fig 4.
 | ||
| Fig. 4 Formation scheme of gel based on Aam/TEPAam (HCl catalyst). | ||
Preparation of copolymer–silica gels in the presence of uranyl nitrate: imprinting (approach II)
The gels were also prepared in the presence of an excess of uranyl nitrate in the gelification solution. The copolymer was dissolved in UO22+ solution (0.1 M) and the pH is then adjusted to 1 by addition of HNO3. The uranyl nitrate was added in excess with respect to the copolymer content (0.5 M). Various amounts of TEOS were added so that the silylated unit/TEOS ratio ranged from 0.01 to 1, then HCl as a catalyst was added. After gelification, the gels were thoroughly leached with water and acetone in order to remove adsorbed uranyl ions before characterization. Finally, these gels can also be treated with nitric acid solution in an ultrasonification procedure to extract all the uranium and to be used further for testing their selectivity in separation. The characteristics of the preparation of these gels are thus the same as for the gel synthesis without uranyl in solution (Table 2).It should be stressed that we did not use any co-solvent such as ethanol, unlike many sol–gel processes,4 since our copolymers are only soluble in water or in THF. Our aim is the direct treatment of aqueous wastes by an easy work-out process. In this way, we cannot decrease extensively the hydrolysis ratio H as defined in eqn (1), since a minimum water volume of 10 ml is required to solubilize the silylated copolymers before adding the TEOS.
 | (1) |
The porosity, pore size distribution and the specific area of the gels were determined using nitrogen adsorption according to the classical BET method.7
Quantification of U in copolymers and gels was by neutron activation analysis (NAA) on 238U followed by gamma spectrometry.
The dynamic leaching tests were run in a Soxhlet extraction device with a volumic ratio of leaching water to gel equal to 50000 corresponding to 24 h non-stop extraction. The quantitative determination of uranyl leached out was carried out by ICP analysis.
Homogeneous gamma irradiation was achieved using a rotating tray irradiator surrounded by 40 rods of 137Cs (40 × 250 Ci, 2000 Gy h−1).
The uranium elution of the gels proceeds as follows: uranium containing gels were initially treated in HNO3 12% (vol.) in an ultrasonic cleaner (20 kHz) for 15 min according to the technique proposed by Saunders et al.1c
We measured that the removal was almost complete i.e. 99 wt% and the matrix remains suitable for the separation experiments.
Separation experiments
In order to evaluate the selectivity of the imprinted gels, Aam/TEPAam gels were immersed in an equimolar aqueous solution of UO22+, Th4+ and La3+ (0.05 M for each element). The cations are present in excess with respect to the gel capacity.After equilibration times ranging from 1 to 70 h without stirring, an aliquot of the solution was analyzed by ICP in order to determine the remaining concentrations of the three cations in the supernatent while considering that the total value remains constant. Then, in order to evidence which cation is preferentially bound to the gel, the three concentration ratios UO22+/Th4+, UO22+/La3+ and La3+/Th4+ fixed in the gel are determined by subtraction of the initial and final concentration of both numerator and denominator.
Results
Complexation of UO22+ by the copolymers
In order to investigate the UO22+ complexation ability of each copolymer, an aqueous solution of uranyl nitrate (0.5 M) was added to a solution of copolymer (0.1 M). Due to their high content of silylated monomer units, the Aam/TEPAam and Aam/TMPAac copolymers precipitate instantaneously, and so the complex is directly recovered by filtration. In the case of the Aam/TEA copolymer, addition of acetone was necessary to precipitate the complex PAam/TEA/UO22+. The molar ratio of fixed uranium to the number of amide functions for the system Aam/TEPAam (nU/ncomplexing functions = 0.5) is the same as that for pure PAam. It thus appears that the alkylsilylated amide function can complex uranyl ion as well as the primary amide of acrylamide (Aam) (Table 3).For the Aam/TEA system, the complexation is less effective. Of course, in this case, the allyltriethoxysilane monomer (TEA) is unable to complex uranyl ion but, in addition, it appears that the introduction of only 3 mol% of this comonomer in PAam decreases the complexing ability of some amide units of the polymer. Most probably, in accordance to the stoichiometry2 of the Aam/UO22+ complex (2∶1), two amide functions should be placed side by side to complex efficiently one uranyl ion. The statistical distribution of the alkylsilylated comonomer should thus perturb this regularity making some amide groups ineffective for complexation. This phenomenon is even more pronounced with the Aam/TMPAac system for which the U/amide molar ratio falls to 0.02; this shows also that the alkylsilylated acrylate does not complex the uranyl in the manner as the amide. This copolymer with a very low complexation ability is not suitable for our purposes. Even if the weight capacity of the PAam/TEA is as high as that of PAam/TEPAam, only few ethoxysilyl functions are available on this copolymer. This feature could be an important drawback for the gel preparation. Results concerning uranyl complexation by copolymers are gathered in Table 3.
Moreover, the stability of the cross-linked PAam/TEPAam/UO22+ to leaching is very important; most probably due to the high cross-linking degree, only 1.7% of the fixed uranium is leached out during the Soxhlet dynamic test. It thus seems that this PAam/TEPAam copolymer is the most promising for gel formation.
Approach I: uranyl binding in copolymer–silica gels
As described above, gels were prepared in aqueous solutions by reaction of the copolymer with TEOS in presence of HCl or NH4F. The Aam/TEA gel did not complex uranyl despite the high Aam content (70 wt%) of the copolymer. With this copolymer, which has a very low content of silylated monomer, the hydrolysis rate of the gel is very high, i.e. 3420. Indeed, the TEA/TEOS ratio is low (0.11) and the amount of water necessary to dissolve the copolymer is high, leading to a high hydrolysis ratio, which is known to hinder the formation of porous materials. After drying, this gel becomes very compact and cannot be swollen in aqueous medium, so that the uranyl ions cannot penetrate the gel and be fixed by the amide groups.The Aam/TEPAac gel revealed only a weak capacity to fix UO22+ ions: (0.1 meq UO22+/g of gel) with respect to the theoretical capacity i.e. the Aam content. Only 25% of the complexing functions react with the metal ions.
Thus, we focussed our attention mainly on the TEPAam/Aam based gel, since this copolymer is the most promising in terms of uranyl fixation.
In all these experiments, the contact time between the uranyl solutions and the gels was fixed at 24 h in order to allow the slow diffusion of the solution into micropores and so to establish contact between uranyl and the complexing sites. Results for various P(Aam/TEPAam) systems are listed in Table 4. For these compounds, the pore diameters ranged from 4 to 20 Å in contrast with data from Broudie et al.4 who reported larger diameters, ca. 40 to 90 Å for malonamide based sol–gels. This difference in pore size distribution arises from the difference in hydrolysis rate which is high for our totally aqueous system (microporous materials) but which is lower for systems prepared with co-solvents (micro-, meso- and macro-porous materials).
| Copolymer | Gel | [TEPAam]/[TEOS] | Copolymer in gel (wt%) | Hydrolysis ratio, H | U (wt% in gel) | U (wt% in copolymer) | Content/meq UO22+ g−1 of gel | Content/meq UO22+ g−1 of copolymer | Content/meq Aam g−1 of gel | n Aam/nUO22+ |
|---|---|---|---|---|---|---|---|---|---|---|
| Aam/TEPAam (25/75) | 1 | 0.063 | 20 | 50 | 0.3 | 1.5 | 0.025 ± 0.001 | 0.13 ± 0.01 | 0.39 ± 0.05 | 32 ± 2 |
| 2 | 0.11 | 55 | 350 | 2.5 | 4.5 | 0.2 ± 0.008 | 0.4 ± 0.04 | 1.1 ± 0.07 | 11 ± 0.4 | |
| 3 | 1.1 | 80 | 2000 | 8.8 | 11.2 | 0.7 ± 0.03 | 0.9 ± 0.1 | 1.6 ± 0.2 | 4 ± 0.3 | |
| 4 | 0.51 | 22 | 200 | 0.3 | 1.4 | 0.02 ± 0.001 | 0.1 ± 0.01 | 0.4 ± 0.05 | 40 ± 3 |
In the mesoporous malonamide based gels,4 the diffusion of the ions inside the matrix was slightly better than the diffusion of uranyl in our polymer based gels. In the best cases, one third of the complexing sites was saturated4cf. one fourth in our case.
From Table 4, it is seen that the greater the copolymer/TEOS ratio in the solution for gel preparation, the greater the copolymer content in the gel. Higher copolymer content in the initial solutions requires more water to dissolve them and thus higher hydrolysis rates occur. Thus gels with the highest copolymer content show the highest hydrolysis rates and the greatest uranium content (as high as 8.8 wt%). In this case, the UO22+/Aam molar ratio for the copolymers entangled in the gels reaches 0.25, cf. 0.5 for the Aam/TEPAam copolymers alone. This result indicates that diffusion is not markedly impeded in these microporous gels.
Nevertheless, the uranyl weight capacity of the gels prepared without uranyl ions in the synthesis bath remains quite low (0.5–9 wt%) when compared with Aam/TEPAam copolymer systems (30–35 wt%).
Approach II: synthesis of the gels in the presence of UO22+
For these nucleophilic reactions, HCl was used as a catalyst. The copolymers Aam/TMPAac (amide and acrylate functions), Aam/TEPAam (two amide functions) appeared of most interest due to their complexing groups. Relevent data are listed in Table 5.
It is found that the uranium content of the insoluble gels exceeds the uranium content of similar gels prepared in the absence of UO22+ ions; shorter solidification times are probably observed because of the increased ionic strength of the medium.6 It is noteworthy that the complexes Aam/UO22+ and TEPAam/UO22+ already form before the gel formation. Soxhlet leaching tests (Table 5) indicate a maximum uranium loss of about 30%. Gels prepared only with TEOS and no copolymer do not fix UO22+ ion (100% leaching after Soxhlet extraction).
000 Gy (2000 ± 10 Gy h−1 over 15 days; 10000 Ci of 137Cs). It is obvious that the gels are degraded. They are found to lose 25–100 wt% of their uranium content after irradiation and subsequent Soxhlet extraction, whereas the non-irradiated gels lost only about 20–30 wt% after leaching. Such data are absent in the literature and so direct comparisons with other systems cannot be made.
Subsequent tests with the irradiated compounds demonstrated that they were capable to fix again UO22+ from aqueous solutions. Unloaded irradiated gels were able to fix uranyl by a subsequent dipping in uranyl solutions and the gels charged in uranyl, which were further irradiated and submitted to dynamic leaching tests, were again able to fix uranyl by dipping. The uranyl capacities of these irradiated gels were slightly decreased (about 10%).
As explained above in the experimental section, Aam/TEPAam imprinted gels (approach II) and non-imprinted gels (approach I) were immersed in an equimolar aqueous solution of UO22+, Th4+ and La3+ (0.05 M for each element). For a sake of comparison, the same experiments were also carried out with the copolymer Aam/TEPAam and the non-imprinted gel prepared without uranyl (Table 6).
| Aam/TEPAam | Specific area (BET)/m2 g−1 | Microporous volume/cm3 g−1 | [UO22+]/[La3+] | [UO22+]/[Th4+] | [La3+]/[Th4+] |
|---|---|---|---|---|---|
| Ternary solution before treatment | 1.07 ± 0.05 | 1.22 ± 0.03 | 1.14 ± 0.05 | ||
| Copolymer | 0.54 ± 0.04 | 0.40 ± 0.01 | 0.72 ± 0.03 | ||
| Non-imprinted gel (approach I) | 427 | 0.230 | 3.3 ± 0.1 | 3.00 ± 0.02 | 0.90 ± 0.04 |
| Uranyl imprinted gel (approach II) | 791 | 0.508 | 10.3 ± 0.4 | 8.6 ± 0.2 | 1.00 ± 0.03 |
It is seen that the copolymer Aam/TEPAam preferentially extracts Th4+ from the ternary solution (Table 6), the high charge density of the Th4+ ion being favourable for a strengthened complexation. By contrast, the PAam/TEPAam gel (pore size: 4–10 Å) synthesized without uranyl (gel 5, Table 5) preferentially fixed UO22+ (Table 6).
This behavior is much more improved for the imprinted gels since the enrichment factor is three times higher.
A typical result is reported in Table 6. It appears that UO22+ is extracted from the solution while the ratio [La3+]/[Th4+] remains unchanged, the enrichment factor reaching one order of magnitude. The pore sizes of the compounds ranged between 4 and 10 Å with a narrow pore size distribution (Fig 5) and a high specific area: 650–790 m2 g−1 (BET measurements). It should be noted that the specific area of the imprinted gels was twice that of the gels prepared without uranyl ions, establishing the benefit of the imprinting technique.
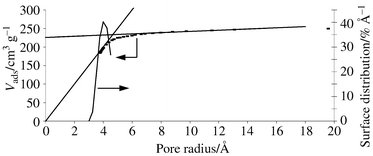 | ||
| Fig. 5 Pore size distribution of the gels. | ||
Clearly the imprinting effect originates from the gel structure since the free copolymer P(Aam/TEPAam) coordinates preferentially Th4+ > La3+ > UO22+. This trend is totally inverted for the imprinted gel, this new matrix becoming much more selective for the templated uranyl ions.
Conclusions
Silylated copolymers
The Aam/TEPAam copolymer has the same molar capacity to immobilize uranium as monomeric PAam. The silylated monomer content is greater than 50%.The complexes are stable enough to resist dynamic leaching tests and therefore, we presume that they are good media for the storage of uranium wastes. The silylated copolymers are interesting matrices for the treatment of LLW. The uranium content (range 2–37 wt%) is rather high when compared to the poly(EGDMA, chloracrylic acid) copolymer composite (5 wt%) described by Saunders et al.1c This difference arises from the level of cross-linking and from the activity of the cross-linking agent for uranyl complexation. In our case, the silylated copolymer plays the role of a cross-linking agent while also acting as a uranyl complexing agent. This feature leads to high uranyl content. In the BNFL process,1c the cross-linking agent ethyleneglycoldimethacrylate (EGDMA) or divinylbenzene are not able to complex uranyl ions.Gels synthesized by approach I (without uranyl)
The molar capacity of these gels with regard to uranium is low being 5 to 50 times lower than that of the corresponding copolymers. Even with a good resistance to leaching, uranium leached in Soxhlet tests does not exceed 20 wt% vs. the initial content, they are not good enough for waste applications. However, a solution may arise from preparing the gel in an organic co-solvent so as to decrease the hydrolysis rate and obtain a more widespread pore size (micro- and meso-pores).Gels synthesized by approach II (in UO22+ pre-existent solutions)
The uranium content in these are still lower than that of single copolymers but five times higher than found for gels via approach I. The resistance to leaching is an additional factor.Approach II: imprinting
Synthesis of the gels in presence of UO22+ (imprinting) doubles the specific area and the number of imprinted cavities, which since they correspond to the volume of UO22+ ions, allows a preferential fixation of uranium from a solution containing different metal ions. The enrichment factor can reach 20 with the Aam/TEPAam based gel, when investigating an equimolar solution of UO22+, Th4+and La3+ is concerned. A drawback is that the totally microporous structure of our gels impedes rapid separation procedures, however, the extension of the pore size distribution to reach the mesoporous region (Broudie et al.4) would be possible by controlling the hydrolysis rate by synthesis in non-aqueous solvents. A compromise should be found between efficacy in imprinting and separation. The kinetics of these separations ought to be improved by increasing the gel porosity.γ Irradiation
The choice of a suitable matrix for the storage of uranium depends on the results of γ radiation damage. Different gels TEPAam/TEOS were intensely irradiated (750000 Gy). We found that gels with a TEPAam/TEOS ratio around or greater than 0.5 were sensitive to irradiation while, with lower ratios, from 0.02 to 0.05 no visible changes were apparent. It is noteworthy that the γ irradiation of our composites corresponds approximately to 1000 Bq g−1. In turn an internal irradiation of 750000 Gy would correspond to a storage time of ∼9 × 1011 years. The leaching of uranium from irradiated gels (20–100%) was greater than that from non-irradiated gels (about 30%). The resistance of the gels towards irradiation was less robust than for more classical organic aromatic polymers such as polypyrrole.8
While the copolymers can fix more uranium than the gels the latter exhibit a better resistance to leaching. At the present state of research, the Aam/TEPAam system appears to be the most promising showing high uranium content and good stability against leaching.
Acknowledgements
D. L. and L. M. thank the IISN for financial support. C. J. thanks the Fonds National pour la Recherche Scientifique (FNRS) for a fellowship. C. J. and R. J. are indebted to the Services Fédéraux des Affaires Scientifiques Techniques et Culturelles for support to the CERM in the framework of the PAI: Supramolecular Catalysis and Supramolecular ChemistryReferences
-
(a) D. Leroy, L. Martinot, P. Mignonsin, F. Caprasse, C. Jérôme and R. Jérôme, Plutonium futures–the Science, Conference transactions, American Institute of Physics conference proceedings, 2000, 532, 84–85 Search PubMed;
(b)
P. H. Castle, P. Hills, J. D. B. Smith and D. C. Philips, Westinghouse
Electric Corp., Pittsburgh, US Pat., 5474688, Oct. 2, 1984 Search PubMed;
(c) D. S. Gregory, P. F. Simon, H. W. Paul, J. J. Malcolm and N. P. Simon, Chem. Commun., 2000, 273–274 RSC;
(d) D. Leroy, L. Martinot, C. Jérôme and R. Jérôme, J. Radioanal. Nucl. Chem., 1999, 240, 240, 867–875 Search PubMed.
- D. Leroy, L. Martinot, C. Jérôme and R. Jérôme, Polymer, 2001, 42, 4589–4596 CrossRef CAS.
- D. Leroy, L. Martinot, M. Debecker, D. Strivay, G. Weber, C. Jérôme and R. Jérôme, J. Appl. Polym. Sci., 2000, 77, 1230–1239 CrossRef CAS.
- J. C. Broudie, O. Conocar, J. E. Moreau, D. Meyer and M. Wong Chi Man, J. Mater. Chem., 1999, 9, 2283–2285 RSC.
- S. N. Port, J. M. Joyce, P. H. Walton and G. D. Saunders, PCT International Publication Number WO 99/15707.
- C. H. Brinker and G. W. Scherer, Sol–Gel Science, Academic Press, London, 1990 Search PubMed.
- S. Brunauer, P. H. Emmett and J. E. Teller, J. Am. Chem. Soc., 1938, 60, 309 CrossRef CAS.
- D. Leroy, Ph. D Thesis, University of Liège, Belgium, 2000.
| This journal is © The Royal Society of Chemistry 2002 |