Experimental determination of the hydrothermal solubility of ReS2 and the Re–ReO2 buffer assemblage and transport of rhenium under supercritical conditions†
Yongliang Xiong*a and Scott A. Wood*b
aDepartment of Geological Sciences, Rutgers, The State University of New Jersey, 610 Taylor Road, Piscataway, NJ 08854-8066, USA. E-mail: xiongyl@rci.rutgers.edu
bDepartment of Geological Sciences , University of Idaho, Box 443022, Moscow, ID 83844-3022, USA. E-mail: swood@uidaho.edu
First published on 11th January 2002
Abstract
To understand the aqueous species important for transport of rhenium under supercritical conditions, we conducted a series of solubility experiments on the Re–ReO2 buffer assemblage and ReS2. In these experiments, pH was buffered by the K–feldspar–muscovite–quartz assemblage; fO2 in sulfur-free systems was buffered by the Re–ReO2 assemblage; and fO2 and fS2 in sulfur-containing systems were buffered by the magnetite–pyrite–pyrrhotite assemblage. Our experimental studies indicate that the species ReCl40 is dominant at 400 °C in slightly acidic to near-neutral, and chloride-rich (total chloride concentrations ranging from 0.5 to 1.0 M) environments, and ReCl3+ may predominate at 500 °C in a solution with total chloride concentrations ranging from 0.5 to 1.5 M. The results also demonstrate that the solubility of ReS2 is about two orders of magnitude less than that of ReO2. This finding not only suggests that ReS2 (or a ReS2 component in molybdenite) is the solubility-controlling phase in sulfur-containing, reducing environments but also implies that a mixing process involving an oxidized, rhenium-containing solution and a solution with reduced sulfur is one of the most effective mechanisms for deposition of rhenium. In analogy with Re, TcS2 may be the stable Tc-bearing phase in deep geological repositories of radioactive wastes.
Introduction
There has been increasing interest in the aqueous geochemistry of rhenium because of its various important applications as summarized in Xiong and Wood.1,2 In our previous publications, the dominant oxidation state of rhenium in high-temperature hydrothermal solutions under geologically reasonable oxygen fugacity conditions was determined experimentally to be +4 by measuring the solubility of ReO2 as a function of oxygen fugacity.1 In the temperature range from 100 to 200 °C, Xiong and Wood2 suggest that the neutral species, formulated as Re(OH)40, is important over a wide range of pH, and is responsible for transport of rhenium leading to the enrichment of rhenium in various environments, including sandstone copper deposits and black shales.In this communication, we present our experimental data on the solubility of rhenium phases under supercritical conditions. The objective of our study is to assess probable rhenium species important under supercritical conditions (up to 510 °C). Based on findings from these experiments, we hope to provide a better understanding of the possible enrichment and deposition mechanisms for rhenium in various high-temperature environments. In particular, our treatment will focus on possible enrichment mechanisms for Re in porphyry copper–molybdenum and skarn molybdenum–(tungsten) deposits. In addition, our experimental results may also provide guidance to applications of the Re–Os system in high-temperature environments.
Methology
The methodology employed in our experiments under supercritical conditions has been described in detail elsewhere.1,3 The key components of the methodology employed are briefly summarized here.In our experiments, self-sealing reaction vessels with a volume of approximately 250 mL, lined with gold, and which permit withdrawal of samples at the temperatures and pressures of interest, were used. The pH and fO2 were buffered by thermodynamically well-characterized solid-phase assemblages such as K–feldspar + muscovite + quartz and Re–ReO2. K–feldspar, quartz, and muscovite were mixed in approximately a 1∶1∶1 mass ratio and the total mass was approximately 15 g. The source of these solid phases and their respective purities are given in detail elsewhere.1,3
Starting materials were metallic rhenium (99.995%, grain size smaller than 149 µm, minimum grain size ≥1.8 µm) and ReO2 (99.9%, grain size smaller than 44 µm, minimum grain size ≥1.8 µm) from Aldrich Chemical Company. The ReS2 (99%, minimum grain size > 1.8 µm) employed was from Johnson Matthey Company. Magnetite, pyrite and pyrrhotite (research grade) were natural samples and were from Ward's Natural Science Establishment, Inc. Starting materials were loaded in silver or gold capsules as in previous work.1
After the reaction vessels were assembled into the furnaces, compressed argon was used to check for leaks in the line. Next, the reaction vessels were purged with compressed argon at least three times to dislodge and remove any residual air in the reaction vessels. The vessels were then heated rapidly to the desired temperature and pressure in nichrome resistance furnaces. The amount of electric power provided to the top and bottom of the furnace could be adjusted separately so that the thermal gradient could be controlled precisely. Details of the temperature control and calibration of type K thermocouples used in our experiments can be found in Xiong and Wood.1,3 To homogenize the solutions in the vessel, convection was induced by maintaining the bottom of the vessel 2 °C higher than the top.
Pressure was measured to within ±10 bar using a pressure transducer and a digital pressure meter from Omega Engineering Company. To minimize the exposure of the transducer to corrosion, the vessel was connected with an Ashcroft Inconel 718 tube analogue gauge in some experimental runs, and the pressure was measured to within ±15 bar. Both the transducer and Ashcroft analogue gauge were calibrated against a Heise analogue gauge.
When sampling, supercritical conditions were assured by setting run temperatures and pressures according to the data of Sourirajan and Kennedy4 for the analogous NaCl system. Sampling details are similar to those of Xiong and Wood.1,3
The concentrations of Re were determined by inductively coupled plasma-atomic emission spectrometry (ICP-AES) with an axial-view torch (Perkin Elmer Optima 3000 XL). In order to minimize matrix effects, blanks and standard calibration solutions were precisely matched to the samples with respect to matrix. The correlation coefficients of the calibration curves were better than 0.999. The conservative detection limit for rhenium is estimated to be better than 14 ppb [∼7.5 × 10−8 mol (kg H2O)−1] based upon at least three replicate analyses of matrix-matched blanks. The analytical precision in terms of the relative standard deviation (RSD) of replicate analyses is better than 1.5%.
After each run, the starting phases, and the pH and fO2 buffer assemblages were examined by X-ray powder diffraction. No components of the buffers were observed to have been exhausted and no new phases appeared during any run, suggesting that buffered conditions were maintained.
In order to facilitate the interpretation of experimental results, the pH values and molar concentrations of major species in the KCl–H2O system at different KCl concentrations were calculated according to the following equations using the EQBRM computer code5 (Table 1). Equilibrium constants for these reactions were obtained from the SUPCRT92 database6 with updated parameters for relevant species for HKF equations from ref. 7–9
| Experimental run number | Oxygen fugacity buffer and experimental parameters | Sample number | Run time/h | Concentration from filtered sample/mol (kg H2O)−1 | Concentration from unfiltered sample/mol (kg H2O)−1 | |
|---|---|---|---|---|---|---|
| a Note: Mt—magnetite, Py—pyrite, Po—pyrrhotite. | ||||||
| Re–0.01MPP | Mt + Py + Po | Re–0.01MPP–1B | 28.75 | Not available | 4.60 × 10−6 | |
| 0.01 mol KCl | Re–0.01MPP–2B | 263.25 | 9.59 × 10−7 | 1.09 × 10−6 | ||
| 400 °C | Re–0.01MPP–3B | 340.5 | 5.13 × 10−7 | 6.23 × 10−7 | ||
| 550 bar | Re–0.01MPP-4B | 455.33 | 9.47 × 10−7 | 1.25 × 10−6 | ||
| ReS2 as starting | Re–0.01MPP–5B | 498 | 4.99 × 10−7 | 6.06 × 10−7 | ||
| Material | Re–0.01MPP–6B | 540.66 | 4.95 × 10−7 | 8.04 × 10−7 | ||
| Re–0.1MPP | Mt + Py + Po | Re–0.1MPP–1B | 71.25 | Not available | 3.92 × 10−4 | |
| 0.1 mol KCl | Re–0.1MPP–2B | 189.25 | 1.57 × 10−6 | 1.50 × 10−6 | ||
| 400 °C | Re–0.1MPP–3B | 238.25 | 6.38 × 10−7 | 1.26 × 10−6 | ||
| 550 bar | Re–0.1MPP–4B | 303.75 | 2.20 × 10−7 | 8.07 × 10−7 | ||
| ReS2 as starting | Re–0.1MPP–5B | 405 | 5.04 × 10−7 | 9.33 × 10−8 | ||
| Material | Re–0.1MPP–6B | 467 | 5.80 × 10−7 | 2.16 × 10−6 | ||
| OsO2–0.1RRO | Re–ReO2 | OsO2–0.1RRO–1B | 12 | Not available | 1.25 × 10−4 | |
| 0.1 mol KCl | OsO2–0.1RRO–2B | 61.75 | 1.67 × 10−4 | 1.88 × 10−4 | ||
| 400 °C | OsO2–0.1RRO–3B | 114.75 | 2.02 × 10−4 | 2.27 × 10−4 | ||
| 550 bar | OsO2–0.1RRO–4B | 179 | 2.76 × 10−4 | 3.21 × 10−4 | ||
| OsO2–0.1RRO–5B | 229.17 | 3.02 × 10−4 | 3.22 × 10−4 | |||
| OsO2–0.1RRO–6B | 276.5 | 2.86 × 10−4 | 2.91 × 10−4 | |||
| OsO2–0.1RRO–7B | 324 | 2.71 × 10−4 | 2.88 × 10−4 | |||
| OsO2–0.1RRO–8B | 394.83 | 3.16 × 10−4 | 3.30 × 10−4 | |||
| Re–0.75/4 | Re–ReO2 | Re–0.75/4–1B | 95.5 | 1.62 × 10−4 | 1.87 × 10−4 | |
| 0.75 mol KCl | Re–0.75/4–2B | 143.5 | 1.89 × 10−4 | 2.07 × 10−4 | ||
| 400 °C | Re–0.75/4–3B | 190.75 | 2.02 × 10−4 | 2.18 × 10−4 | ||
| 550 bar | Re–0.75/4–4B | 239.75 | 2.29 × 10−4 | 2.35 × 10−4 | ||
| Re–0.75/4–5B | 289.5 | 1.77 × 10−4 | 1.94 × 10−4 | |||
| Re–0.75/4–6B | 330 | 1.63 × 10−4 | 1.72 × 10−4 | |||
| Re–0.5 | Re–ReO2 | Re–0.5–1B | 121 | Not available | 3.92 × 10−6 | |
| 0.5 mol KCl | Re–0.5–2B | 193 | Not available | 4.97 × 10−6 | ||
| 410 °C | Re–0.5–3B | 247 | Not available | 3.31 × 10−6 | ||
| 350 bar | Re–0.5–4B | 295 | Not available | 5.66 × 10−6 | ||
| — | Re–0.5–5B | 361 | Not available | 1.08 × 10−5 | ||
| Re–0.5–6B | 457 | Not available | 1.18 × 10−5 | |||
| Re–0.5–7B | 528.5 | Not available | 9.22 × 10−6 | |||
| Re–0.5–8B | 607 | Not available | 1.36 × 10−5 | |||
| Re–0.5–9B | 654 | Not available | 1.43 × 10−5 | |||
| Re–0.5–10B | 751 | Not available | 9.54 × 10−6 | |||
| Re–0.5–11B | 796.5 | Not available | 1.22 × 10−5 | |||
| Re–1.0S | Re–ReO2 | Re–1.0S–1B | 143.83 | Not available | 4.95 × 10−5 | |
| 1.0 mol KCl | Re–1.0S–2B | 233.83 | Not available | 2.54 × 10−4 | ||
| 410 °C | Re–1.0S–3B | 333.60 | Not available | 3.10 × 10−4 | ||
| 800 bar | Re–1.0S–4B | 410.83 | Not available | 4.11 × 10−4 | ||
| Approaching | Re–1.0S–5B | 488.98 | Not available | 3.46 × 10−4 | ||
| Equilibrium | Re–1.0S–6B | 538.57 | Not available | 4.61 × 10−4 | ||
| From | Re–1.0S–7B | 679.23 | Not available | 5.30 × 10−4 | ||
| Lower Temp. | Re–1.0S–8B | 732.9 | Not available | 4.79 × 10−4 | ||
| Re–1.0S–9B | 795.58 | Not available | 5.09 × 10−4 | |||
| Re–1.0S–10B | 846.73 | Not available | 5.89 × 10−4 | |||
| Re–1.0S–11B | 942.82 | Not available | 2.80 × 10−4 | |||
| Re–1.0S–12B | 990.82 | Not available | 3.57 × 10−4 | |||
| Re–1.0S–13B | 1033.82 | Not available | 5.58 × 10−4 | |||
| Re–1.0S–14B | 1081.98 | Not available | 4.56 × 10−4 | |||
| Re–1.0S | Re–ReO2 | Re–1.0S–15B | 64.5 | Not available | 4.59 × 10−4 | |
| 1.0 mol KCl | Re–1.0S–16B | 136.67 | Not available | 2.50 × 10−4 | ||
| 410 °C | Re–1.0S–17B | 236.5 | Not available | 3.85 × 10−4 | ||
| 800 bar | Re–1.0S–18B | 303 | Not available | 2.09 × 10−4 | ||
| Approaching | Re–1.0S–19B | 353 | Not available | 3.59 × 10−4 | ||
| Equilibrium | Re–1.0S–20B | 423.67 | Not available | 5.62 × 10−4 | ||
| From | Re–1.0S–21B | 544 | Not available | 5.09 × 10−4 | ||
| Higher temp. | Re–1.0S–22B | 616 | Not available | 5.08 × 10−4 | ||
| Re–1.0S–23B | 663.5 | Not available | 4.68 × 10−4 | |||
| Re–1.0S | Re–ReO2 | Re–1.0S–24B | 167.5 | Not available | 5.82 × 10−4 | |
| 1.0 mol KCl | Re–1.0S–25B | 213.17 | Not available | 5.56 × 10−4 | ||
| 450 °C | Re–1.0S–26B | 262.5 | Not available | 5.47 × 10−4 | ||
| 800 bar | Re–1.0S–27B | 309.5 | Not available | 4.97 × 10−4 | ||
| Re–1.0S–28B | 359 | Not available | 3.78 × 10−4 | |||
| Re–1.0S–29B | 406.5 | Not available | 5.44 × 10−4 | |||
| Re–1.0S–30B | 454 | Not available | 5.54 × 10−4 | |||
| Re–1.0S–31B | 526 | Not available | 5.62 × 10−4 | |||
| Re–1.0S–32B | 574 | Not available | 5.06 × 10−4 | |||
| Re–1.0S–33B | 622 | Not available | 4.02 × 10−4 | |||
| Re–1.0S–34B | 670.25 | Not available | 3.41 × 10−4 | |||
| Re–1.0S–35B | 718.08 | Not available | 5.43 × 10−4 | |||
| Re–1.0S–36B | 722 | Not available | 7.32 × 10−4 | |||
| Re–0.5 | Re–ReO2 | Re—0.5–12B | 65.3 | Not available | 2.28 × 10−5 | |
| 0.5 mol KCl | Re–0.5–13B | 143.57 | Not available | 2.10 × 10−5 | ||
| 460 °C | Re–0.5–14B | 193.15 | Not available | 1.54 × 10−5 | ||
| 550 bar | Re–0.5–15B | 333.82 | Not available | 8.40 × 10−6 | ||
| Re–0.5–16B | 388.5 | Not available | 6.49 × 10−6 | |||
| Re–0.5–17B | 451.15 | Not available | 8.60 × 10−6 | |||
| Re–0.5–18B | 502.32 | Not available | 9.41 × 10−6 | |||
| Re–0.5–19B | 598.32 | Not available | 8.79 × 10−6 | |||
| Re–0.5–20B | 646.32 | Not available | 8.57 × 10−6 | |||
| Re–0.5–21B | 689.32 | Not available | 6.88 × 10−6 | |||
| Re–0.5–22B | 737.49 | Not available | 6.99 × 10−6 | |||
| Re–0.01RRO | Re–ReO2 | Re–0.01RRO–1B | 5 | Not available | 1.86 × 10−4 | |
| 0.01 mol KCl | Re–0.01RRO–2B | 9 | Not available | 3.14 × 10−4 | ||
| 500 °C | Re–0.01RRO–3B | 47 | 3.11 × 10−4 | 3.13 × 10−4 | ||
| 550 bar | Re–0.01RRO–4B | 94.5 | 2.91 × 10−4 | 3.05 × 10−4 | ||
| Re–0.01RRO–5B | 143.5 | 2.40 × 10−4 | 2.41 × 10−4 | |||
| Re–0.01RRO–6B | 190.5 | 2.44 × 10−4 | 2.52 × 10−4 | |||
| Re–0.01RRO–7B | 238.5 | 3.27 × 10−4 | 3.22 × 10−4 | |||
| Re–0.1RRO | Re–ReO2 | Re–0.1RRO–1B | 52 | Not available | 5.52 × 10−4 | |
| 0.1 mol KCl | Re–0.1RRO–2B | 69.5 | Not available | 5.89 × 10−4 | ||
| 500 °C | Re–0.1RRO–3B | 74.75 | Not available | 5.63 × 10−4 | ||
| 550 bar | Re–0.1RRO–4B | 93.5 | Not available | 6.68 × 10−4 | ||
| Re–0.1RRO–5B | 211.25 | 1.24 × 10−4 | 3.19 × 10−4 | |||
| Re–0.1RRO–6B | 328 | 1.89 × 10−4 | 2.02 × 10−4 | |||
| Re–0.1RRO–7B | 379.5 | 7.70 × 10−5 | 2.05 × 10−4 | |||
| Re–0.1MPP/2 | Mt+Py+Po | Re–0.1MPP/2–1B | 52.83 | Not available | 1.18 × 10−5 | |
| 0.1 mol KCl | Re–0.1MPP/2–2B | 70.33 | Not available | 5.76 × 10−6 | ||
| 500 °C | Re–0.1MPP/2–3B | 75.58 | Not available | 3.14 × 10−6 | ||
| 550 bar | Re–0.1MPP/2–4B | 94.33 | 7.75 × 10−7 | 8.44 × 10−7 | ||
| ReS2 as starting | Re–0.1MPP/2–5B | 212.08 | Below detection limit | Below detection limit | ||
| Material | Re–0.1MPP/2–6B | 328.83 | Below detection limit | Below detection limit | ||
| Re/Os–1.0RRO | Re–ReO2 | Re–1.0RRO–1B | 10.5 | Not available | 1.82 × 10−4 | |
| 1.0 mol KCl | Re–1.0RRO–2B | 99.5 | 1.83 × 10−4 | 2.04 × 10−4 | ||
| 500 °C | Re–1.0RRO–3B | 141.5 | 1.26 × 10−4 | 1.51 × 10−4 | ||
| 550 bar | Re–1.0RRO–4B | 202 | 1.01 × 10−4 | 2.07 × 10−4 | ||
| Re–1.0RRO–5B | 274 | 2.09 × 10−4 | 1.94 × 10−4 | |||
| Re/Os–1.5 | Re–ReO2 | Re/Os–1.5–1B | 172 | Not available | 4.67 × 10−4 | |
| 1.0 mol KCl | Re/Os–1.5–2B | 216.25 | Not available | 5.49 × 10−4 | ||
| 500 °C | Re/Os–1.5–3B | 264.67 | Not available | 8.44 × 10−4 | ||
| 800 bar | Re/Os–1.5–4B | 314 | Not available | 1.00 × 10−3 | ||
| Re/Os–1.5–5B | 359.67 | Not available | 1.17 × 10−3 | |||
| Re/Os–1.5–6B | 409 | Not available | 9.02 × 10−4 | |||
| Re/Os–1.5–7B | 456 | Not available | 1.07 × 10−3 | |||
| Re/Os–1.5–8B | 505.5 | Not available | 1.36 × 10−3 | |||
| Re/Os–1.5–9B | 552 | Not available | 1.41 × 10−3 | |||
| Re/Os–1.5–10B | 599.5 | Not available | 7.49 × 10−4 | |||
| Re/Os–1.5–11B | 648.5 | Not available | 1.30 × 10−3 | |||
| Re/Os–1.5–12B | 696.5 | Not available | 7.18 × 10−4 | |||
| Re/Os–1.5–13B | 744.5 | Not available | 7.46 × 10−4 | |||
| Re/Os–1.5–14B | 792.75 | Not available | 5.95 × 10−4 | |||
| Re/Os–1.5–15B | 840.58 | Not available | 6.93 × 10−4 | |||
| Re/Os–1.5–16B | 888.41 | Not available | 5.79 × 10−4 | |||
| Re/Os–1.5–17B | 913 | Not available | 1.00 × 10−3 | |||
| Re/Os–1.5–18B | 937.25 | Not available | 7.51 × 10−4 | |||
| Re-0.5 | Re–ReO2 | Re–0.5–23B | 53.5 | Not available | 2.52 × 10−5 | |
| 0.5 mol KCl | Re–0.5–24B | 122 | Not available | 8.17 × 10−6 | ||
| 510 °C | Re–0.5–25B | 169.83 | Not available | 5.84 × 10−6 | ||
| 750 bar | Re–0.5–26B | 216.5 | Not available | 5.25 × 10−6 | ||
| Re–0.5–27B | 288.67 | Not available | 8.78 × 10−6 | |||
| Re–0.5–28B | 388.5 | Not available | 7.21 × 10−6 | |||
| Re–0.5–29B | 456 | Not available | 9.35 × 10−6 | |||
| Re–0.5–30B | 506 | Not available | 1.04 × 10−5 | |||
Mass-action expressions were:
H2O(l)
![[left over right harpoons]](https://www.rsc.org/images/entities/char_21cb.gif) H+
+ OH− H+
+ OH− | (1) |
| KCl0
K+
+ Cl− | (2) |
| HCl0
H+
+ Cl− | (3) |
| KOH0
K+
+ OH− | (4) |
| 1.5 K–spar + H+
0.5 Mus + 3 Qtz + K+ | (5) |
| mH+ + mK+ = mCl− + mOH− | (6) |
| mΣCl = mCl− + mKClo + mHClo | (7) |
| log γi = −AZi2 I0.5/(1 + åiBI0.5) + bI | (8) |
In those experiments assessing the solubility of rhenium sulfide (ReS2, purity 99.5%, Johnson Matthey Co.) in sulfur-containing environments, fO2 and fS2 were buffered by the magnetite(Mt)–pyrite(Py)–pyrrhotite (Po) assemblage, as shown by the following reactions:
| 2FeS2
(Py)
2FeS (Po)
+ S2
(g) | (9) |
| 3FeS2
(Py)
+ Fe3O4
(Mt)
6FeS (Po)
+ 2O2
(g) | (10) |
The pH values fixed by the assemblage KMQ in our experiments range from 4.5 to 5.7 at around 400 °C and from 4.6 to 5.7 at around 500 °C, respectively (Table 1) (for reference, neutral pH at 500 °C and 800 bar is ∼6.0). The pH of the KMQ buffer in a 0.1 mol KCl solution at 500 °C and 550 bar is calculated to be 5.5 (neutral pH at 500 °C and 550 bar is ∼6.6) and in 0.1 mol KCl solution at 400 °C and 550 bar is calculated to be 5.0 (neutral pH at 400 °C and 550 bar is ∼5.6), respectively. Therefore, the experiments are all in the near-neutral to slightly acidic pH range.
Experimental design
The purpose of the present experiments was to determine the solubility of rhenium as a function of chloride concentration and pH. As we have demonstrated that, at geologically reasonable oxygen fugacity the dominant oxidation state of rhenium is +4,1 the general solubility reactions can be expressed as one of the following, the same as those suggested by Xiong and Wood,1 using ReO2 as starting material:| ReO2 (s) + nCl− + 4H+ = ReCln4 − n + 2H2O | (11) |
| ReO2 (s) + nCl− + 4(1 − 0.25p)H+ = Re(OH)pCln4(1 − 0.25p) − n + 2(1 − 0.5p)H2O | (12) |
| ReO2 (s) + nCl− + (2 − p) H+ = ReOCln(OH)p2 − p − n + (1 − p) H2O | (13) |
Using reaction (12) as an example, if the equilibrium constant for reaction (12) is expressed in logarithmic form and rearranged when temperature, pressure, and concentrations of chloride are constant, in log ΣRe versus pH space, the ligand number p for hydroxide can be evaluated according to the slope:
| (∂ log ΣRe/∂ pH)T,P,mCl− = −(4 − p) | (14) |
| (∂ log ΣRe/∂ log mCl−)T,P,pH = n | (15) |
Experimental results and interpretations
The results of experiments conducted under supercritical conditions are tabulated in Table 1 and plotted in Fig. 1. The molal concentrations of major species in each system as calculated using EQBRM (see above) are listed in Table 2. | ||
| Fig. 1 Rhenium concentration in equilibrium with the K–feldspar–muscovite–quartz and Re–ReO2 assemblages/ReS2 in KCl solutions. A Results at 400 °C unless otherwise labeled; B results at 450 °C unless otherwise labeled; and C results at 500 °C unless otherwise labeled. | ||
| KCl concentration, temperature, and pressure | pH | pOH | log mK+ | log mCl− | log mKClo | log mHClo | log mKOHo |
|---|---|---|---|---|---|---|---|
| 0.01 mol KCl, 400 °C, 550 bar | 5.7 | 5.6 | −2.0 | −2.0 | −3.1 | −5.4 | −5.3 |
| 0.1 mol KCl, 400 °C, 550 bar | 5.0 | 6.3 | −1.1 | −1.1 | −1.7 | −4.1 | −4.3 |
| 0.75 mol KCl, 400 °C, 550 bar | 4.5 | 6.8 | −0.23 | −0.23 | −0.80 | −3.2 | −5.9 |
| 0.5 mol KCl, 410 °C, 550 bar | 4.6 | 6.7 | −0.41 | −0.41 | −0.94 | −3.3 | −3.2 |
| 1.0 mol KCl, 410 °C, 550 bar | 4.5 | 6.8 | −0.096 | −0.096 | −0.71 | −3.1 | −2.4 |
| 1.0 mol KCl, 450 °C, 550 bar | 5.0 | 7.0 | −0.067 | −0.067 | −0.85 | −2.8 | −0.16 |
| 0.5 mol KCl, 460 °C, 550 bar | 5.0 | 6.9 | −0.43 | −0.43 | −0.90 | −2.8 | −1.4 |
| 0.01 mol KCl, 500 oC, 550 bar | 5.7 | 6.2 | −2.3 | −2.3 | −2.4 | −4.0 | −5.2 |
| 0.1 mol KCl, 500 °C, 550 bar | 5.2 | 6.7 | −1.5 | −1.5 | −1.2 | −2.8 | −4.6 |
| 1.0 mol KCl, 500 °C, 800 bar | 4.6 | 7.3 | −0.59 | −0.59 | −0.14 | −1.8 | −3.0 |
| 1.5 mol KCl, 500 °C, 800 bar | 4.6 | 7.3 | −0.38 | −0.38 | 0.026 | −1.6 | −2.5 |
| 0.5 mol KCl, 510 °C, 750 bar | 4.9 | 7.2 | −1.1 | −1.1 | −0.39 | −2.0 | −4.0 |
From Fig. 1 it is clear that equilibrium was attained after 400 h at 400 °C and 200 h at 500 °C. In comparison, it should be recalled that our experimental results presented elsewhere2 under subcritical conditions were buffered with respect to oxygen fugacity and pH by a fixed partial pressure of hydrogen and by aqueous pH buffers, respectively. These buffers equilibrate more quickly than solid assemblages adopted both in the present experiments and those involving platinum group elements (PGE) under supercritical conditions presented elsewhere.3 It can be seen from Fig. 1A and 1C that the concentrations of rhenium in experimental runs using ReS2 buffered by the Mt–Py–Po assemblage are about two orders of magnitude lower than those using the Re–ReO2 assemblage at the same temperature and chloride concentration. Experimental results show that there is good agreement of concentrations between filtered and unfiltered samples from experiments involving the Re–ReO2 assemblage (Fig. 1A–1C), implying that rhenium is truly in solution. However, there is some scatter between the concentrations from filtered samples and those from unfiltered samples in the experimental run involving ReS2 buffered by the Mt–Py–Po assemblage (Fig. 1A). This may be due to the fact that experimental runs involving ReS2 have lower concentrations of rhenium than those using the Re–ReO2 assemblage, resulting in a larger analytical uncertainty.
The average equilibrium concentrations (±two standard deviations) are listed for each experiment in Table 3. All of the data after 200 h and 400 h are included in the calculation of the average equilibrium concentrations at 500 and 400 °C, respectively.
| Experimental run number | T /°C | Experimental conditions | Average equilibrium concentration (mol/kg H2O) with 2σ |
|---|---|---|---|
| Re–0.01MPP | 400 °C | Mt + Py + Po buffer, 0.01 mol KCl, 550 bar, ReS2 as starting material | 7.5 ± 2.8 × 10−7 |
| Re–0.1MPP | 400 °C | Mt + Py + Po buffer, 0.1 mol KCl, 550 bar, ReS2 as starting material | 1.2 ± 0.6 × 10−6 |
| OsO2–0.1RRO | 400 °C | Re–ReO2 buffer, 0.1 mol KCl, 550 bar | 3.0 ± 0.4 × 10−4 |
| Re–0.75/4 | 400 °C | Re–ReO2 buffer, 0.75 mol KCl, 550 bar | 1.9 ± 0.4 × 10−4 |
| Re–0.5 | 410 °C | Re–ReO2 buffer, 0.5 mol KCl, 350 bar | 1.2 ± 0.4 × 10−5 |
| Re–1.0S | 410 °C | Re–ReO2 buffer, 1.0 mol KCl, 800 bar | 5.1 ± 0.6 × 10−4 |
| Re–1.0S | 450 °C | Re–ReO2 buffer, 1.0 mol KCl, 800 bar | 5.2 ± 2.2 × 10−4 |
| Re–0.5 | 460 °C | Re–ReO2 buffer, 0.5 mol KCl, 550 bar | 8.0 ± 2.0 × 10−6 |
| Re–0.01RRO | 500 °C | Re–ReO2 buffer, 0.01 mol KCl, 550 bar | 2.9 ± 0.6 × 10−4 |
| Re–0.1RRO | 500 °C | Re–ReO2 buffer, 0.1 mol KCl, 550 bar | 2.1 ± 1.1 × 10−4 |
| Re–0.1MPP/2 | 500 °C | Mt + Py + Po buffer, 0.1 mol KCl, 550 bar, ReS2 as starting material | 3.2 ± 2.0 × 10−6 |
| Re/Os–1.0RRO | 500 °C | Re–ReO2 buffer, 1.0 mol KCl, 550 bar | 2.0 ± 0.1 × 10−4 |
| Re/Os–1.5 | 500 °C | Re–ReO2 buffer, 1.5 mol KCl, 800 bar | 9.1 ± 2.0×10−4 |
| Re–0.5 | 510 °C | Re–ReO2 buffer, 0.5 mol KCl, 750 bar | 8.2 ± 3.6 × 10−6 |
Ideally, the chloride- and pH-dependence of the solubility of rhenium is independently evaluated according to eqn. (11) and (12), as we did under subcritical conditions.2 However, because the KMQ buffer was adopted in our experiments under supercritical conditions, pH and chloride dependence cannot be evaluated independently; i.e., the coefficient p in reaction (12) cannot be evaluated by keeping chloride concentration constant while pH is changed independently. In this communication, we assumed that mixed chloride complexes with hydroxyl suggested in reaction (12) or (13) are not important in the present experiments considering the conditions of the high temperatures, relatively high concentration of chloride, and slightly acidic to near-neutral pHs. This assumption is mainly based on the findings of previous studies which demonstrated increased stability of chloride complexes, simplified speciation, and the unimportance of mixed complexes at high temperatures (i.e., 500 °C).16,17 Therefore, the interpretation of chloride dependence is based on reaction (11). However, it should be cautioned that this assumption is subject to experimental verification in the future.
To evaluate the chloride-dependence of the solubility of the assemblage Re–ReO2, log mΣRe is plotted versus log mCl− at constant pH in accordance with eqn. (15). For this purpose, the rhenium concentration data from Re–0.5, Re–0.75/4 and Re–1.0S (Table 3) are plotted against log mCl− (Fig. 2). Note that the pH is almost constant (from 4.5 to 4.6) in these experimental runs (Table 1), but the measured Re concentrations were corrected for pH differences assuming the pH-dependence indicated by reaction (11). As the slope of a straight line fit to the data in Fig. 2 is close to 4 (3.94 ± 0.35, R2 = 0.99), the dominant species appears to be ReCl40 according to reaction (16):
| ReO2 (s) + 4Cl− + 4H+ = ReCl40 + 2H2O | (16) |
 | ||
| Fig. 2 Plot of logarithmic rhenium concentration (corrected for pH variations) versus logarithmic chloride concentration at 400 °C. Only experimental runs involving Re–ReO2 assemblage were considered and pH was almost constant in these runs. | ||
It should be mentioned that we attach considerable uncertainty to the interpretation of the chloride dependence of the experimental data at 500 °C because the variation in pH is 0.3 log units for these experiments. The solubility correction introduced by such pH differences should be more than one log unit according to reaction (11), which may result in large uncertainties in the chloride ligand number when determined as described in the preceding paragraph. The slope determined with the pH correction according to reaction (11) is 1.28 ± 0.16 (R2 = 0.985) (Fig. 3), whereas the slope determined without the pH correction is 2.99 ± 0.016 (R2 = 0.99997) (Fig. 4). Judging from the results at 400 °C and at lower temperatures,2 a slope of ∼3 (corresponding to ReCl3+) is more likely than a slope of ∼1 (corresponding to ReCl+3) at 500 °C. This interpretation is also supported by the relatively small variation of calculated equilibrium constants as a function of ionic strength (see below) . However, as the pH differences are relatively large in these experiments, more experiments are certainly needed to refine the interpretations at 500 °C.
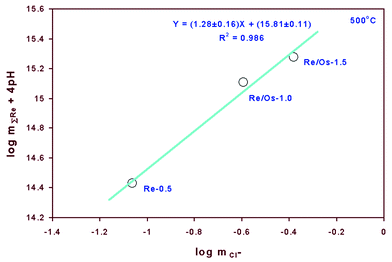 | ||
| Fig. 3 Plot of logarithmic rhenium concentration (corrected for pH) versus logarithmic chloride concentration for solutions in equilibrium with Re and ReO2 at 500 °C. | ||
 | ||
| Fig. 4 Plot of logarithmic rhenium concentration (without pH corrections) versus logarithmic chloride concentration for solutions in equilibrium with Re and ReO2 at 500 °C. | ||
It is of interest to note that there is a negative dependence on temperature in the temperature range from 400 to 500 °C in the same solution, implying that there is a retrograde solubility of the Re–ReO2 buffer assemblage. This can be demonstrated by examining the results from OsO2–0.1RRO at 400 °C and Re–0.1RRO at 500 °C. The former has an average equilibrium concentration of 3.0 ± 0.4 × 10−4 molal, whereas the latter has an average equilibrium concentration of 2.1 ± 1.1 × 10−4 molal. This trend is also suggested by the Re–0.5 runs at 410 and 510 °C, and by the Re–1.0S run (1.0 mol KCl) at 410 °C and the Re/Os–1.0RRO run (1.0 mol KCl) at 500 °C. In addition to higher calculated pH at 500 than at 400 °C, this may be due to the combination of decreasing free chloride concentration with decreasing chloride ligand number with temperature. For example, in a 1.0 mol KCl solution, the concentration of free chloride decreases from about −0.096 log units at 410 °C to about −0.59 log units at 500 °C (Table 1), i.e., a decrease of ∼0.6 log units. This decrease in the concentration of free chloride with increasing temperature may also be responsible for the apparent decrease in chloride ligand number as temperature increases from 400 to 500 °C (see the preceding paragraph). On the other hand, the solubility of ReS2 shows a slight increase in solubility between 400 and 500 °C (cf., Re–0.1MPP at 400 °C and Re–0.1MPP/2 at 500 °C).
Derivation of equilibrium constants. In order to calculate thermodynamic equilibrium constants according to reaction (16), the activity coefficient of the neutral species, ReCl40, is assumed to be unity. As mentioned before, the activities of other species were calculated by using the extended Debye–Hückel equation and EQBRM code. The thermodynamic equilibrium constants for reaction (16) at 400 °C at different ionic strengths are listed in Table 4. At 500 °C, the species ReCl3+ was identified tentatively as the predominant species. The thermodynamic equilibrium constant for reaction (17) at 500 °C
| ReO2 (s) + 3Cl− + 4H+ = ReCl3+ + 2H2O | (17) |
| Reaction | Temperature | Experimental run | Ionic strength | log K |
|---|---|---|---|---|
| ReO2 (s) + 4Cl− + 4H+ = ReCl4o + 2H2O | 400 °C | Re-0.5 | 0.39 | 17.96 |
| Re–0.75/4 | 0.59 | 18.48 | ||
| Re–1.0S | 0.80 | 18.73 | ||
| Average | 18.39 ± 0.64 (2σ) | |||
| ReO2 (s) + 3Cl− + 4H+ + H2O = ReCl3+ + 2H2O | 500 °C | Re–0.5 | 0.086 | 18.69 |
| Re/Os–1.0RRO | 0.25 | 18.26 | ||
| Re/Os–1.5 | 0.42 | 18.62 | ||
| Average | 18.52 ± 0.47 (2σ) |
Implications
The above results indicate that, in high-temperature environments, appreciable amounts of rhenium can be dissolved in chloride-containing (log mCl− = −0.4 to −0.1) aqueous fluids as the neutral chloride species, ReCl40. It is also of importance to note that ReS2 is much less soluble than ReO2 (about two orders of magnitude lower). These conclusions have bearing on the transport and deposition of rhenium in various environments and our data are the first step in the quantitative elucidation of origins of rhenium deposits in these environments. In the following, we apply our results to various environments and explore their implications.As mentioned above, the solubility of ReS2 is about two orders of magnitude lower than that of ReO2. This has important implications. First of all, it is apparent that rhenium will be stabilized as a sulfide phase(s) in environments where reduced sulfur is present. This explains why rhenium sulfides occur in a wide range of environments including mafic and ultramafic complexes such as the Stillwater Complex18 and fumarolic deposits from volcanoes,19 whereas sulfur-free rhenium phases have limited occurrences. Second, it can be inferred that samples which have been subject to alteration by oxidizing fluids are likely to have experienced rhenium remobilization, and therefore they are not good candidates for Re–Os radiogenic investigations. Third, it can be deduced that reducing fluids containing sulfur have much less capacity for transporting rhenium, and therefore environments dominated by reducing fluids are not favorable to formation of rhenium deposits of importance. To form rhenium deposits of significance, oxidizing fluids must be operative at some stage(s) of ore formation. On the other hand, reducing fluids have the least ability to disturb the Re–Os system. In the following, transport of rhenium by hydrothermal fluids is addressed quantitatively.
Quantitative assessment of potential size of rhenium mineralization in porphyry copper–molybdenum and skarn tungsten–molybdenum systems
Molybdenite in porphyry copper–molybdenum and skarn tungsten–molybdenum systems has been observed to be enriched in Re.20–28 It is of special interest to note that existing reserves of rhenium indicate that porphyry copper–molybdenum systems are very important.23 In the following, we first present field observations on rhenium mineralization in skarn tungsten–molybdenum and porphyry copper–molybdenum systems and then compare these observations with our quantitative predictions based on our experimental data.Field observations: The Little Boulder Creek skarn molybdenum deposit in Idaho can be used as an example to estimate the size of skarn Mo–(W) systems and the amount of rhenium mineralization. The reserves of ores with a grade of 0.15 wt% of MoS2 are 167 million tons.29 Although the rhenium concentration in molybdenite in this deposit is not available, we may assume that the rhenium concentration in molybdenite in this deposit is similar to the rhenium concentration in other skarn Mo–(W) deposits. For example, Stein et al.29 determined that the rhenium concentration in molybdenite in the Schwartz, Klee and Meyer skarn Mo–W deposits in the Pitkaranta district, Russia, ranges from 130 to 479 ppm with an average of 224 ppm. Assuming the rhenium concentration in molybdenite in the Little Boulder Creek deposit is of the order of 100 to 200 ppm, the mass of rhenium in this deposit is estimated to be from ∼2.5 × 107 to ∼5 × 107 grams (Table 5).
| Predictions and field observations | T/°C | Solubility controlling phase | logfO2 | logfS2 | pH | log aCl− | Log aΣS | Transportable re assuming saturation with solid phase in grams | Remarks and references for thermodynamic parameters |
|---|---|---|---|---|---|---|---|---|---|
| a Magnetite–hematite buffer.b PP: pyrite–pyrrhotite buffer.c KMQ: K–feldspar–muscovite–quartz buffer.d MPP: magnetite–pyrite–pyrrhotite buffer.e WQ: wollastonite–quartz buffer, which should buffer pH values in near-neutral range as mentioned in the text.f Converted from salinity ranging from >60 wt% NaCl eq. to ∼8 wt% NaCl eq.g As log mFeSO4, King Island. | |||||||||
| This study | 400 to 500 | ReS2 | −26.9 to −21.6 | −7.6 to −4.8 | 5.0, KMQ to 5.2, KMQ | −1.5 to −2.3 | −1. 2 to 0 | Typical porphyry Cu–Mo systems: ∼2 × 108 to ∼6 × 108 (∼2 × 1010 to ∼6 × 1010 for giant porphyry Cu–Mo systems) | Log fO2, logfS2 and log aΣS are calculated from MPP bufferd |
| Skarn W–Mo systems: ∼4 × 107 to ∼1.2 × 108 | |||||||||
| Porphyry copper–molybdenum system | 350 to 550 | Solid solution of ReS2 and MoS2 | −26.5 to −16.5a | PP bufferb | KMQ bufferc | −1.0 | −1.0 | ∼4 × 1010 (El Teniente, giant porphyry Cu–Mo) | ref. 40 |
| ∼8 × 106 (La Disputada, typical porphyry Cu–Mo) | |||||||||
| Skarn tungsten–molybdenum system | 400 to 650 | Solid solution of ReS2 and MoS2 | −24.1 to −22.1 | −8.2 to −6.25 | KMQ and WQ bufferse | 0.17 to >1.4 (log mNaCl)f | ∼−0.4g | ∼4 × 107 | ref. 32 |
In the El Teniente porphyry Cu–Mo mine, the grade of rhenium in ores is 10 ppm (10 grams tonne−1).23 As ore reserves are estimated to be about 3.5 billion tonnes, the total mass of rhenium in the ore reserves is about 3.5 × 1010 grams.23 In addition, this deposit has about 4 × 108 tonnes of tailings in its waste dumps with a grade of 9 grams tonne−1 of rhenium.23 Therefore, the total rhenium in this deposit is ∼4 × 1010 grams (Table 5).
Predictions: In order to determine, from our experimental solubility data, the total amount of Re that can be transported in porphyry Cu–Mo and skarn Mo–W deposit-forming systems, the total amount of fluid involved in these systems must be estimated. The total amount of Re predicted based on our experimental results can then be compared to the amounts calculated in the preceding section from grade and tonnage data.
The amount of fluid involved in a typical porphyry Cu–(Mo) system has been estimated to be ∼1015 grams using the average tonnage of copper (∼1 million tonnes) and the copper concentrations in ore-forming fluids (1000 ppm).3 In giant porphyry Cu–Mo systems such as El Teniente porphyry Cu–Mo mine, the total mass of fluid involved may be one to two orders of magnitude higher than that in a typical porphyry Cu–Mo system. For example, the tonnage of copper in El Teniente porphyry Cu–Mo mine is at least 31.5 million tonnes.24 Therefore, a total mass of fluids of ∼1017 grams may be reasonable. The pH of porphyry Cu–(Mo) systems is likely to be buffered by the KMQ assemblage.3 Other thermodynamic parameters in porphyry Cu–(Mo) systems employed in the present predictions have been described in Xiong and Wood.3
In order to estimate the total mass of fluids involved in the formation of a typical skarn tungsten–molybdenum system, the calculated scheelite solubility (39 ppm) in 1.0 mol NaCl solution at 600 °C and 2000 bar of Wood and Samson31 is used. This calculated value has been demonstrated to be in excellent agreement with an experimentally measured value.31 The Molyhill skarn W–Mo deposit in the Northwestern Territory, Australia, has reserves of 1.5 million tons of ores with a WO3 grade of 1.0 wt%.32 Apparently, the mass of tungsten in the Molyhill deposit is 1.2 × 1010 grams. The total mass of fluid involved should be ∼3.0 × 1014 grams according to the solubility of scheelite cited above, assuming that all the tungsten in the fluid is deposited. The Osgood Mountains W–Mo skarns in Humbolt County, Nevada, USA, have reserves of 1.4 million tons of ores with a WO3 grade of 0.45 wt%.32 The mass of tungsten in this deposit is 5 × 109 grams. Therefore, the estimated total mass of fluid involved should be ∼1.3 × 1014 grams. Consequently, based on the estimated total mass of fluids involved in these two deposits, we assume that the total mass of fluid involved in the formation of a typical W–Mo skarn system should be in the neighborhood of ∼2 × 1014 grams.
The pH of skarn W–Mo systems is likely to be buffered by various assemblages such as the wollastonite–quartz assemblage, which is common in the distal zone of W–Mo skarns:
| CaSiO3 (wollastonite) + 2H+ = Ca+2 (aq) + SiO2 (quartz) + H2O | (18) |
Although the wollastonite–quartz assemblage has not been deliberately calibrated as a pH buffer, it has been reasoned that this assemblage should buffer pH values in the near-neutral range,33 which is close to our experimental pH values. In the endoskarn zone, where major W–Sn mineralization occurs, the pH of initial mineralizing fluids exsolved from crystallizing (granitoid) magmas should be buffered by mineral assemblages of granitoid rocks such as quartz + K–feldspar + muscovite. For example, in the endoskarn of the Sangdong deposit, Korea, biotite skarn and muscovite skarn are dominant.32 As the quartz + K–feldspar + muscovite assemblage was employed as a pH buffer in our experiments, the pH values in our experiments should be most likely to resemble those in the early stages of mineralizing fluids.
It has been documented that fluids responsible for proximal skarns with which W–Mo mineralization is generally associated have salinities ranging from >60 wt% NaCl eq. to ∼8 wt% NaCl eq.,32 which correspond to concentrations of NaCl ranging from ∼1.5 M to higher than ∼26 M. Fluids responsible for distal skarns commonly have lower salinities ranging from ∼12.5 to ∼1.25 wt% NaCl eq.,32 corresponding to concentrations of NaCl from ∼0.2 to ∼2.5 M. Obviously, these salinities, especially those in fluids responsible for proximal skarns, are generally higher than those in our experiments. In this respect, our solubility data yield minimum estimates of the amount of Re that can be transported by these hydrothermal systems (but see comments below regarding the occurrence of ReS2 in solid solution).
Assuming the solubility-controlling phase is pure ReS2 and using the estimated total masses of fluids in typical porphyry Cu–(Mo) and skarn W–Mo systems mentioned above, when we apply our solubility data solutions with total chloride concentration of 0.1 M (Table 3) at 400 °C (1.2 × 10−6 mol kg−1 H2O) and at 500 °C (3.2 × 10−6 mol kg−1 H2O) to the above systems, the rhenium that can be transported by typical porphyry Cu–Mo and skarn W–Mo systems ranges from ∼2 × 108 to ∼6 × 108 and from ∼4 × 107 to ∼1.2 × 108 grams, respectively (Table 5). It should be emphasized that the experimental conditions are comparable to those conditions prevailing in porphyry Cu–Mo and skarn W–Mo systems (Table 5), except that the salinities in our experiments are somewhat lower. Therefore, direct application of our experimental results using ReS2 as the starting material and magnetite–pyrite–pyrrhotite assemblage to buffer fO2 and fS2 is well justified. However, the ReS2 used in our experiments is a pure end-member. In the natural systems, it is likely that a solid solution between MoS2 and ReS2 would control the solubility of rhenium as suggested in Table 5. It is well known that the solubility of a component in solid solution is generally less than that of the pure end-member phase because the activity of the solid solution is less than unity.34 Although the concentrations of rhenium in molybdenite (MoS2) have been determined in several deposits as mentioned above, the activity coefficients of ReS2 in MoS2–ReS2 solid solution system are not known. Therefore, the activity of ReS2 in such solid solutions cannot be quantitatively evaluated at present. However, it can be expected that pure ReS2 should have higher solubility than the solid solution between MoS2 and ReS2. Notice that our predicted tonnage of rhenium mineralization for skarn W–Mo systems is on the same order of magnitude as the observed tonnage in a typical skarn W–Mo systems (Table 5). Our predicted tonnage of rhenium mineralization for porphyry Cu–Mo systems is about one order of magnitude higher than the observed tonnage in typical porphyry systems (such as La Disputada, Chile), but is on the same order of magnitude as the tonnage observed in the giant porphyry Cu–Mo systems (e.g., El Teniente, Chile) (Table 5). Therefore, when the fact that solid solution of MoS2 and ReS2 controls solubility is taken into account, our experimental results are in excellent agreement with field observations and our results for giant porphyry Cu–Mo systems (Table 5) predict that the El Teniente deposit-forming fluid was probably close to saturation with the ReS2 component in solid solution with molybdenite.
In the Chenmanshan skarn and porphyry deposit (where molybdenite is observed) in Juijiang, Jiangxi Province, China, the grade of rhenium is 6 × 10−5%.35 The reserves of Cu in this deposit were 2.1 × 106 tons36 and the grade of Cu was 0.75%.35 Therefore, the total amount of ore is estimated to be ∼2.8 × 1014 grams. Hence, the amount of Re that had been transported is computed to be ∼108 grams. In comparison with the above minimum mass of Re that can be transported (∼2 × 108 to ∼6 × 108 grams) in a typical porphyry system, the Chenmanshan porphyry deposit-forming fluid may have also been saturated with ReS2 or its solid solution with other sulfides.
Deposition of rhenium
As the stabilities of the predominant rhenium complexes deduced from our experiments, i.e., ReCl40 and ReCl3+, have strong dependences on chloride concentration, we can infer that dilution owing to mixing of high-concentration chloride fluids with low-chloride fluids is one of most effective mechanisms for the deposition of rhenium from porphyry systems. It also needs to be realized that, according to reactions (16), (17), (19) and (20)| ReS2 + 4H+ + nCl− ↔ ReCln4 − n + 2H2S0 | (19) |
| ReS2 + 2H+ + nCl− ↔ ReCln4 − n + 2HS− | (20) |
The slight prograde solubility of ReS2 in the temperature range from 400 to 500 °C means that more rhenium sulfide is dissolved at higher temperatures. This is different from the Re–ReO2 assemblage in sulfur-free environments, which shows a slight retrograde solubility in the temperature range from 400 to 510 °C. The slight prograde solubility of ReS2 might explain the puzzling observation that molybdenite deposited at higher temperatures has lower rhenium concentration (i.e., lower content of ReS2).20,37 This explanation is contingent upon a weak or retrograde temperature dependence of molybdenite solubility over the above temperature range. However, regarding the solubility of molybdenite in hydrothermal solutions, a consistent picture has not emerged.38
The much lower solubility of ReS2 compared to that of the Re–ReO2 assemblage also has implications to disposal of fissiogenic 99Tc, for which Re is a good analogue.39 As ReS2 is the stable phase in most subsurface geological environments, TcS2 should be the preferred phase in deep geological repositories for radioactive wastes. This can ensure that even if radioactive wastes in deep geological repositories interact with solution at elevated temperatures in the worst-case scenario, release of 99Tc would be at a minimum. To achieve this goal, radioactive wastes containing 99Tc should be sequestered as TcS2.
Acknowledgements
Research grants from the National Science Foundation (EAR-9614773) to S.A.W. and from the Geological Society of America to Y.-L.X. are gratefully acknowledged. This paper benefitted from the constructive reviews of two referees for Geochemical Transactions, which significantly improved our presentation. Thanks are extended to Dr Martin Schoonen, the editor of this special collection of papers honoring Dr Hubert Barnes, for handling our manuscript. We would also like to take this opportunity to express our appreciation of Dr Hubert Barnes, one of the founders of experimental hydrothermal geochemistry, for the tremendous impact he and his colleagues have had on the field of hydrothermal geochemistry.References
- Y.-L. Xiong and S. A. Wood, Chem. Geol., 1999, 158, 245 CrossRef CAS.
- Y.-L. Xiong and S. A. Wood, Econ. Geol., 2001, 96, 1429 Search PubMed.
- Y.-L. Xiong and S. A. Wood, Mineral. Petrol., 2000, 68, 1 Search PubMed.
- S. Sourirajan and G. C. Kennedy, Am. J. Sci., 1962, 260, 115 Search PubMed.
- G. M. Anderson and D. A. Crerar, Thermodynamics in Geochemistry, Oxford University Press, New York, 1993, p. 588. Search PubMed.
- J. W. Johnson, E. H. Oelkers and H. C. Helgeson, Comput. Geosci., 1992, 18, 899 CrossRef.
- D. A. Sverjensky, J. J. Hemley and W. M. D'Angelo, Geochim. Cosmochim. Acta, 1991, 55, 989 CrossRef CAS.
- V. A. Pokrovskii and H. C. Helgeson, Geochim. Cosmochim. Acta, 1997, 61, 2175 CrossRef CAS.
- B. R. Tagirov, A. V. Zotov and N. N. Akinfiev, Geochim. Cosmochim. Acta, 1997, 61, 4267 CrossRef CAS.
- R. W. Henley, A. H. Truesdell, P. B. Barton Jr. and J. A. Whitney, Fluid—mineral Equilibria in Hydrothermal Systems, Reviews in Economic Geology Volume 1, The Economic Geology Publishing Company, El Paso, Texas, 1984, p. 267 Search PubMed.
- H. C. Helgeson and D. H. Kirkham, Am. J. Sci., 1974, 274, 1199 Search PubMed.
- D. K. Nordstrom and J. L. Munoz, Geochemical Thermodynamics, Blackwell Scientific Publications, Boston, 1986, pp. 477 Search PubMed.
- H. C. Helgeson, D. H. Kirkham and G. C. Flowers, Am. J. Sci., 1981, 281, 1249 Search PubMed.
- J. Burgess, Metal ions in solution, Ellis Horwood Limited, Chichester, UK, 1978, p. 481 Search PubMed.
- C. F. Baes Jr. and R. E. Mesmer, The Hydrolysis of Cations, John Wiley and Sons, New York, 1976, p. 489. Search PubMed.
- S. A. Wood, D. A. Crerar and M. P. Borcsik, Econ. Geol., 1987, 82, 1864 Search PubMed.
- G. H. Brimhall and D. A. Crerar, in Thermodynamic Modeling of Geological Materials: Minerals, Fluids and Melts, Reviews in Mineralogy, Volume 17, ed. I. S. E. Carmichael and H. P. Eugster, 1987, p. 235. Search PubMed.
- M. Tarkian, R. M. Housley and A. Volborth, Eur. J. Mineral., 1991, 3, 977 CAS.
- M. A. Korzhinsky, S. Tkachenko, K. I. Shmulovich, Y. A. Taran and G. S. Steinberg, Nature, 1994, 369, 51 CrossRef CAS.
- M. Fleischer, Econ. Geol., 1959, 54, 1406 Search PubMed.
- L. Paganelli, Geochim. Cosmochim. Acta, 1963, 27, 401 CrossRef CAS.
- G. H. Ripley, Geochim. Cosmochim. Acta, 1967, 31, 1489 CrossRef.
- A. Sutulov, Molybdenum and rhenium recovery from porphyry coppers. University of Concepcion, Chile, 1970, p. 259. Search PubMed.
- A. Sutulov, Copper porphyries. University of Concepcion, Chile, 1975, p. 206. Search PubMed.
- Y. M. Poplavko, F. F. D'yakonova and L. V. Mel'nikova, Geochem. Int., 1984, 21(2), 40 Search PubMed.
- D. Huang, Y. Wang and F. Nie, Chin. J. Geochem., 1988, 7(2), 136 Search PubMed.
- T. E. McCandless, J. Ruiz and A. R. Campbell, Geochim. Cosmochim. Acta, 1993, 57, 889 CrossRef CAS.
- H. J. Stein, R. J. Markey, J. W. Morgan, A. Du and Y. Sun, Econ. Geol., 1997, 92, 827 Search PubMed.
- L. D. Meinert, Geosci. Can., 1992, 19, 145 Search PubMed.
- H. J. Stein, R. J. Markey, J. W. Morgan, K. Sundblad and A. Larin, in The seventh international symposium on Rapakivi granites and related rocks, ed. I. Haapala, O. T. Ramo and P. Kosunen, abstract volume, 1996, p. 68. Search PubMed.
- S. A. Wood and I. M. Samson, Econ. Geol., 2000, 95, 143 Search PubMed.
- T. A. P. Kwak, W-Sn Skarn Deposits and Related Metamorphic Skarns and Granitoids, Elsevier Science Publishers B. V., Amsterdam, The Netherlands, 1987, p. 451. Search PubMed.
- Z.-X. Xie and J. V. Walther, Geochim. Cosmochim. Acta, 1993, 57, 1947 CrossRef CAS.
- W. Stumm and J. J. Morgan, Aquatic Chemistry, John Wiley and Sons, Inc., New York, 1970, p. 583. Search PubMed.
- Y.-F. Chang, X.-P. Liu and Y.-C. Wu, The copper–iron belt of the middle and lower reaches of the Yangtse River (Changjian River), Geological Publishing House, Beijing, 1991, 379 Search PubMed.
- Y.-S. Zhai, Y.-L. Xiong, S.-Z. Yao and X.-D. Lin, Ore Geol. Rev., 1996, 11, 229 CrossRef.
- D. L. Giles and J. H. Schilling, 24th International Geological Congress, Montreal, Section 10, 1972, p. 145..
- S. A. Wood and I. M. Samson, in Techniques in Hydrothermal Ore Deposits: Reviews in Economic Geology, ed. J. Richards and P. Larson, 1998, v. 10, p. 33–80. Search PubMed.
- D. G. Brookins, Appl. Geochem., 1986, 1, 513 CrossRef CAS.
- D. A. Crerar and H. L. Barnes, Econ. Geol., 1976, 71, 772 Search PubMed.
Footnote |
| † Presented during the ACS Division of Geochemistry symposium Biogeochemical Consequences of Dynamic Interactions Between Benthic Fauna, Microbes and Aquatic Sediments, San Diego, April 2001. |
| This journal is © The Royal Society of Chemistry 2002 |