Zero-valent metal accelerators for the dechlorination of pentachlorophenol (PCP) in subcritical water
William D. Marshall*a, Alena Kubátováb, Arnaud J.M. Lagadecb, David J. Millerb and Steven B. Hawthorneb
aDept. of Food Science and Agricultural Chemistry, Macdonald Campus of McGill, 21,111 Lakeshore Road, Ste-Anne-de-Bellevue, Qué., Canada H9X 3V9. E-mail: marshall@macdonald.mcgill.ca
bEnergy and Environmental Research Center, University of North Dakota, Grand Forks, North Dakota 58202-9018, USA
First published on 16th January 2002
Abstract
Preliminary trials with 0.5 mg pentachlorophenol (PCP) in the presence/absence of 50 mg of 325-mesh Ag0/Fe0 (2% w/w) granules, demonstrated that the extent of dechlorination increased with both increased reaction time (0.5–2 h) and increased temperature (200–350 °C) but that recoveries of products were incomplete. Whereas reaction (200 °C) in the absence of metal accelerator furnished only unreacted PCP after 2 h and a mixture of tetrachloro species and substrate (∼50%) after 4 h of reaction, timed trials in the presence of Fe0 or 2% (w/w) Ag0/Fe0 bimetallic mixture resulted in more extensive dechlorinations. Only sequential dechlorinations were observed for iron-based accelerators and the dechlorination route was consistent with relief of steric strain as the dominant influence that determined the decomposition products. There was no appreciable difference in the course of the dechlorinations between the Fe0 and the 2% (w/w) Ag0/Fe0 bimetallic mixture but differences in the rates of reaction were evident. Dechlorinations with magnesium-based bimetallic accelerators were more extensive, more rapid and both concerted and stepwise reactions were observed. With 100 mg Pd0/Mg0, 63% of products consisted of phenol plus cyclohexanone and a further 9% was o-chlorophenol yet 13% were tetrachloro species. By contrast, an equal quantity of Mg0 alone mediated only partial dechlorination. The dechlorinations represent a detoxification—the toxiforic chloro substituents of the substrate are reduced to innocuous chloride while the sacrificial Mg metal is oxidised to Mg2+.
Green ContextDechlorination of heavily chlorinated aromatics is required for waste water treatment. Here a straightforward combination of Mg and subcritical water is used to effect an efficient destruction of these compounds using a relatively innocuous medium.DJM |
Introduction
Water, at ambient temperature and pressure (dielectric constant, ε ∼801), is extremely polar. Whereas increasing temperature decreases the dielectric constant appreciably, increasing pressures cause only modest increases in this parameter. The net effect is that under supercritical conditions [>374 °C and >22 MPa (220 bar)] the dielectric constant of the fluid is reduced to <10 (similar to dichloromethane). The corrosivity of water at near critical conditions (higher density) has prompted researchers to systematically investigate water under subcritical conditions. Fortunately, the polarity of water can be decreased appreciably at relatively mild pressures (sufficient to maintain the liquid state) and temperatures in the range of 100–350 °C. Thus, ε = 27–29 for T = 250 °C and pressures of 5.1–25.3 MPa (51–253 bar). With these conditions, subcritical water resembles a polar organic solvent (for ethanol ε = 24 and for methanol ε = 33). Dechlorination rate limitations that result from the sparing solubilities of non-polar chlorinated substrates in water (at ambient conditions) can be overcome.A popular approach to abiotic dehalogenations has been to mediate the reduction in aqueous media in the presence of a sacrificial metal in its elemental form. Zero-valent iron2–9 and iron–palladium10–12 bimetallic mixtures have become especially popular for this purpose. In anoxic aqueous media, free metal ions, chloride ion and hydrogen gas were produced by reaction at the surface of metal particles and protons were consumed. The process kinetics were dependent on solution pH, surface area of the metal particle, substrate concentration, buffer selection and solvent composition.7 Dechlorination approximately followed first-order kinetics and rate coefficients tended to increase with time, that possibly resulted from an increased surface area of reactive metal due to cathodic depolarization and pitting.4
Based on the low concentration of chlorinated degradation products in the solution phase it was suggested that most of the substrate remained sorbed to the iron surface until complete dechlorination had been achieved.6 Prolonged exposure of the Fe0 or Pd0/Fe0 surface to a saturated solution of aqueous organochlorine compounds resulted in the growth of a hydroxylated iron oxide film that deactivated the Pd0/Fe0 surface. However, the activity of the metal surface was restored by washing with a dilute acid.3,12,13
The dehalogenated of aromatics with zero-valent metals have also been studied. Copper metal has been reported to dechlorinate DDT14 and its derivatives. Catalytic hydroprocessing over molecular hydrogen that mediates the reduction of aryl-chlorine substituents to chloride at relatively low temperatures has been reported with Ni-, Pd-, Pt- and Rh-based catalysts.15–20 The liquid phase catalytic hydrodechlorination of chlorophenols over Pt/C21 and in the gas phase over Ni0/SiO222,23 and over Ni0/zeolite22 has also been examined. Hydrogen treatment of HOArCl isomers in the range of 150–300 °C, yielded phenol as the only appreciable product.22 Under these conditions, phenol was hydrogenated to cyclohexanol and cyclohexanone with benzene being formed at T >250 °C. The continuous gas phase hydrodechlorination of pentachlorophenol (PCP) was studied in H2 at 200–350 °C over 1.5 or 15.2% Ni/SiO2 and Ni/zeolite.23 With these conditions, dechlorination has been shown22,24 to proceed via an electrophilic mechanism involving spillover hydrogen and associated chloroaromatic. There is persuasive evidence25,26 for the co-existence of charged (H+) and uncharged (H atoms) spillover hydrogen on silica where the former is considered to be the reactive specie in catalytic hydrodechlorination.
Analogous continuous dechlorinations have involved the use of steam or supercritical carbon dioxide. In the steam reforming process,27 liquid substrate was merged with steam and pyrolysed over a commercial Ni catalyst [Ni0/CaAl2O4 (23 wt%)] or 0.5% Pt/γ-Al2O3 at 600–800 °C. Aryl chloride was converted to CO + H2 + HCl in a steam reforming reaction and the CO plus water was transposed to CO2 and hydrogen with a second water gas shift reaction. In another process, PCBs28 or PCP29 was merged with supercritical CO2 and dechlorinated efficiently over (2% w/w) Ag0/Fe0 at ∼450 °C. Decreases in the catalyst activity with time were restored to their original values with a methanol–water wash. Zero-valent iron and bimetallic mixtures have also been evaluated (in batch processes) for PCP30,31 and for PCB32,33 dechlorinations. For subcritical water, Hinz et al.33 have proposed that the magnetite [Fe(OH)2] formed by Fe0 corrosion of water is converted thermally to Fe3O4 with the liberation of H2.
The objectives of the current study were to evaluate selected zero-valent metals or bimetallic mixtures for their ability to accelerate the dechlorination of PCP in hot pressurised water. If differences were evident, it was anticipated that changes in the product distribution with time would provide insights into the mechanistic course of the dechlorination.
Experimental
Chemicals
Pentachlorophenol (PCP, nominally 99% pure), trichlorophenols (1,2,3-, 1,2,4-, 1,2,5-, 2,3,4-, 2,3,5-), dichlorophenols (2,3-, 2,4-, 2,5-, 2,6-, 3,4-, 3,5-) monochlorophenol (2-, 3-, and 4-) and phenol were purchased from Aldrich Chemical Co., Milwaukee, WI, USA). Tetrachlorophenols (2,3,4,5-, 2,3,4,6- and 2,3,5,6-) were purchased from Supelco, Bellefonte, PA, USA. Acids [HCl (15 M) and HNO3 (17% v/v)], sodium bicarbonate and solvents (dichloromethane and acetone) were purchased from Fisher Scientific Co., Fair Lawn, NJ, USA. Copper powder (electrolytic grade, 40-100 mesh, nominally 99.5% purity), iron (40 mesh, 100 mesh or 325-mesh, nominally 99.5% purity), magnesium (∼20-mesh), nickel (50–100 mesh, nominally 99.5% purity), zinc particles (100 mesh, nominally 99.9% purity), were purchased from Aldrich Chemical Co., or Alfa Aesar (Ward Hill, NJ, USA). Chemicals were ACS-Reagent grade (unless noted otherwise) and were used as received.Dechlorinations
Timed trials were performed under static (no flow) conditions in 4 mL capacity stainless steel cells (64 mm long by 6.3 mm inner diameter, fitted with threaded (npt) screw-top end caps, Parker Hannifen Corp., Columbus, OH). Each reactor was partilly filled by adding 2.7 mL water (that had been purged with N2 for 2 h) to 50 mg test accelerator that had been amended with 0.5 mg PCP (contained in 10 μL acetone). Each reactor was sealed immediately and added to a clamping device mounted on the circumference of a circular disk. The disk was mounted on a metal plate (that approximated the dimensions of the door of the gas chromatographic oven) and could be rotated to mix the contents of each reactor (in end over end fashion). For reactions at 200 °C, the oven door was opened and the loaded rack–metal plate assembly was substituted quickly into the oven that had been pre-heated to 250 °C. The oven set-point temperature was maintained at 250 °C until the oven temperature had increased to 190 °C then the temperature set-point of the chromatograph was lowered to the desired 200 °C. An analogous procedure was used for other experimental reaction temperatures. Companion trials in which a thermocouple extended into the aqueous reaction mixture had indicated that the interior cell temperature reached 190 °C after 4 min. The rack was rotated continuously (∼30 revolutions min−1) and timed trials were considered to have commenced as soon as the oven had returned to operating temperature. Upon completion of each timed trial, the reaction was stopped by cooling each sealed cell rapidly in cold running water.Safety note
It is imperative to maintain sufficient headspace above the reaction mixture so that the internal pressure is governed by the steam/water equilibrium and excessive pressure is avoided. In the range 105–350 °C, the pressure for steam/water1 ranges between 1.3 and 170 bar (∼2500 psi) substantially below the 517 bar (7500 psi) pressure rating of the cells.Reaction work up
The contents of each reactor was acidified with five drops of conc. HCl or HNO3 then transferred to a 7 mL vial and extracted three sequential times with 1 mL CH2Cl2. The organic extracts were combined and supplemented with 0.3 mg 2-chloronaphthalene (in 10 μL acetone) that acted as an internal standard and analysed by GC-MS.Ag0 or Pd0/metal0 bimetallic mixture
The general preparations described by Zhang et al.34 were followed. Ag0/Fe0 and Pd0/Fe0 were prepared from 325- or 100-mesh iron granules that had been washed copiously with 6 M HNO3 and rinsed with distilled water. Sufficient aqueous AgNO3 or K2PdCl6 to result in a 2% or 0.2–0.5% (w/w) surface coverage, respectively, of the iron, was added to the aqueous metal suspension that was gently mixed on a rotary evaporator for 12 h. Ag0/Ni0 was prepared in similar fashion by reaction of aqueous AgNO3 [sufficient to provide a 2% (w/w) surface coverage] with aqueous suspension of pre-washed 50–100 mesh nickel granules during 12 h. For magnesium granules, the reaction with aqueous AgNO3 or K2PdCl6 was more rapid and only 10 min were required.Pd0/Mg0
Mg0 (25 or 10 g of 12–50 mesh granules) was combined with ∼50 mL H2O in a 250 mL round bottom flask (RBF). An aqueous solution of K2PdCl6 (188 mg/∼10 mL) was added and the flask was swirled vigorously under cold running water to mix the reagents. The reaction was exothermic and the supernatant liquid rapidly turned black. The RBF was maintained on the rotary evaporator to maximise mixing. After 10 min, the aqueous supernatant fraction was decanted off and discarded. The granules were washed (∼30 mL) three times with water and a further three times with acetone. The solids were then dried under reduced pressure on the rotary evaporator and stored in a sealed jar.Ag0/Mg0
In similar fashion, AgNO3 (788 mg) dissolved in ∼25 mL H2O was combined with the 25 g Mg0 granules suspended in ∼50 mL H2O and reacted for 10 min. An identical work up furnished the Ag0/Mg0 bimetallic mixture.Gas chromatography–mass spectrometry
Chromatographic separations were performed on an Agilent Technologies model 5890 series II gas chromatograph fitted with a model 5970 mass selective detector and an autoinjector system. Injections were performed in the splitless mode into a DB-5 capillary column (30 m × 0.25 mm i.d. and 0.25 μm film thickness, J & W Scientific, Rancho Cordova, CA, USA) and eluted with helium (column head pressure, 10 psi). After an initial hold for 1 min at 40 °C, the column was ramped, at 10 °C min−1, to 320 °C and held for a further 5 min prior to cooling. The temperature of the injector and transfer line were maintained at 300 and 280 °C, respectively. Quantitations were based on relative response factors of individual standards to the internal standard (2-chloronaphthalene). Similarly, the identification and quantitation of degradation products were based on comparisons of mass spectra and retention times of pure standards with those of the sample components (unless noted otherwise).Chloride ion determination
Chloride ion in aqueous fractions of reaction mixtures that had been charged with 5 mg substrate were performed in triplicate or quadruplicate with a chloride ion selective electrode (Cole Parmer, Vernon Hills, IL, USA).Results and discussion
Because water that was added to each reactor had been deaerated for 2 h with nitrogen prior to use it was anticipated that the dominant reactions would be hydrolytic rather than oxidative. For the preparation of bimetallic metal mixtures, it had been established previously that only traces of silver or palladium remained in the aqueous supernatant fraction after reaction with the Fe0 or Mg0. In preliminary ranging trials, it was also established that the presence of zero-valent metals did accelerate dechlorinations relative to companion runs that contained only PCP. However, neither the copper, zinc, nickel nor a 2% (w/w) Ag0/Ni0 bimetallic mixture were as efficient as iron metal (Fe0) at mediating dechlorinations. As has been observed by others,9,12 it was corroborated that a reduced particle size (325- vs. 100- vs. 40-mesh) of the Fe0 granules did increase the rate/extent of PCP dechlorination in this medium as well.Iron-based accelerators
Initial trials that were performed with 0.5 mg PCP (1.88 μmol) in the presence/absence of 50 mg of 325-mesh Ag0/Fe0 granules (∼0.9 mmol), demonstrated that the extent of dechlorination increased with both increased reaction time (0.5–2 h) and increased temperature (200–350 °C). However, recoveries were incomplete (ranging from 20–27%) so that the values recorded in Table 1 represent the molar percent distribution among the products that could be determined by GC/MS with the extraction procedure. The low recoveries were repeatable and not surprising given the tendency6,35 of chlorinated substrates to sorb strongly to metal surfaces and the probable formation of ring hydroxylation products that might not be detected with the recovery procedure. Chlorobenzenes and PCBs have been reported to form ring-hydroxylated products in subcritical H2O. Dechlorinations also occurred in the absence of accelerator (Table 1) but were appreciably less extensive.| At 200 °C | Substrate | Tetra-Cl | Tri-Cl | Di-Cl | Mono-Cl | Phenol |
|---|---|---|---|---|---|---|
| a ± One relative standard deviation based on two or three replicate runs.b N.D. = none detected. | ||||||
| PCP, no catalyst, 0.5 h | 96 | 0.5 | N.D.b | N.D. | N.D. | N.D. |
| Ag0/Fe0, 0.5 h (3 runs) | N.D. | 4 ± 1 | 65 ± 3 | 27 ± 2 | N.D. | N.D. |
| PCP, no catalyst, 1 h | 78 | 17 | 3 | N.D. | N.D. | N.D. |
| Ag0/Fe0,1 h (2 runs) | N.D. | N.D. | 17 ± 1 | 59 ± 4 | 8 ± 2.0 | 3 ± 0.4 |
| PCP, no catalyst, 1 h | 77 | 17 | 4 | N.D.b | N.D. | N.D. |
| Ag0/Fe0,1 h (2 runs) | N.D. | N.D. | 19 ± 1 | 67 ± 4 | 9 ± 2 | 2 ± 0.4 |
| PCP, no catalyst, 1.5 h | 64 | 32 | N.D. | N.D. | N.D. | N.D. |
| Ag0/Fe0, 1.5 h (3 runs) | N.D. | N.D. | 1 ± 2 | 56 ± 2 | 32 ± 0.4 | 9 ± 0.5 |
| PCP, no catalyst, 2 h | N.D. | 40 | 54 | N.D. | 3 | 5 |
| Ag0/Fe0, 2 h (3 runs) | N.D | N.D. | N.D. | 42 ± 2 | 51 ± 2 | 1 ± 0.9 |
| For 1 h | Substrate | Tetra-Cl | Tri-Cl | Di-Cl | Mono-Cl | Phenol |
| PCP, no catalyst, 200 °C | 77 | 17 | 4 | N.D. | N.D. | N.D. |
| Ag0/Fe0, 200 °C (2 runs) | N.D. | N.D. | 22 ± 1 | 64 ± 4 | 9 ± 2 | 2 ± 0.4 |
| PCP, no catalyst, 250 °C | 66 | 5 | 15 | N.D. | 4 | 9 |
| Ag0/Fe0, 250 °C (3 runs) | N.D. | 3 ± 4 | 71 ± 5 | 17 ± 3 | N.D. | N.D. |
| PCP, no catalyst, 300 °C | N.D. | 1 | 33 | 1 | 3 | 52 |
| Ag0/Fe0 1 h, 300 °C (3 runs) | N.D. | N.D. | 7 ± 0.5 | 12 ± 0.1 | 12 | 48 ± 7 |
| PCP, no catalyst, 350 °C | 0.2 | N.D. | 0.2 | 2 | 32 | 46 |
| Ag0/Fe0, 350 (3 runs) | N.D. | N.D. | N.D. | N.D. | 10 ± 2 | 82 ± 2 |
In a subsequent series of trials, the influences of bimetallic Ag0/Fe0 mixtures and Fe0 on the dechlorination rates were compared. Dechlorinations of 0.5 mg PCP in the presence of 200 mg Ag0/Fe0 (Fig. 1A), 100 mg Ag0/Fe0 (Fig. 1B) or 100 mg Fe0 (Fig. 1C) granules for up to 20 h were compared. In all cases, dechlorination reactions at 200 °C in this medium proceeded in stepwise fashion and none of the dechlorinations was complete in any of these timed trials. Monochloro species accumulated rapidly within the 200 mg trials (◆, Fig. 1A) and was virtually quantitative (>95%) within 3 h of reaction whereas the accumulation of monochloro analytes was somewhat slower for the treatments with 100 mg of the bimetallic mixture (Fig. 1B) and was maximized at 10 h. By contrast, tetrachloro species (●, Fig. 1C) comprised >60% of the product mixture at 0.5 h for the Fe0 accelerated reaction (Fig. 1C), but only traces of these compounds were observed in the 100 mg Ag0/Fe0mediated dechlorinations (Fig. 1B) and none was detected in 200 mg trials (Fig. 1A). Neither monochlorinated products nor phenol were detected in the Fe0 catalysed dechlorinations but were the only components at later sampling times for the Ag0/Fe0 accelerated reaction. The fact that the Ag0/Fe0 mixture and Fe0 continued to react after 5 and 10 h, respectively, seems to mitigate against complete accelerator inactivation at this time. It seems more likely that the rates of reaction for different substrates are appreciably different. Fig. 1 corroborates the observations of others12 that the rates of zero-valent metal mediated dechlorinations are accelerated by the presence of multiple chlorine substituents.
 | ||
| Fig. 1 Variations with time in the product distribution for the hydrolytic dechlorination of pentachlorophenol in the presence of A, 200 mg of Ag0/Fe0; B, 100 mg of Ag0/Fe0 or C, 100 mg of Fe0. | ||
Product speciation
Reaction at 200 °C in the absence of accelerator furnished only unreacted PCP after 2 h and a mixture of 2,3,4,5- and 2,3,5,6-tetrachlorophenol (10:1 ratio) and substrate (∼50%) after 4 h of reaction. The dominant chloro-species that were accumulated with time in the iron-accelerated trials are summarised in Fig. 2. For short reaction periods (0.5–1.5 h) in the presence of Fe0, one tetrachloro specie [2,3,4,5-tetrachlorophenol, (2,3,4,5-TeCP)] dominated the product distribution (∼60% at 0.5 h). Two trichloro species [2,4,5- and 2,3,4-trichlorophenol (TCP)] were also present, in an approximate 2:1 ratio at the 0.5 h sampling time. Traces of 2,3,5-trichlorophenol were also observed and only at short reaction times. Whereas the quantity of 2,3,4-trichloro specie remained a minor component (∼10%) of the product mixture, the 2,4,5-trichloro specie rapidly increased with time to dominate the mixture (∼60%) at 3.5 h and only decreased gradually with longer reaction times to be replaced with 3,4-dichlorophenol (3,4-DCP). Post the initial ortho-dechlorination to form the 2,3,4,5- species, the second dechlorination occurred virtually exclusively at the meta position and resulted in the 2,4,5- and 2,3,4- species. The subsequent o-dechlorination resulted in the 3,4-dichloro product. One possible explanation for the selectivity might be that the interaction of the substrate PCP with the surface of the metal accelerator occurred predominantly via the π-electron system of the aromatic ring so that all the Cl substituents were accessible to the catalyst. The ortho–para directing character of the hydroxy substituent does not seem to explain the relatively high level of selectivity that is associated with the observed sequential dechlorination process. To the extent that aryl dechlorination is an electrophilic (E1) process, dechlorination at the o- or p-positions would stabilize the intermediate cation and favour dechlorination at these sites. If resonance stabilization were the dominant influence the product distribution would be predicted to consist of 2,3,5-TCP, 2,3-DCP, 2,5-DCP and 3-MCP. As an alternative explanation, the removal of bulky chlorine substituents from the ring would provide relief of steric strain. The initial loss of Cl from the C-2 or C-6 position (ortho to the hydroxy and a chlorine substituent) decreases steric strain and repulsion more than the loss of a m-Cl substituent. The loss of a second chlorine substituent occurs at a position with two nearest Cl neighbours and the loss of the third chlorine again occurs from the position that is ortho to the hydroxyl group.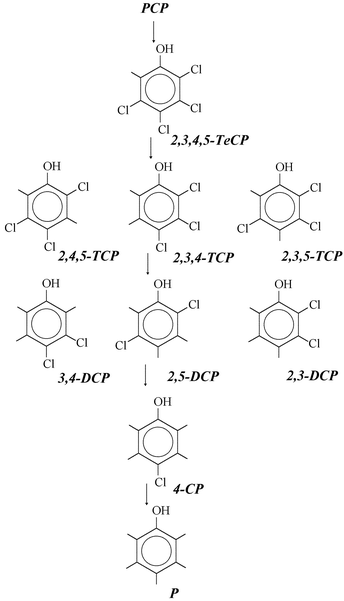 | ||
| Fig. 2 Reaction pathway for the dechlorination of PCP mediated by Fe0 or an Ag0/Fe0 bimetallic mixture. | ||
For dechlorinations with an equivalent quantity (100 mg) of an Ag0/Fe0 mixture, only traces of 2,3,4,5-tetrachlorophenol were observed (0.5 h). Four trichlorophenols (2,3,4- and 2,4,5-) each accounted for ∼20% and the 2,3,5- isomer accounted for ∼10% of the products after 0.5 h of reaction. The 2,3,6- isomer was observed in only one of the replicate trials and only at the 0.5 h sampling time. Chlorinated phenols 2,3- (∼8%) and 3,4-dichlorophenol (∼25%) and 4-chlorophenol (∼5%) accounted for the remainder of the products. Whereas trichloro species were absent at 2 h, the dichlorinated phenols persisted throughout much of the trial and the 4-chlorophenol dominated the products after 2 h. Phenol was only detected intermittently and only in the latter stages of the trial.
For reaction with 200 mg of 2% (w/w) Ag0/Fe0, (Table 2) lesser quantities of 2,4,5- (∼28%), and traces of 2,3,5- (7%) and 2,3,4- (4%) were recovered from the reaction mixture at shorter reaction times (0.5 or 1 h). Dichlorinated species were more prevalent and more persistent with time. Small quantities of 2,3-dichloro (10%), and 2,5-dichloro (∼10%) did not accumulate with time but the 3,4-dichloro species (initially ∼40%) increased to 62% at 1.5 h then subsequently decreased gradually to 10% at 5 h. The only monochlorophenol to accumulate was the 4-chloro specie that had increased to 86% at 2.5 h and increased further to 92% at 20 h. The only other product detected at longer reaction times (>2 h) was phenol (3–8%). Seemingly, there was no appreciable difference in the course of the reaction for the Fe0 and the 2% (w/w) Ag0/Fe0 bimetallic mixture but differences in the rates of dechlorination were clearly evident (Fig. 1).
| Time/h | 2,3,4-TriCl | 2,3,5-TriCl | 2,4,5-TriCl | 2,3-DiCl | 2,5-DiCl | 3,4-DiCl | 4-Cl | Phenol |
|---|---|---|---|---|---|---|---|---|
| a ± One relative standard deviation based on two or three replicate runs.b N.D. = none detected. | ||||||||
| 0.5 | 4 ± 1 | 7 ± 4 | 28 ± 11 | 10 ± 3 | 38 ± 7 | 6 ± 4 | 2 ± 3 | N.D.b |
| 1.0 | 2 ± 3 | 4 ± 4 | 21 ± 8 | 10 ± 10 | 22 ± 24 | 42 ± 21 | N.D. | N.D. |
| 1.5 | N.D. | N.D. | 8 ± 11 | 6 ± 8 | 13 ± 1 | 62 ± 13 | 12 ± 13 | N.D. |
| 2.0 | N.D. | N.D. | N.D. | N.D. | N.D. | 50 ± 7 | 50 ± 11 | N.D. |
| 2.5 | N.D. | N.D. | N.D. | N.D. | 5 ± 8 | 4 ± 8 | 86 ± 16 | 3 ± 2 |
| 3.0 | N.D. | N.D. | N.D. | N.D. | 8 ± 14 | N.D. | 90 ± 13 | 2 ± 4 |
| 3.5 | N.D. | N.D. | N.D. | N.D. | N.D. | 12 ± 17 | 82 ± 9 | 6 ± 9 |
| 5.0 | N.D. | N.D. | N.D. | N.D. | N.D. | 10 ± 9 | 82 ± 6 | 8 ± 3 |
| 10.0 | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. | 95 ± 3 | 5 ± 3 |
| 20.0 | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. | 92 ± 5 | 8 ± 5 |
Magnesium-based accelerators
Dechlorinations with magnesium-based accelerators were appreciably more extensive and more rapid. As with Fe0, Mg0 is known to hydrolyse water exothermically to form Mg(OH)2 and H2; a white powder was observed in all crude product mixtures but companion trials with the thermocouple did not detect any influence of the temperature of the reaction mixture on the dechlorinations. In the presence of excess K2PdCl6 (25 μmol), PCP (1.88 μmol) was dechlorinated quantitatively to phenol and cyclohexanol (Table 3). Increased quantities of Mg0 (50 vs. 100 vs. 200 mg) in the reaction mixture favoured hydrogenation of the ring and subsequent reduction to cyclohexanol post dechlorination to phenol. However, the rates of formation of specific intermediates did not depend linearly as the quantity of added accelerator. By contrast, Mg0 alone mediated only partial dechlorination to the 2,3,5,6- and 2,3,4,6-tetrachloro species and 2,3,4- and 2,3,5-trichlorophenol. A pre-formed bimetallic mixture (Pd0/Mg0) at 0.2% (w/w) loading or 2% (w/w) Ag0/Mg0 was less efficient than the co-addition of Mg0 + K2PdCl6. Again, the extent of dechlorination was dependant on the quantity of accelerator (50 vs. 100 mg) added to the reaction. For 50 mg Pd0/Mg0, 16% of the products were totally dechlorinated yet 63% were tetrachloro species indicating that this pre-formed accelerator mediated both concerted and stepwise dechlorinations. For 100 mg Pd0/Mg0, 63% of products consisted of phenol plus cyclohexanone and a further 9% was o-chloro phenol yet 13% were tetrachloro species. The Ag0/Mg0 bimetallic mixture, at 50 or 100 mg, proved to be less efficient than the Pd0/Mg0 mixture; only partial dechlorination was observed and this mixture was abandoned.| Treatment | PCP | Tetra-Clb | Tri-Clc | Di-Cld | Mono-Cle | Phenol | Cyclo-olf | Cyclo-oneg |
|---|---|---|---|---|---|---|---|---|
| a ± One relative standard deviation based on two or three replicate runs.b 2,3,4,5-Tetrachlorophenol and 2,3,5,6-tetrachlorophenol.c 2,3,4-; 2,3,5- and 2,4,5-Trichlorophenol.d 2,3-; 2,5-; and 2,6-Dichlorophenol.e o-Chlorophenol and p-chlorophenol.f Cyclohexanol.g Cyclohexanone. | ||||||||
| 50 mg Mg0 + 25 μmol K2PdCl6, 1 h | N.D. | N.D. | N.D. | N.D. | N.D. | 60 ± 6 | 40 ± 6 | Trace |
| 100 mg Mg0 + 25 μmol K2PdCl6, 1 h | N.D. | N.D. | N.D. | N.D. | N.D. | 15 ± 21 | 85 ± 21 | N.D. |
| 200 mg Mg0 + 25 μmol K2PdCl6, 1 h | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. | 100 | N.D. |
| 50 mg Mg0, 1 h | N.D. | 69 ± 2 | 31 ± 0.1 | N.D. | N.D. | N.D. | N.D. | N.D. |
| 50 mg, Pd0/Mg0, (0.2% w/w), 1 h | N.D. | 63 ± 2 | 21 ± 8 | trace | 4 ± 4 | 4 ± 5 | N.D. | 12 ± 4 |
| 100 mg, Pd0/Mg0, (0.2% w/w), 1 h | N.D. | 13 ± 1 | 11 ± 1 | 4 ± 6 | 9 ± 2 | 38 ±1 | N.D. | 25 ± 5 |
| 50 mg Ag0/Mg0, (2% w/w), 1 h | 16 ± 11 | 55 ± 4 | 26 ± 12 | 3 ± 4 | N.D. | N.D. | N.D. | N.D. |
| 100 mg Ag0/Mg0, (2% w/w) 1 h | N.D. | N.D. | 7 ± 2 | 84 ± 4 | 7 ± 3 | N.D. | N.D. | N.D. |
| 25 mg Pd0/Mg0 (0.5% w/w), 0.5 h | 10 ± 14 | 8 ± 2 | 2 ± 3 | N.D. | 1 ± 2 | 75 ± 35 | N.D. | 4 ± 5 |
| 50 mg Pdo/Mg0 (0.5% w/w), 0.5 h | N.D. | N.D. | 4 ± 5 | N.D. | N.D. | 83 ± 3 | N.D. | 12 ± 2 |
| 100 mg Pdo/Mg0 (0.5% w/w), 0.5 h | N.D. | N.D. | N.D. | N.D. | 1 ± 1 | 76 ± 5 | N.D. | 21 ± 4 |
| 25 mg Pd/Mg (0.5% w/w), 1 h | 18 ± 23 | 36 ± 16 | 2 ± 2 | Trace | 3 ± 0.5 | 38 ± 5 | N.D. | 3 ± 0.5 |
| 50 mg Pd/Mg (0.5% w/w), 1 h | N.D. | 0.3 ± 0.4 | 6 ± 8 | N.D. | 4 ±0.9 | 78 ± 5 | N.D. | 9 ± 2 |
| 100 mg Pd/Mg (0.5% w/w), 1 h | N.D. | N.D. | N.D. | N.D. | 7 ± 7 | 86 ± 5 | N.D. | 8 ± 4 |
| 25 mg Pd0/Mg0 (0.5% w/w), 2 h | 18 ± 23 | 36 ± 17 | 2 ± 2 | Trace | 3 ± 0.5 | 38 ± 5 | N.D. | 3 ± 0.5 |
| 50 mg Pd0/Mg0 (0.5% w/w), 2 h | N.D. | N.D. | N.D. | N.D. | 6 ± 8 | 91 ± 13 | N.D. | 3 ± 5 |
| 100 mg Pd0/Mg0 (0.5% w/w), 2 h | N.D. | N.D. | N.D. | N.D. | N.D. | 96 ± 6 | N.D. | 5 ± 7 |
| 50 mg Mg0, 2 h | 38 ± 9 | 52 ± 8 | 10 ± 14 | N.D. | N.D. | N.D. | N.D. | N.D. |
Increased reaction time (0.5 vs. 1.0 vs. 2.0 h) also increased the extent of dechlorination modestly however the effect was not monotonic. Substrate PCP was observed among the products at all three reaction times only for the 25 mg loading of 0.5% (w/w) accelerator. Increased loadings of accelerator were generally more efficient at mediating dechlorinations than extended reaction times.
Increased PCP loadings
In a final series of trials, 100 mg of Pd0/Mg0 were reacted with 5 mg (18.8 μmol) or 50 mg (187.7 μmol) of PCP for 1 h at 200 °C. In both cases, the principal product was phenol. Compared to the original PCP loading, phenol was found at 97 ± 10 mol% and 23 ± 1 mol% for low and high quantities of PCP, respectively. Additionally, several minor products were observed for both concentrations of PCP (Table 4). For 5 mg of substrate the products included cyclohexanone (∼3 mol%), monochlorophenols (∼4 mol%) represented mainly by o-Cl phenol, dichlorophenols (∼2 mol%), trichlorophenols (∼2 mol%), tetrachlorophenols (∼3 mol%) and unreacted PCP (∼4 mol%). Of the four dichlorophenols isomers, 2,3- and 3,4- were major products and only traces of 2,5- and 2,6-isomers were observed. For trichlorophenols, 2,3,4- and 2,3,5-isomers were major and traces of 2,3,6- and 2,4,5-isomers were observed. All three tetrachlorophenol isomers were observed with the 2,3,4,6-isomer as the most abundant. These distributions have been calculated relative to the 5 mg (18.8 μmol) of substrate PCP that were added to the reaction mixture. Thus, 111 ± 14 mol% of the substrate was accounted for among the products, 100 ± 12 mol% as non-chlorinated and a further 11 ± 2 mol% of partially dechlorinated materials.| Initial substrate loading | 18.8 μmol | 187.7 μmol |
|---|---|---|
| a Mean ± one relative standard deviation based on two or three replicate runs. | ||
| Cyclohexanone | 3.2 ± 2.5 | 0.3 ± 0.2 |
| Phenol | 97 ± 10 | 23 ± 1 |
| MCPs | 3.8 ± 2.6 | 2.2 ± 0.5 |
| DCPs | 2.4 ± 2.1 | 4.2 ± 1.3 |
| TCPs | 1.6 ± 0.9 | 2.5 ± 1.0 |
| TeCPs | 2.9 ± 2.4 | 2.3 ± 2.4 |
| Unreacted PCP | 3.7 ± 3.4 | 2.3 ± 3.9 |
At the higher substrate loading (187.7 μmol) the recovery of phenol (23 ± 1 mol%) was decreased; only an additional 14 ± 2 mol% of substrate was found in chlorinated materials, and the recovery of unreacted PCP was only 2 mol%. Lower recoveries for the higher loading might be explained by an exhausted accelerator and/or sorption of the substrate and products to the surface of the accelerator.
Efforts were also made to determine the chlorine balance at the termination of the Pd0/Mg0 trials at higher PCP loadings (5 or 50 mg). The crude reaction product mixtures were acidified with nitric acid) prior to extraction with organic solvent. The chlorine mass balance at the 5 mg substrate loading (Table 5) indicated incomplete release of chloride into the aqueous phase ∼20% and a further ∼5% was accounted for among the organochlorine products that had been quantified. It seems probable that at least a portion of the remainder consisted of smaller chlorinated fragments that were not extracted or detected by GC/MS. The highest substrate loading (187.7 μmol) proved to be similar. Again, ∼10% of the chlorine in the PCP substrate was detected as chloride and a further ∼10% was accounted for among the chlorinated organics fraction. Although strong acid has been employed for the extraction,31 the recovery of chloride and/or of organochlorine products was insufficient to obtain mass balance for chlorine. In a companion trial, 5 mg (18.8 μmol) PCP was reacted with 100 mg of 325-mesh Fe0. Despite a work up that included sufficient H+ to solubilise the surfaces of the Fe0 particles and to acidify the crude product mixture to pH ∼1, product recoveries were disappointingly low; only ∼12% of the chlorine was accounted for.
| Wt% of chlorine | ||||
|---|---|---|---|---|
| Accelerator/mg | Initial PCP massb/mg | Cl− Ion | Organochlorine | PCP |
| a ± One standard deviation based on three or four replicate determinations.b Reactions were performed for 1 h at 200 °C. | ||||
| Fe0 (100 mg) | 5 | 7 ± 2 | 6 ± 3 | 0 |
| Pd0/Mg0 (50 mg) | 5 | 20 ± 4 | 5 ± 4 | 0.1 ± 0.2 |
| Pd0/Mg0 (100 mg) | 5 | 21 ± 6 | 4 ± 2 | 3 ± 3 |
| Pd0/Mg0 (50 mg) | 50 | 10 ± 2 | 8 ± 5 | 2 ± 1 |
| Pd0/Mg0 (100 mg) | 50 | 11 ± 1 | 6 ± 2 | 5 ± 7 |
Mg0-mediated dechlorinations
Mg0-based accelerators followed a reaction course somewhat different from the Fe0 mediated dechlorinations for which only stepwise dechlorinations had been observed. For bimetallic Pd0/Mg0, phenol was the major product yet with limited loadings of this accelerator, the other major products were tetrachloro species (Table 3). Based on the accumulation of limited quantities of partially dechlorinated compounds in the product mixture, Fig. 3 is proposed as a probable dechlorination scheme. All three tetrachloro species were present in a ∼2∶1∶1 ratio with the 2,3,4,6-isomer predominating. Two congeners dominated the trichloro fraction 2,3,5- and 2,4,5- (∼5∶4 ratio), two dichloro species (2,3- and 3,4- in approximately equal concentrations) dominated the dichloro fraction and two monochlorophenols ortho and para were present among the products in a ∼2∶1 ratio. In addition to the dechlorination products isolated from Fe0-mediated dechlorinations (Fig. 2), the remaining chlorinated products are similar to the distribution of products reported by Shin and Keane23 (labelled with an * in Fig. 3) that included 2,3,5-TCP, 2,4,5-TCP and 2,5-DCP over Ni/zeolite and 2,3-DCP and 2-CP (but not 3,5-DCP) over Ni/SiO2. Small quantities of cyclohexanone also accumulated in the presence of the pre-formed bimetallic mixture whereas a larger excess of K2PdCl6 together with Mg0 resulted only in cyclohexanol. Mg0 or Ag0/Mg0 were less efficient accelerators and caused only limited dechlorination. Although reaction conditions remain to be optimised, the results with Pd0/Mg0 in these trials are encouraging. Dechlorination in this case represents a detoxification in which the toxiforic chloro substituents of the substrate are reduced to innocuous chloride while the sacrificial Mg metal is oxidised to Mg2+. The released Mg2+ ion is also considered to be innocuous. The reaction is rapid and extensive at relatively low temperature and there does not appear to be any reason why other reducible substituents including other halogens or aryl-nitro group would not be behave similarly. | ||
| Fig. 3 Reaction pathway for the dechlorination of PCP mediated by Mg0 or Pd0/Mg0 bimetallic mixture. | ||
References
- L. Harr, J. S. Gallagher and G. S. Kell, National Bureau of Standards/National Research Council Steam Tables, Hemisphere Publishing Corporation, Bristol, PA, 1984 Search PubMed.
- R. W. Gillham and S. F. O’Hannesin, Ground Water, 1994, 32, 958 Search PubMed.
- L. J. Matheson and P. G. Tratnyek, Environ. Sci. Technol., 1994, 28, 2045 CAS.
- B. R. Helland, P. J. J. Alvarez, J. L. Schnoor and L. Jerald, J. Hazard. Mater., 1995, 41, 205 CrossRef CAS.
- K. D. Warren, R. G. Arnold, T. L. Bishop, L. C. Lindholm and E. A. Betterton, J. Hazard. Mater., 1995, 41, 217 CrossRef CAS.
- W. S. Orth and R. W. Gillham, Environ. Sci. Technol., 1995, 30, 66 CrossRef.
- T. L. Johnson, M. M. Scherer and P. G. Tratnyek, Environ. Sci. Technol., 1996, 30, 2634 CrossRef CAS.
- C.-B. Wang and W.-x. Zhang, Environ. Sci. Technol., 1997, 31, 2154 CrossRef CAS.
- G. D. Sayles, G. W. You, M. Wang and M. J. Kupferle, Environ. Sci. Technol., 1997, 31, 3448 CrossRef CAS.
- R. Muftikian, Q. Fernando and N. Korte, Water Res., 1995, 29, 2434 CrossRef CAS.
- C. Grittini, M. Malcomson, Q. Fernando and N. Korte, Environ. Sci. Technol., 1995, 29, 2898 CAS.
- R. Muftikian, K. Nebesny, Q. Fernando and N. Korte, Environ. Sci. Technol., 1996, 30, 3593 CrossRef CAS.
- B. A. Balko and P. G. Tratnyek, J. Phys. Chem. B, 1998, 102, 1459 CrossRef CAS.
- G. D. Sayles, G. You, M. Wang and M. J. Kupferle, Environ. Sci. Technol., 1997, 31, 3448–3454 CrossRef CAS.
- A. R. Suzdorf, S. V. Morozov, N. N. Anshits, S. I. Tsiganova and A. G. Anshits, Catal. Lett., 1994, 29, 49 Search PubMed.
- F. Gioia, E. J. Gallagher and V. Famiglietti, J. Hazard. Mater., 1994, 38, 277 CrossRef CAS.
- R. B. LaPierre, L. Guczi, W. L. Kranich and A. H. Weiss, J. Catal., 1978, 52, 218 CrossRef CAS.
- B. Coq, G. Ferrat and F. Figueras, J. Catal., 1986, 101, 434 CrossRef CAS.
- E. J. Creyghton, M. H. W. Burgers, J. C. Jansen and H. van Bekkum, Appl. Catal. A, 1995, 128, 275 CrossRef CAS.
- J. B. Hoke, G. A. Gramiccioni and E. N. Balko, Appl. Catal. B, 1992, 1, 285 CrossRef CAS.
- S. Chon and D. T. Allen, AIChE J, 1991, 37, 1730 Search PubMed.
- E.-J. Shin and M. A. Keane, Appl. Catal. B, 1998, 18, 241 CrossRef.
- E.-J. Shin and M. A. Keane, Catal. Lett., 1999, 58, 141 Search PubMed.
- G. Tavoularis and M. A. Keane, J, Mol. Catal. A, 1999, 142, 187 CrossRef CAS.
- F. Roessner and U. Roland, J. Mol. Catal. A: Chem., 1996, 112, 401 CrossRef CAS.
- U. Roland, T. Braunschweig and F. Roessner, J. Mol. Catal. A: Chem., 1997, 127, 61 CrossRef CAS.
- N. Couté and J. T. Richardson, Appl. Catal. B, 2000, 26, 217 CrossRef CAS.
- Q. Wu, W. D. Marshall and A. Majid, Green Chem., 2000, 2, 127 RSC.
- A. Kabir and W. D. Marshall, Green Chem., 2001, 3, 47 RSC.
- C. Grittini, G. A. Romeo, Jr., Q. Fernando and N. E. Korte, Complete Dechlorination of Pentachlorophenol, Book of Abstracts, 212th ACS National Meeting, Orlando, FL, August 25–29, ENVR-049, Publisher: American Chemical Society, Washington, D.C, 1996 Search PubMed.
- Y.-H. Kim and E. R. Carraway, Environ. Sci. Technol., 2000, 34, 2014 CrossRef CAS.
- H. M. Yak, B. W. Wenclawiak, I. F. Cheng, J. G. Doyle and C. M. Wai, Environ. Sci. Technol., 1999, 33, 1307 CrossRef CAS.
- D. C. Hinz, C. M. Wai and B. W. Wenclawiak, J. Environ. Monit., 2000, 2, 45 RSC.
- W.-x. Zhang, C.-B. Wang and H. L. Lien, Catal. Today, 1998, 40, 387 CrossRef CAS.
- F. X. M. Casey, S. K. Ong and R. Horton, Environ. Sci. Technol., 2000, 34, 5023 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2002 |