Field measurement of lead in workplace air and paint chip samples by ultrasonic extraction and portable anodic stripping voltammetry†
Aaron
Sussell
and
Kevin
Ashley
*
US Department of Health and Human Services, Centers for Disease Control and Prevention‡, National Institute for Occupational Safety and Health, 4676 Columbia Parkway, Cincinnati, OH 45226-1998, USA. E-mail: KAshley@cdc.gov
First published on 10th January 2002
Abstract
On-site measurement of lead in workplace air filter samples and paint chip samples by ultrasonic extraction and anodic stripping voltammetry (UE-ASV) was evaluated in the field during renovation and remodeling activities in residences having leaded paint. Aerosol and paint samples were collected using standard techniques, and the samples were analyzed on-site for lead content by portable UE-ASV. Lead in sample extracts was subsequently determined by atomic absorption (AA) spectrometry in a fixed-site laboratory. The remaining sample extracts plus undissolved material (air filters or paint particles) were then subjected to hot plate digestion in concentrated nitric acid–30% hydrogen peroxide prior to AA analysis for lead. Field UE-ASV lead data were thereby compared to UE-AA and hot plate digestion-AA results from fixed-site laboratory lead measurement. Determination of lead in air filter samples by UE-ASV (over the range of 5 µg to ∼800 µg Pb per sample) was extremely well correlated with lead measurement by UE-AA and hot plate digestion-AA procedures. However, a significant negative bias associated with ASV measurement was observed, and this was attributed to a matrix effect. Lead measurement in paint chip samples by UE-ASV (over the range of ∼10 to ∼550 µg Pb g−1) was well correlated with lead measurement by UE-AA and hot plate digestion-AA procedures. However, correlation and precision were lower for lead measurement in paint samples as compared to aerosol samples, and a negative bias was also observed. Lead measurements by UE-AA were compared to lead determinations by hot plate digestion-AA; these data were highly correlated and demonstrated no significant bias. Thus it was concluded that the ultrasonic extraction procedure performed equivalently to hot plate digestion. It was reasoned that matrix effects due to the preparation and analysis of paint chip particles resulted in greater imprecision as well as negative bias by ASV measurement. Despite significant negative bias in this sample set, UE-ASV offers promise for on-site measurement of lead in samples of interest in occupational and environmental health.
Introduction
Renovators, painters, and lead hazard reduction contractors commonly encounter lead-containing paint in their work on buildings and other structures. During work activities, lead in paint coatings can be disturbed, and workers may be exposed to aerosols containing excessively high levels of lead.1–3 Also, disturbed lead-containing paint left over from work activities can contaminate the home in the form of settled paint particles on floors, window sills, furniture, etc.1 Lead hazards are of concern not only to residential renovation contractors, but also to homeowners and occupants who do renovation and remodeling work on their own houses. Settled dust resulting from renovation and painting activities, if not adequately cleaned up, can be perilous to children occupying the home.The ability to measure lead concentrations in media such as paint coatings and aerosols in the field would allow for rapid assessment of worker exposures to this toxic metal. On-site analysis results would enable industrial hygienists or other public health professionals to take immediate corrective actions for the avoidance of excessive worker exposures. Hence, a field-portable analytical method has been investigated on-site for the determination of lead in paint and airborne particulate samples collected during renovation and repainting activities. Field-portable methods are especially of interest in renovation, remodeling, painting, maintenance, and other construction activities where jobs are often short-term and the workforce is frequently transient in nature.
In this study, an on-site analytical method involving the use of ultrasonic extraction (UE) of paint and personal and area air samples, followed by field-portable anodic stripping voltammetry (ASV) for the determination of lead, was investigated in the field during renovation and painting activities in residences. Several previous studies have demonstrated the potential of the portable UE-ASV method for lead monitoring in samples of interest for environmental and occupational health purposes.4–8 These efforts have resulted in the promulgation of a US National Institute for Occupational Safety and Health (NIOSH) method for on-site lead measurement,9 as well as ASTM standard procedures for sample ultrasonication10 and electroanalysis11 in the field. It was the purpose of the present study to investigate the on-site performance of field-portable UE-ASV for the measurement of lead in personal and area air samples, as well as paint samples, obtained during work activities. In this way, the UE-ASV procedure was field-tested under realistic conditions, and the performance of the method could be assessed by comparison with results from fixed-site laboratory analysis using atomic absorption (AA) spectrometry. Moreover, the results of this work could be evaluated in terms of NIOSH guidelines for method evaluation.12
Experimental
Sampling
All air samples were collected during lead hazard reduction tasks at two single-family residences in Oshkosh, WI, USA. These residences were over forty years old and of similar age, with unknown painting history. Paint samples from various exterior building components (e.g., siding, window frames, sills, baseboards) were collected prior to initiation of work tasks. The two residences contained significant amounts of lead-containing paint in various stages of deterioration.Workplace air samples were taken in accordance with NIOSH sampling methods.9,13 Both personal and area air samples were obtained, and these included short-term, task-based samples as well as long-term samples. The air samples were collected on pre-loaded 0.8 µm pore size, 37 mm diameter mixed cellulose ester (MCE) membrane filters which were housed in 3-piece (closed-face) plastic sampling cassettes (SKC, Eighty Four, PA, USA). Air samples were collected using a flow rate of 3.0 (±0.1) L min−1 with battery-powered personal sampling pumps (SKC Model 224-PCXR7). Sampling times varied from 10 to 202 min, enabling a wide range of particulate loadings on the filters. Nearly 100 air samples were collected during the study.
Paint samples were collected in accordance with an ASTM standard procedure.14 The paint chip samples (dimensions 2.5 cm × 2.5 cm) were obtained from areas where the paint was peeling or, if there was no peeling paint, by cold scraping with a stainless steel scraper. Inclusion of substrate was minimized to the extent possible. Collected paint samples were placed into 50 mL plastic centrifuge tubes (Becton Dickinson, Lincoln Park, NJ, USA) and capped. Over 40 paint samples were collected during the course of the investigation.
Field sample preparation
Air filter samples were prepared on-site in accordance with NIOSH9 and ASTM10 field extraction procedures. Following sampling, the filters were removed from the sampling cassettes and were placed in 50 mL centrifuge tubes (Becton Dickinson). The filters were then subjected to sonication for 30 min in 10 mL of 10% (v/v) spectroscopic grade nitric acid (Aldrich, Milwaukee, WI, USA) using a laboratory ultrasonic bath (Sonicor Model SC-150/H, Farmingdale, NY, USA). After sonication, the samples were allowed to settle; air filter samples were not diluted prior to analysis.Paint samples were prepared in accordance with ASTM field sample preparation protocols.10,15 The paint chips were crushed using the following procedure.7,15 First the centrifuge tubes containing the samples were cooled with dry ice (CO2(s), purchased locally) for several minutes in order to make the paint brittle. A clean, hard plastic rod was also cooled with dry ice. This rod was then used to crush the super-cooled paint sample within the centrifuge tube. Using the rod, the paint chip samples were ground as finely as possible. Some paint samples could be ground to a fine powder, while other samples which could not be as easily ground were crushed and homogenized until a consistency resembling that of ground black pepper was achieved. Grinding times per sample ranged from ∼5 to ∼15 min. After grinding, the paint samples were weighed on a portable, battery-powered analytical balance (Acculab Pocket Pro C/50, Newtown, PA, USA) to a precision of ±0.002 g. The weighed samples were reintroduced quantitatively into the original 50 mL centrifuge tubes, and were then subjected to sonication (Sonicor Model SC-150/H ultrasonic bath) for 30 min in 10 mL of 25% (v/v) spectroscopic grade nitric acid (Aldrich).10 After sonication, the samples were allowed to settle prior to dilution with deionized water to 50 mL.
Field analysis
Sonicated air filter and paint sample extracts were analyzed on-site for lead content by portable ASV in accordance with NIOSH9 and ASTM11 procedures. A 5 mL aliquot of each extracted sample was placed into a ∼6 mL plastic vial, and a sample preparation tablet (Palintest USA SP-B, Erlanger, KY, USA) was introduced into the sample aliquot. Each tablet (which consisted of supporting electrolyte and oxygen scavenger) was crushed with a clean plastic stirring rod, and the sample aliquot was then capped and mixed thoroughly. Lead was then determined in each sample aliquot (after uncapping) with a portable, battery-powered ASV device (Model SA-5000, Palintest USA) which uses disposable screen-printed electrodes (Type SE-1, Palintest USA). The lower reporting limits (limits of quantitation) for lead content were 5 and 25 µg of lead per sample for air filter and paint samples, respectively.Following field analysis by UE-ASV,16 sample aliquots which had been measured for lead content on-site were recombined into the original sample extracts (contained in 50 mL centrifuge tubes). These samples were then transported to a fixed-site laboratory for atomic spectrometric lead measurement (see below).
Fixed-site laboratory analysis
Aliquots of sonicated extracts (∼5 mL) from air filter and paint samples were analyzed directly for lead content by AA spectrometry using the 283.3 nm lead absorption line. Samples were first analyzed by flame atomic absorption spectrometry (FAAS; Perkin-Elmer Model 5000, Bodenseewerk, Germany); samples giving lead results below the detection limit by FAAS were subsequently analyzed by graphite furnace atomic absorption spectrometry (GFAAS; Perkin-Elmer Model 5100ZL). The remainders of the prepared samples (which consisted of extract solutions plus undissolved filters or paint particles) were digested and analyzed using a modification of NIOSH methods13 in the following manner. (1) The entire contents of the 50 mL centrifuge tubes were quantitatively transferred to 100 mL borosilicate beakers (Cole-Parmer, Vernon Hills, IL, USA). (2) Instrumental grade concentrated nitric acid (EM Science, Gibbstown, NJ, USA), 20 mL, was introduced to each beaker containing the transferred samples. (3) The beakers were then placed on a hot plate (Type 2200, Thermolyne, Dubuque, IA, USA) having a surface temperature of 140 (±5) °C until the sample volume was reduced to approximately 10 mL. (4) Sample solutions were allowed to cool to room temperature, at which point 5 mL of 30% (v/v) low trace metal grade hydrogen peroxide (Curtin Matheson, Houston, TX, USA) was added. (5) These samples were returned to the hot plate and taken to dryness. (6) Digested samples were dissolved in 10 mL of 5% (v/v) nitric acid. (7) Aliquots (∼5 mL) of the digested samples were analyzed for lead content by FAAS (and then by GFAAS if the FAAS result was below the detection limit). The lower reporting limits of lead content for FAAS and GFAAS analysis were 5 µg and 0.1 µg Pb per sample, respectively.Quality control
Matrices for quality control (QC) samples consisted of one representative certified reference material (CRM) each for air filters and paints (National Institute of Standards and Technology [NIST] Standard Reference Materials® [SRMs], Gaithersburg, MD, USA): NIST SRM 3087a (Metals on Filter Media) and NIST 1579a (Lead-Based Paint). QC samples were processed by both field and fixed-site laboratory methods, in the same manner as field samples, at a minimum frequency of 5% (i.e., at least one QC sample per twenty field samples). Acceptance criteria for QC data were as described in pertinent ASTM procedures for on-site17 and fixed-site18 lead measurements.Results
Air filter samples
Fig. 1 shows a plot of results from on-site UE-ASV analysis of workplace air filter samples vs. data from AA analysis of ultrasonic extracts of the same samples in a fixed-site laboratory. Figures of merit from linear regression analysis of lead-in-air measurements are summarized in Table 1. Where applicable, the UE-AA results were volume-corrected to the original volume of 10 mL in order to account for any solution losses from sample transport and handling. The data shown in Fig. 1 are those results for which paired values above the lower reporting limits for both UE-ASV and UE-AA were obtained. The range of lead loadings observed in the air filter samples was ∼2 µg to ∼800 µg Pb per sample (but only to ∼100 µg Pb per sample for house B; see Fig. 1).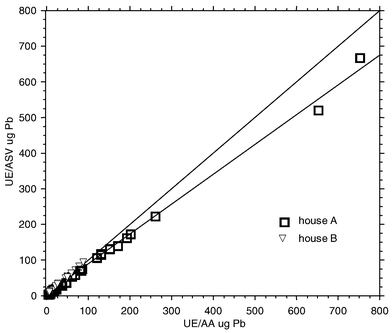 | ||
| Fig. 1 Field measurement of lead in workplace air filter samples: plot of UE-ASV on-site data vs. UE-AA fixed-site laboratory results. (The diagonal line through the graph shows the ideal y = x 1∶1 correspondence line, while the line through the data points represents the best fit regression line.) | ||
A similar analysis of field UE-ASV results against results from AA analysis of samples digested using a hot plate and concentrated nitric acid–30% hydrogen peroxide is also summarized in Table 1. Table 1 also summarizes results from AA analysis of ultrasonic extracts vs. AA results from hot-plate digested samples. The data from air filter sample digestates were volume-corrected in order to account for loss of solution due to aliquots previously used for AA analysis of ultrasonic extracts. Again, only results giving values above the reporting limits for the respective analytical methods were treated by linear regression, and similar ranges of lead-in-air concentrations were observed. Since the AA reporting limits for both sonicated and hot-plate digested samples are lower than for ASV results, regression analysis results from more paired data points after AA analysis are presented.
Paint chip samples
Fig. 2 shows a plot of results from on-site UE-ASV analysis of paint chip samples vs. data from AA analysis of ultrasonic extracts of the same samples in a fixed-site laboratory. Where applicable, the UE-AA results were volume-corrected to 50 mL in order to account for any solution losses from sample transport and handling. All paint chip samples had measurable lead concentrations by both UE-ASV and UE-AA techniques. The range of lead concentrations observed in the paint samples was 11 µg g−1 to 530 µg g−1 Pb, and similar ranges of lead concentrations in paint samples were observed from both houses where the work was carried out. The mean lead concentrations in the two houses were 22% Pb vs. 37% Pb by weight for houses A and B, respectively; this difference was statistically significant. But since no site-specific statistically significant differences in the paint lead concentration ranges were observed, no effort was made to separately analyze the data sets from the two houses.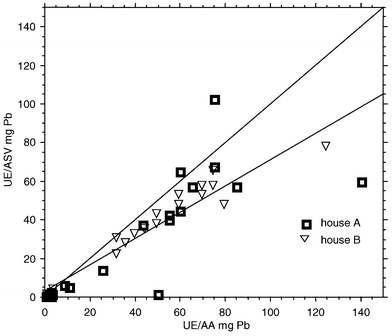 | ||
| Fig. 2 Field measurement of lead in ground paint chip samples: plot of UE-ASV on-site data vs. UE-AA fixed-site laboratory results. (The diagonal line through the graph shows the ideal y = x 1∶1 correspondence line, while the line through the data points represents the best fit regression line.) | ||
Regression analysis of field UE-ASV results against results from AA measurement of samples digested using a hot plate and concentrated nitric acid–30% hydrogen peroxide is summarized in Table 2. Table 2 also summarizes linear regression data from AA analysis of ultrasonic paint chip extracts vs. AA results from extracted paint samples subjected to subsequent hot plate digestion in concentrated nitric acid–30% hydrogen peroxide. Where applicable, results were volume-corrected in order to account for any solution losses from sample transport and handling. All paint chip samples had measurable lead concentrations by all sample preparation and analysis techniques which were used.
QC samples
Analytical results from QC samples (i.e., NIST SRMs 3087a and 1579a) are given in Table 3. For SRM 1579a, the mass of each sample was between 0.15 and 0.25 g.| NIST SRMa | Percent recovery | ||
|---|---|---|---|
| UE-ASVb | UE-AAc | Hot plate-AAd | |
| a Certified lead levels are 40.38 µg Pb/filter and 11.83% Pb by mass for SRMs 3087a and 1579a, respectively. b Ultrasonic extraction (in 10% or 25% (v/v) HNO3 for SRMs 3087a and 1579a, respectively) and anodic stripping voltammetry. c Ultrasonic extraction (in 10% or 25% (v/v) HNO3 for SRMs 3087a and 1579a, respectively) and atomic absorption spectrometry. d Hot plate digestion (in concentrated HNO3/30% H2O2) and atomic absorption spectrometry. | |||
| 3087a (n = 6) | 94.8 (6.6) | 99.0 (1.5) | 98.2 (3.4) |
| 1579a (n = 3) | 95.4 (5.9) | 102.2 (3.7) | 92.9 (8.3) |
Discussion
The data of Fig. 1 and Table 1 show that the results from on-site UE-ASV of workplace air filter samples are extremely well correlated with AA results from fixed-site laboratory analysis. Observed correlation coefficients (r2 values) are very close to unity (Table 1), and are remarkably high for field-based analyses. Also, the y-intercepts of both UE-ASV vs. UE-AA and hot plate-AA regression lines are near the origin, which is indicative of high method selectivity for lead. Additionally, the magnitudes of the standard errors about the calibration lines in the data of Table 1 are quite small. Such a high precision level is an attribute of repeatable measurement techniques. It is also important to note that the apparent recovery of lead by UE-ASV is significantly greater than the 75% criterion specified by NIOSH.12It is noteworthy, however, that the slopes of the regression lines for ASV vs. AA analysis (Table 1) show significant departures from unity. These significant differences from ideal x = y behavior (i.e., slope = 1.0) were confirmed statistically by t-tests of the estimated slopes (p < 0.0001).19 Such an observation is indicative of either low analytical recoveries, or systematic instrumental errors, which in turn could result from calibration errors or matrix effects. Yet in view of the acceptable QC data which are presented in Table 3, calibration errors are ruled out. If calibration errors were the cause of the observed regression lines in Fig. 1 and Table 1 having slopes significantly lower than one, results from QC samples would likely be outside the expected limits (i.e., 100% ± 15% recovery17,18).
Another possible culprit is the extraction procedure. If low lead recoveries resulted from ultrasonic extraction, one would expect a plot of UE-AA vs. hot plate digestion-AA results to show a similar diminished slope. However, this was not observed; rather, the slope of the regression line comparing UE-AA vs. hot plate-AA (Table 1) is not statistically different than 1.0 (p < 0.001), which demonstrates nearly equivalent recovery by the sonication technique as compared to hot plate digestion. Thus it appears here that acceptable lead recovery is obtained by using ultrasonic extraction, which is in agreement with earlier studies.4–8,20–22 Additional leaching of lead from the sample matrix after ultrasonic extraction, i.e., during transport and storage, is another potential confounder which could conceivably result in the observed negative bias in on-site measurements. However, no such time-dependence effects on recoveries have been observed in previous work on UE in performance evaluation materials nor on real-world samples,5–7,22,23 hence significant additional leaching after extraction is considered unlikely.
Having eliminated calibration error and low recovery from ultrasonic extraction as possible causes of negative bias, it appears that a systematic error arising from the ASV measurement may be causing the analytical result to be biased somewhat low. Such an observation could be ascribed to a matrix effect. In this case, a matrix influence might be due to the nature of the paint-derived aerosol particles which arise from the disturbance of painted surfaces during work activities. It is interesting that no such matrix effect has been observed previously.6 But owing to a wide variety of paint formulations24 and substrate materials,7 certain compounds may be present which could cause a diminution in the observed Pb signal from ASV analysis. Indeed, surface poisoning of electrodes by adsorptive species has been observed in electroanalytical techniques (such as ASV) which have been employed historically for industrial hygiene monitoring purposes.25
Fig. 1 shows that the range of measured lead loadings in workplace air filter samples was much greater for work tasks at house A vs. house B. However, the mean lead concentration in paint was greater for house B than for house A. Although different ranges and means of lead loadings were observed from the two houses where the work was performed, these differences were attributed to the fact that more high-exposure paint-removal tasks (e.g., dry scraping1) were carried out at house A. Both houses had mean lead paint concentrations which were very high (considering an action level of 0.5% by weight26), and disturbing this paint could easily give rise to high airborne lead concentrations.3
In view of the data presented in Fig. 1, it is of interest to examine ASV results at the low end of the graph (i.e., between 5 and ∼100 µg Pb). If only those results are considered (n = 65), the slope of the best fit regression line between UE-ASV and UE-AA data increases to 0.929 (compare to slopes of Table 1). Also, the intercept decreases in magnitude (to 1.30 µg), the standard error changes slightly (to 0.011 from 0.006), and the correlation coefficient remains close to unity (0.992). While a negative bias is still present in the low-end data, the magnitude of this bias is about 10% less than for the full data set. Hence it appears that the greatest contribution to negative bias in the full data set is due to results significantly greater than 100 µg Pb per sample. It stands to reason that higher concentrations of interferent might be present in air samples having greater lead particulate loadings, and this may be responsible for the observed negative bias.
The UE-ASV vs. AA data from paint samples, shown in Fig. 2 and Table 2, yielded correlation coefficients (r2 values) which departed significantly from unity. Departure from ideal correlation in Fig. 2 and Table 2 is probably due primarily to a few data points falling significantly off the ideal y = x correlation line. However, no analytical results were excluded from the data sets, and no outlier tests were conducted. The intercepts of the regression lines in Fig. 2 and Table 2 are near the origin, which are indicative of method selectivity for lead. The slopes of the regression lines pertaining to UE-ASV of paint samples are significantly less than unity (p < 0.0001), suggesting either low extraction recovery or an instrumental influence (akin to that described above for air filter samples). Also, there is less precision in UE-ASV lead measurement from paint samples when compared to air samples (compare Figs 1 and 2, Tables 1 and 2), suggesting a matrix influence. The results shown in Fig. 2 and Table 2 are similar to UE-ASV results previously published for on-site determination of lead in paint samples by this field method.7
Results from a comparison of UE-AA vs. hot plate-AA analyses (Table 2) illustrate that AA data from sonicated and digested paint samples are well correlated and unbiased (observed slope is equivalent to 1.0; p < 0.05). The implication is that the sonication procedure is not responsible for the apparent low recoveries of lead from paint chip samples. Also, it is reiterated that QC samples (Table 3) showed no significant biases, thereby eliminating calibration error in UE-ASV measurement as a cause of departure from ideal correlation. Hence, as in the air sample results discussed previously, an electrode surface interference to electrochemical measurement by ASV is suspected to be responsible for the apparent low recoveries.
Additional leaching of lead from the paint sample matrix after ultrasonic extraction, i.e., during transport and storage, is another potential confounder which could conceivably result in the observed negative bias in on-site measurements. As mentioned earlier, such an effect is felt to be unlikely in air filter samples, which have a very small particle size range. However, longer leaching times may be required for ground paint samples in order to achieve complete lead recovery.27 Compared to air filter samples, ground paint samples have a mean particle size which is orders of magnitude larger.28 Yet another variable which cannot be eliminated is the type of paint and/or overlayer coating, which can also influence the ASV measurement by introducing an interference effect.27 Further study is needed in order to investigate these possible contributions to method bias in UE-ASV analysis of paint samples.
The differences in precision between lead determination in aerosol samples and paint samples, regardless of analytical method, are noteworthy (again compare Figs 1 and 2, Tables 1 and 2). It is apparent that the extraction procedure, whether it be sonication or hot plate digestion, more effectively dissolves lead in a reproducible fashion from air filter samples vs. ground paint samples. Such an observation is not surprising, given that aerosol particles captured by air samplers are significantly smaller than paint particles which are prepared on-site, and are therefore more easily attacked by the extraction medium. Some paint particles cannot be ground well,28 and in some cases the lead within certain paint matrices is insoluble regardless of the extraction procedure.7 In other instances, sonication may give greater recovery of lead from paint samples (e.g., lead chromate paints) as compared to hot plate digestion.29 AA analyses can be susceptible to matrix influences,30 although the primary contribution to imprecision in the paint data is probably larger and variable particle size. The extract matrix can also influence ASV measurement by the presence of adsorptive species, as mentioned previously.
If ASV results from paint samples are separated by house, regression analysis yields similar slopes and intercepts: slope = 0.678 for house A (n = 23), and slope = 0.669 for house B (n = 18); the y-intercepts are 2.26 and 4.73 µg Pb g−1 for houses A and B, respectively. However, standard errors (Sy2) and correlation coefficients (r2) are very different for the two sites: Sy2 = 0.096 for house A vs. 0.046 for house B, and r2 = 0.704 for house A vs. 0.930 for house B (these differences were statistically significant). Thus it appears that most of the observed variability in the ASV paint data arises from paint samples collected at house A. But no such differences were observed in the air filter data from the two different houses (see e.g., Fig. 1). Similar differences in UE-ASV results from paint samples have been observed previously in paint samples collected from different substrates.7 In practice, it is very difficult to diminish contributions to variability associated with on-site UE-ASV measurement of lead in paint samples.
Weighted regression was performed on UE-ASV vs. UE-AA and UE-ASV vs. hot plate-AA data (Fig. 2 and Table 2) in order to minimize the influence of apparent outliers on the calibration lines.31 Correlation coefficients (r2 values) for these data comparisons increased to 0.890 for both data set comparisons. The slopes obtained from the weighted regression lines for these data sets also increased significantly (to 0.785 and 0.815, respectively). It is interesting to note that the slopes of the weighted regression lines from paint sample analyses are similar to the slopes obtained from the air filter data of Fig. 1 and Table 1. This indicates a similar bias for the data sets from both aerosol samples and paint samples, suggesting a similar matrix effect for the two sample types. Such an observation may be rationalized since the air particulate samples were derived from the painted surfaces which were disturbed. So it stands to reason that similar matrices were obtained for air filter and paint samples. But the mean particle sizes for the two types of samples are very different, and this apparently resulted in disparities in analytical precision for aerosol and paint samples.
In conclusion, the on-site determination of lead in workplace air filter samples by portable UE-ASV during lead hazard reduction activities was found to satisfy NIOSH accuracy and recovery criteria of ±25% and >75%, respectively.12 Method precision was remarkably high for aerosols generated from lead-containing paints, although a negative bias, attributed to the electroanalytical measurement, was observed. However, UE-ASV method bias was much less over a lower range of lead loadings (to ∼100 µg Pb per air filter sample). For these low-end UE-ASV lead-in-air results, a negative bias of about 7% was estimated by examination of the slope of the best fit regression line. On-site measurement of lead in paint chip samples was less precise and more negatively biased as compared to aerosol samples, but such observations are not surprising in view of matrix influences. Compared to portable X-ray fluorescence (XRF),7,32 the UE-ASV results from this work demonstrated much better precision and similar bias for lead measurements in both matrices studied. Moreover, the detection limits for Pb in air filter and paint samples by UE-ASV are lower than for portable XRF. Higher lead-in-air exposures resulted at one site not because of higher mean lead concentration in the paint that was disturbed, but rather on account of more high-exposure tasks, such as dry scraping.1–3 Although the study is limited in that it was carried out in only two houses, this work has shown that on-site lead measurement by field-portable UE-ASV offers promise for field screening purposes in occupational health applications.33 The method is especially useful for on-site determination of lead in workplace air samples over a range of (at least) 5 µg to ∼800 µg Pb per sample.
Acknowledgements
Technical help from CDC/NIOSH co-workers (G. Piacitelli, T. Wise, B. King, Z. Chaudre, and J. McKernan) is gratefully acknowledged. We thank R. Song for assistance with statistical analyses. R. Streicher, A. Echt, L. Jaycox, and E. Kennedy critically reviewed the draft manuscript. We appreciate the thorough review and commentary offered by the referees.References
- US National Institute for Occupational Safety. and Health (NIOSH), Protecting Workers Exposed to Lead-Based Paint Hazards–A Report to Congress (DHHS [NIOSH] Publ. No. 98–112), ed. A. Sussell, NIOSH, Cincinnati, OH, 1997, revised 1998 Search PubMed.
- A. Sussell, J. Gittleman and M. Singal, Appl. Occup. Environ. Hyg., 1998, 13, 770 CAS.
- A. Sussell, C. Hart, D. Wild and K. Ashley, Appl. Occup. Environ. Hyg., 1999, 14, 177 CrossRef CAS.
- K. Ashley, Electroanalysis, 1995, 7, 1189 CAS.
- US Environmental Protection Agency (EPA), Evaluation Of The Performance Of Reflectance And Electrochemical Technologies For The Measurement Of Lead In Characterized Paints, Bulk Dusts, And Soils, (EPA 600/R-95-093), EPA, Research Triangle Park, NC, 1996 Search PubMed.
- K. Ashley, K. J. Mapp and M. Millson, Am. Ind. Hyg. Assoc. J., 1998, 59, 671 CrossRef CAS.
- K. Ashley, M. Hunter, L. H. Tait, J. Dozier, J. L. Seaman and P. F. Berry, Field Anal. Chem. Technol., 1998, 2, 39 Search PubMed.
- K. Ashley, R. Song, C. A. Esche, P. C. Schlecht, P. A. Baron and T. J. Wise, J. Environ. Monit., 1999, 1, 459 RSC.
- NIOSH Manual Of Analytical Methods, NIOSH Method 7701, ed. M. E. Cassinelli and P. F. O'Connor. NIOSH, Cincinnati, OH, 4th edn., 1998, [2nd Suppl.] Search PubMed.
- Annual Book of ASTM Standards, ASTM E1979, American Society of Testing and Materials (ASTM), West Conshohocken, PA, 2000, Vol. 04.11 Search PubMed.
- Annual Book of ASTM Standards, ASTM E2051, ASTM, West Conshohocken, PA, 2000, Vol. 04.11 Search PubMed.
- E. R. Kennedy, T. J. Fischbach, R. Song, P. M. Eller and S. A. Shulman, Guidelines For Air Sampling And Analytical Method Development And Evaluation, (DHHS [NIOSH] Publ. No. 95-117), NIOSH, Cincinnati, OH, 1995 Search PubMed.
- NIOSH Manual of Analytical Methods, NIOSH Methods 7082 and 7300, ed. M. E. Cassinelli and P. F. O'Connor, NIOSH, Cincinnati, OH, 4th edn., 1994 Search PubMed.
- ASTM Standards On Lead Hazards Associated With Buildings, ASTM E1729, ASTM, West Conshohocken, PA, 1998 Search PubMed.
- ASTM Standards On Lead Hazards Associated With Buildings, ASTM E1645, ASTM, West Conshohocken, PA, 1998 Search PubMed.
- K. Ashley, Appl. Occup. Environ. Hyg., 1998, 13, 94 CAS.
- ASTM Standards On Lead Hazards Associated With Buildings, ASTM E1775, ASTM, West Conshohocken, PA, 1998 Search PubMed.
- ASTM Standards On Lead Hazards Associated With Buildings, ASTM E1613, ASTM, West Conshohocken, PA, 1998 Search PubMed.
- J. C. Miller and J. N. Miller, Statistics For Analytical Chemistry, Ellis Horwood, Chichester, UK, 1984, Ch. 4 Search PubMed.
- K. Ashley, Trends Anal. Chem., 1998, 17, 366 CrossRef CAS.
- K. Ashley, T. J. Wise, W. Mercado and D. B. Parry, J. Hazard. Mater., 2001, 83, 41 CrossRef CAS.
- K. Ashley, R. N. Andrews, L. Cavazos and M. Demange, J. Anal. At. Spectrom., 2001, 16, 1147 RSC.
- M. Millson and K. Ashley, in Measurement Of Toxic And Related Air Pollutants, Vol. 1, Air & Waste Management Association, Pittsburgh, PA, 1997, p. 304 Search PubMed.
- D. Greninger, V. Kollonitsch, C. H. Kline, L. C. Willemsens and J. F. Cole, Lead Chemicals. International Lead Zinc Organization, New York, 1975 Search PubMed.
- K. Ashley, Electroanalysis, 1994, 6, 805 CAS.
- US Department of Housing and Urban Development (HUD), Guidelines for the Evaluation and Control of Lead-Based Paint Hazards in Housing. HUD, Washington, DC, 1995 Search PubMed.
- W. J. Rossiter, M. G. Vangel, M. E. McKnight, A. Signor and W. E. Byrd, Ultrasonic Extraction/Anodic Stripping Voltammetry for Determining Lead in Household Paint: A Laboratory Evaluation (NISTIR 6571). National Institute of Standards and Technology (NIST), Gaithersburg, MD, 2001 Search PubMed.
- S. L. Harper and W. F. Gutknecht, J. Environ. Monit., 2001, 3, 335 RSC.
- G. L. Tinklenberg, personal communication, 2001.
- J. A. Dean, Analytical Chemistry Handbook. McGraw-Hill, New York, 1995, Ch. 7 Search PubMed.
- N. R. Draper and H. Smith, Applied Regression Analysis, 2nd edn. Wiley, New York, 1981, pp. 108–116 Search PubMed.
- J. C. Morley, C. S. Clark, J. A. Deddens, K. Ashley and S. Roda, Appl. Occup. Environ. Hyg., 1999, 16, 306 CrossRef CAS.
- R. Song, P. C. Schlecht and K. Ashley, J. Hazard. Mater., 2001, 83, 29 CrossRef CAS.
Footnotes |
| † This article was prepared by US Government employees as part of their official duties, and legally may not be copyrighted in the United States of America. |
| ‡ Disclaimer: Mention of company names or products does not constitute endorsement by the Centers for Disease Control and Prevention. |
| This journal is © The Royal Society of Chemistry 2002 |