The role of GC-MS and LC-MS in the discovery of drinking water disinfection by-products
Susan D. Richardson
United States Environmental Protection Agency, National Exposure Research Laboratory, Ecosystems Research Division, 960 College Station Rd., Athens, GA 30605, USA. E-mail: richardson.susan@epa.gov
First published on 9th November 2001
Abstract
Gas chromatography-mass spectrometry (GC-MS) has played a pivotal role in the discovery of disinfection by-products (DBPs) in drinking water. DBPs are formed when disinfectants, such as chlorine, ozone, chlorine dioxide or chloramine, react with natural organic matter in the water. The first DBP known—chloroform—was identified by Rook in 1974 using GC-MS. Soon thereafter, chloroform and other trihalomethanes were found to be ubiquitous in chlorinated drinking water. In 1976, the National Cancer Institute published results linking chloroform to cancer in laboratory animals, and an important public health issue was born. Mass spectrometry and, specifically, GC-MS became the key tool used for measuring these DBPs in water and for discovering other DBPs that were formed. Over the last 25 years, hundreds of DBPs have been identified, mostly through the use of GC-MS, which has spawned additional health effects studies and regulations. Early on, GC with low resolution electron ionization (EI)-MS was used, together with confirmation with chemical standards, for identification work. Later, researchers utilized chemical ionization (CI)-MS to provide molecular weight information and high resolution EI-MS to aid in the determination of empirical formulae for the molecular ions and fragments. More recently, liquid chromatography-mass spectrometry (LC-MS) with either electrospray ionization (ESI) or atmospheric pressure chemical ionization (APCI) has been used to try to uncover highly polar DBPs that most experts believe have been missed by earlier GC-MS studies. Despite 25 years of research in the identification of new DBPs, new ones are being discovered every year, even for chlorine which has been the most extensively studied.
 Susan D. Richardson Susan D. Richardson |
1 Introduction
Disinfection by-products (DBPs) are formed when a disinfectant, such as chlorine or ozone, reacts with organic matter that is naturally present in the water.1 This natural organic matter (NOM) is in nearly all source waters that are used for drinking water, including lakes, rivers and even many groundwaters. NOM is a complex, heterogeneous mixture of substances, such as humic acids, fulvic acids, amino acids, carbohydrates, lipids, lignins, waxes and organic acids. Fulvic and humic acids have been shown to be the primary precursor materials for the formation of DBPs.2–4Fig. 1 shows an example of one of the structural models for fulvic acid.5 Due to its complex nature, and the ability of this material to form complex aggregates, it has not been possible to determine an exact structure for a particular fulvic or humic acid. Other recently characterized portions of NOM, ‘hydrophilic and transphilic fractions’, which are characterized in composition by carbohydrates, amino-sugars, alcohols and organic acids, are also proving to be important DBP precursors, as well as the so-called ‘colloidal fraction’, characterized by carbohydrates, amino-sugars, deoxyribonucleic acid (DNA), polyhydroxybutyrates and fatty acids.6 | ||
| Fig. 1 Proposed average structural model of Suwannee River fulvic acid.5 | ||
The reaction between the disinfectant and NOM occurs because most disinfectants used for treating drinking water are also powerful oxidants; they oxidize (and some also halogenate) NOM, in addition to killing harmful microorganisms in the water. It is vitally important to disinfect water because, prior to the use of disinfectants for treating drinking water, millions of people died from water-borne illnesses, such as cholera and typhoid. With the advent of disinfection in the early 1900s, these deaths virtually ceased in developed nations. However, there is concern about the chemical DBPs formed, because some have been linked to cancer in laboratory animals,7–17 and many others still remain to be studied. There are also more recent concerns about possible reproductive and developmental effects from exposure to DBPs.18–29 DBPs that are now regulated by the US Environmental Protection Agency (EPA) are listed in Table 1;30,31 the maximum contaminant levels (MCLs) shown in Table 1 represent the maximum concentrations allowed under the terms of the regulations. Also listed in Table 1 are the World Health Organization (WHO) guidelines for DBPs and the European Union DBP standards.
| US EPA regulations | |
|---|---|
| DBP | MCL/µg L−1 |
| Total THMs | 80 |
| Five haloacetic acids | 60 |
| Bromate | 10 |
| Chlorite | 1000 |
| World Health Organization (WHO) guidelines | |
|---|---|
| DBP | Guideline value/µg L−1 |
| Chloroform | 200 |
| Bromodichloromethane | 60 |
| Dibromochloromethane | 100 |
| Bromoform | 100 |
| Dichloroacetic acid | 50b |
| Trichloroacetic acid | 100b |
| Bromate | 25b |
| Chlorite | 200b |
| Chloral hydrate (trichloroacetaldehyde) | 10b |
| Dichloroacetonitrile | 90b |
| Dibromoacetonitrile | 100b |
| Trichloroacetonitrile | 1b |
| Cyanogen chloride (as CN) | 70 |
| 2,4,6-Trichlorophenol | 200 |
| Formaldehyde | 900 |
| European Union standards | |
|---|---|
| a The total THMs represent the sum of the concentrations of the THMs—chloroform, bromoform, bromodichloromethane and dibromochloromethane. They have been regulated in the USA since 1979,30 but their MCL was recently lowered from 100 to 80 µg L−1 under the Stage 1 Disinfectants/DBP (D/DBP) Rule.31 WHO guidelines on THMs state that the sum of the ratio of the concentration of each THM to its respective guideline value should not exceed unity. The five haloacetic acids represent the sum of the concentrations of monochloro-, dichloro-, trichloro-, monobromo- and dibromoacetic acid. These haloacetic acids, together with bromate and chlorite, were regulated for the first time under the Stage 1 D/DBP Rule.31 WHO guidelines can be found at www.who.int/water_sanitation_health/GDWQ/Summary_tables/Tab2d.htm. European Union drinking water standards can be found at www.nucfilm.com/eu_water_directive.pdf.b Provisional guideline value.c Where possible, without compromising disinfection, member states should strive for a lower value. This value must be met, at the latest, 10 calendar years after issue of the Directive (November 3, 1998); within 5 years of the Directive, a value of 25 µg L−1 must be met. | |
| DBP | Standard value/µg L−1 |
| Total THMs | 100 |
| Bromate | 10c |
Mass spectrometry started to play a key role in the analysis of drinking water in the early 1970s. Gas chromatography-mass spectrometry (GC-MS) allowed researchers to separate complex mixtures and identify the individual pollutants/analytes. GC-MS involves the introduction of a small amount of sample extract (usually 1–2 µL) into a heated injection port, where the chemical mixture is vaporized, and introduced onto a chromatographic column. The individual compounds are separated on this column and elute through the column into the mass spectrometer, where they are ionized and analyzed. A more complete discussion of GC-MS is available elsewhere.32–34 Packed GC columns were used initially—capillary GC columns were not widely available until the mid-1970s to early 1980s. Early capillary GC experiments involved glass capillary columns, which could be easily broken. The development of fused silica columns helped to make GC more robust, and brought a proliferation of various stationary phases that could be used to separate a variety of different chemical mixtures. Fused silica columns have since remained the standard of GC analysis.
Yet, even with the earlier GC and GC-MS equipment and techniques of the 1970s, an amazing amount of information was gained during this time. In particular, Rook identified the first DBP ever known—chloroform—in 1974,35 as well as the other trihalomethanes (bromoform, bromodichloromethane and dibromochloromethane). In the following year, the US EPA published the results of a national survey which showed that chloroform and the other trihalomethanes were ubiquitous in chlorinated drinking water,36 and, in 1976, the National Cancer Institute published results linking chloroform to cancer in laboratory animals.37 As a result, an important public health issue was born.
With the advent of capillary GC columns and a variety of stationary phases that could be used to separate different compounds, as well as computer-assisted analysis, GC-MS increasingly played a pivotal role in the discovery of drinking water DBPs. Over the last 25 years, more than 500 DBPs have been identified.1 The identification of unknown DBPs continues to be important because, even with the tremendous amount of work that has already taken place, more than 50% of the total organic halide (TOX) formed in chlorinated drinking water remains unknown.38 Similarly, over 60% of the assimilable organic carbon (AOC) formed in ozonated drinking water remains unknown.39Fig. 2 illustrates what we currently know about the TOX from chlorinated drinking water and AOC from ozonated drinking water.
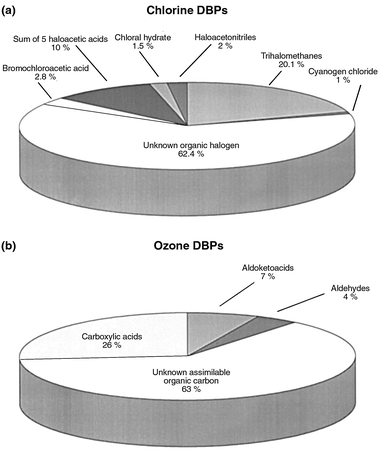 | ||
| Fig. 2 (a) Relative amounts of halogenated DBPs as a proportion of total organic halogen (TOX) in chlorinated drinking water.38 (b) Relative amounts of ozone DBPs in drinking water as a proportion of the total assimilable organic carbon (AOC).39 TOX and AOC data are representative values from a particular chlorination plant and an ozonation demonstration plant, respectively. Data collected by Stuart W. Krasner, Metropolitan Water District of Southern California. Adapted with permission. | ||
Chlorine is still the most widely used disinfectant and has been the most extensively studied. However, the so-called alternative disinfectants, including ozone, chlorine dioxide and chloramine, are gaining in popularity, due to stricter regulations and concerns about chlorine DBPs and also the superior disinfection of ozone and chlorine dioxide for inactivating resistant pathogens. Thus, more recent efforts have focused on identifying the DBPs from the alternative disinfectants, about which less is known.40–64 Combinations of disinfectants have also been studied, as disinfectants such as ozone or chlorine dioxide are almost never used alone in drinking water treatment.40,41,45,47,48 For example, ozone has a relatively short half-life and, in order to maintain disinfection throughout the distribution system, a small dose of chloramine or chlorine is typically used at the end of the plant (after most microorganisms have been killed) to prevent the regrowth of bacteria and other microorganisms in the system.
The objectives of this paper are to review the history of drinking water DBPs and the role that GC-MS and liquid chromatography-mass spectrometry (LC-MS) have played in their identification. Also addressed in this paper are the current research needs for DBPs, and suggestions for the future direction of research in this area.
2 Identifying DBPs with GC-MS
Fig. 3 shows a typical GC-MS chromatogram for a drinking water extract. In any given extract, there may be as many as 300 compounds detected. Many of these will be naturally occurring compounds or pollutants that were already present in the source water prior to disinfection, and many will be DBPs. Because drinking water extracts are usually complex mixtures, such as that shown in Fig. 3, careful background subtraction is important to obtain a ‘clean’ mass spectrum (one that does not contain ions from another closely eluting compound). Table 2 lists examples of DBPs that have been identified for chlorine disinfection. These DBPs include halogenated chemicals, such as the trihalomethanes, haloacetic acids, haloketones, haloacetonitriles, haloaldehydes, haloalcohols, haloamides and halonitromethanes, and non-halogenated chemicals, such as carboxylic acids, aldehydes and ketones.1 Most of these identifications were the result of the use of GC-MS, with careful interpretation of the mass spectra. Before mass spectral library databases (such as NIST or Wiley) were available, mass spectra had to be interpreted ‘by hand’, without the advantage of having a library match. Even today, it is amazing how many DBPs are not found in the large spectral libraries (that contain more than 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 spectra). It is also common to find many similar library entries for a single unknown compound or to find only one isomer in the library for an unknown compound that has several isomer possibilities. As a result, mass spectral interpretation is still a valuable tool.
000 spectra). It is also common to find many similar library entries for a single unknown compound or to find only one isomer in the library for an unknown compound that has several isomer possibilities. As a result, mass spectral interpretation is still a valuable tool. | ||
| Fig. 3 A typical GC-MS chromatogram of a drinking water extract. Each peak represents a different compound; some are DBPs formed by the treatment, some are naturally occurring compounds or pollutants present in the source water prior to disinfection. | ||
| Halogenated DBPs | Examples |
|---|---|
| Trihalomethanes | Chloroform, bromodichloromethane, bromochloroiodomethane |
| Other haloalkanes/alkenes | Dibromomethane, pentachloropropene |
| Halomonocarboxylic acids | Trichloroacetic acid, dichloro-hydroxy-benzoic acid |
| Halodicarboxylic acids | Chloro-hydroxy-dicarboxylic acid |
| Halotricarboxylic acids | 2-Chloro-3-dicarboxy-2-butenoic acid |
| MX and analogs | 3-Chloro-4-(dichloromethyl)-5-hydroxy-2(5H)-furanone (MX), (E)-2-chloro-3-(dichloromethyl)-4-oxobutenoic acid (EMX) |
| Haloketones | 1,1,1-Trichloropropanone |
| Halonitriles | Dichloroacetonitrile, cyanogen chloride |
| Haloaldehydes | Trichloroacetaldehyde (chloral hydrate) |
| Haloalcohols | Chloroisobutanol |
| Haloamides | 2,2-Dichloroacetamide |
| Haloesters | 1-Chloroethanol acetate |
| Halophenols/aromatics | 2-Chlorophenol, chlorobenzene |
| Halonitromethanes | Chloropicrin (trichloronitromethane) |
| Halothiophenes | Tetrachlorothiophene |
| Non-halogenated DBPs | Examples |
|---|---|
| Mono- and dicarboxylic acids | Hexanoic acid, propanedioic acid, benzoic acid |
| Heterocyclic carboxylic acids | 5-Methyl-2-furancarboxylic acid |
| Cyano-carboxylic acids | 3-Cyanopropanoic acid |
| Nitriles | Benzeneacetonitrile |
| Phenols/aromatics | Methylphenol, benzene |
| Aldehydes | Formaldehyde, benzaldehyde |
Typical GC-MS analyses use only one type of MS: low resolution electron ionization (EI). This technique is adequate for identifying most regulated compounds or commonly measured compounds whose mass spectra can be matched with the library database spectra and for which the GC retention time is known. For those cases where the library match is not present or the match is inconclusive, it is helpful to use additional MS techniques, in addition to extensive interpretation of the traditional low resolution EI mass spectrum. High resolution MS can be coupled to GC to obtain exact mass data for the unknowns, which can provide the empirical formula for the unknown structure and empirical formula information for the fragments of the molecule. For example, high resolution MS can be used to determine the number of carbons, hydrogens, oxygens and nitrogens present in the overall structure of the unknown compound and in fragments of the compound, thereby significantly narrowing down the number of structural possibilities. In addition, whenever the ion representing the complete molecule (the molecular ion) is not present by EI-MS, chemical ionization (CI)-MS can reveal it. GC-infrared (IR) spectroscopy can also be used in addition to GC-MS to determine functional group information in the molecule (e.g. whether an oxygen present in the molecule is due to an alcohol, aldehyde, ketone or ether group). Putting this information together to identify the structure of an unknown compound is very much like putting a puzzle together, and analyzing this information is often a back-and-forth process. Finally, when chemical standards are available, the tentative identifications made by GC-MS can be confirmed by a match with the unknown's mass spectrum and GC retention time.
An example of how MS has been used to identify drinking water DBPs is illustrated with the identification of dibromonitromethane. The identification of this DBP was reported in 1999,40,41 and it has recently been shown to be extremely cytotoxic and genotoxic to mammalian cells.65,66 Dibromonitromethane is formed by treatment with chlorine or chloramine and also by treatment with ozone–chlorine and ozone–chloramine.40,41Fig. 4 shows the GC-EI-MS spectrum of this compound. It was not present in either the NIST or Wiley library databases. Halogenated DBPs (containing chlorine or bromine), such as this one, are usually a little easier to identify due to the halogenated patterns they exhibit in their mass spectra. For example, the ions at m/z 171, 173 and 175 form a triplet that is indicative of the presence of two bromines in the structure. This pattern is always the same, no matter how many carbons, hydrogens, oxygens or nitrogens are present in the structure. Similarly, one bromine gives a distinctive pattern, as do three bromines, two chlorines, etc. This is because of the significant natural isotopes of chlorine and bromine. Chlorine has two isotopes at m/z 35 and 37, with a ratio of 3∶1 in abundance. Bromine also has two isotopes at m/z 79 and 81, but they are close to equal in abundance, being nearly 1∶1. The triplet pattern for two bromines comes from the distribution of the bromine isotopes: the first peak in the triplet at m/z 171 arises from the combination of two 79Br isotopes, the second peak at m/z 173 from the combination of one 79Br and one 81Br, and the third peak at m/z 175 from the combination of two 81Br isotopes (the remaining mass comes from the carbon and hydrogen present in the structure). The patterns are formed similarly for various combinations of chlorines and bromines. So, immediately from the mass spectrum, it was evident that there were at least two bromines in the unknown's structure.
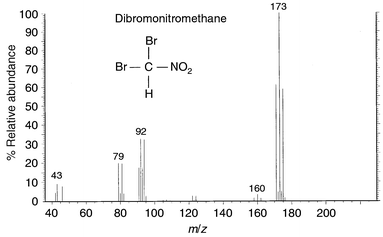 | ||
| Fig. 4 EI mass spectrum of a DBP identified as dibromonitromethane. | ||
Knowing that m/z 171 contained two bromines, it was likely that the missing mass of 13 was due to CH. This provided evidence that the cluster of ions at m/z 171, 173 and 175 was not the molecular ion; hence, the molecular weight information and the overall composition of the structure were missing from the mass spectrum. The dibromonitromethane molecule had completely fragmented under the high energy EI conditions. The absence of the molecular ion is not uncommon. When molecular ions are missing, a ‘softer’ (lower energy) ionization technique, CI-MS, can be used to generate a pseudo-molecular ion. CI-MS involves the initial electron ionization of a gas, such as methane, isobutane or ammonia. In practice, the gas is introduced into the mass spectrometer ion source and is ionized prior to the introduction of the sample.33 The gas forms ions and collides with the sample molecules, imparting a charge to them. Generally, (M + H)+ ions are formed under positive ion conditions, so that if the original molecular weight was 200, the ion formed under CI conditions would be m/z 201. CI-MS is successful in most cases for revealing the pseudo-molecular ion. However, in the case of dibromonitromethane, a molecular ion could not be produced, probably due to the strong electronegativity of this molecule.
However, when the high resolution EI data (at 10000 resolution) were analyzed, the missing half of the molecule was evident in the lower part of the mass spectrum. The ion at m/z 43, which most often is C3H7, was clearly the more unusual CHNO fragment, as indicated by its exact mass of 43.006 Da. The ion at m/z 46 was determined to be NO2, with an exact mass of 45.993 Da. Therefore, from the remaining ions identified as CHNOBr (m/z 122/124), Br2
(m/z 158/160/162) and CHBr2
(m/z 171/173/175), the complete structure was postulated to be dibromonitromethane. In this case, GC-IR was also used to confirm the presence of the NO2 group, and a standard was synthesized to confirm its identity (a chemical standard was not commercially available).
The identification of the DBP, MX, is also a classic example of how MS and GC-MS have been used to identify DBPs. The formal name for MX is 3-chloro-4-(dichloromethyl)-5-hydroxy-2(5H)-furanone; however, it is present in a ring-opened, oxo-butenoic acid form at the pH of drinking water (Fig. 5). This single DBP has been shown to be responsible for 20–50% of the mutagenicity of chlorinated drinking water (in the bacterial Ames test).67 At the time it was identified, MX was the most mutagenic DBP ever identified in drinking water, and it has also been recently shown to be an animal carcinogen.17 The identification of this potent mutagen eluded researchers for quite some time because of its low concentration and its polarity. In fact, MX got its name from being called ‘Mutagen X’ until it was finally identified. It was originally identified in pulp mill effluent,68,69 but was later found in chlorinated drinking water from a number of samples taken around the world.67,70–73 Other analogs of MX were also later identified in chlorinated drinking water, including its geometric isomer (EMX),67,71 oxidized and reduced forms of MX (ox-MX and red-MX),74 as well as brominated analogs (the so-called BMXs).75,76
 | ||
| Fig. 5 Closed and open forms of MX (a pH-sensitive equilibrium). | ||
The initial identification of MX in pulp mill effluent involved the use of high resolution MS and IR spectroscopy, after extensive purification by fractionation.68 MX, in its original form, cannot be analyzed by GC. However, GC and GC-MS analyses of MX can be made if MX is methylated or derivatized by other means.64,69 Today, MX analyses are commonly made by methylating drinking water extracts and analyzing them by GC-MS.
3 Limitations of GC-MS
Although GC-MS has been and continues to be the most effective analytical technique for identifying unknown DBPs in drinking water, its use is limited to the lower molecular weight fraction of DBPs. As the molecular weight of a molecule increases, generally it becomes less volatile, and is not amenable to GC. A study by Khiari et al.77 has shown that a significant portion of the unidentified TOX in chlorinated drinking water is greater than 1000 Da in molecular weight. This fraction would probably not be volatile enough to permit analysis and identification by GC-MS. Thus, this is one limitation of GC-MS.Another limitation involves the analysis of ionic DBPs. Some ionic DBPs, such as haloacetic acids, can still be extracted from water and analyzed by GC-MS if the pH is lowered sufficiently below their pKa value to render them neutral. Other ionic DBPs, such as bromate, which is a DBP from ozone and is a potent carcinogen in rats,16 must be analyzed and identified by techniques other than GC-MS. Ion chromatography and inductively coupled plasma (ICP)-MS techniques are the common methods used for ionic DBPs.78–82
Also, highly polar, hydrophilic compounds are not amenable to direct analysis by GC-MS. First, they are generally very difficult or impossible to extract from water. Extraction into an organic solvent is necessary for GC-MS analysis because water cannot, in general, be injected directly onto a GC, and samples need to be concentrated so that the low levels (ng L−1 to µg L−1) of DBPs can be detected. In addition, polar compounds do not chromatograph well by GC. For example, carboxylic acids tend to ‘smear out’ across the GC column, producing broad peaks that can coelute with other compounds and can be difficult to detect.
One way to overcome these obstacles is through derivatization of the polar DBP and subsequent GC-MS analysis. This has been successfully performed for carboxylic acid DBPs: when they are methylated, the once broad GC peaks become sharp and defined, allowing better detection and good separations. Derivatizations have also enabled the analysis and identification of polar carbonyl DBPs from ozone disinfection. A popular derivatizing agent, pentafluorobenzylhydroxylamine (PFBHA), has been successfully used to react with polar carbonyl compounds, such as formaldehyde, cyanoformaldehyde, propanal and glyoxal (Fig. 6, top). This reaction takes place directly in water and renders the polar compounds non-polar, allowing them to be extracted from water and analyzed by GC-MS.40–43,83 The PFBHA derivatives also exhibit a base peak in their mass spectra at m/z 181, which enables all of the derivatives to be quickly distinguished from surrounding, non-derivatized chemicals in the complex mixture. In some cases, so-called double derivatizations have been used to enable the analysis of polar DBPs by GC-MS. For example, Xie and Reckhow84 have used PFBHA derivatization, followed by methylation, to enable the GC-MS detection of aldo- and keto-acids, such as glyoxylic acid and pyruvic acid.
 | ||
| Fig. 6 Top: PFBHA derivatization procedure that aids in the extraction and identification by GC-MS of polar carbonyl DBPs in drinking water. PFBHA derivatives show a base peak at m/z 181 in their mass spectra. Bottom: DNPH derivatization procedure that aids in the identification of highly polar carbonyl DBPs in drinking water by LC-MS. The derivatives are not initially ionized, which allows for preconcentration by conventional C18 solid phase extraction. The nitro groups cause the NH group to be acidic and ionize under electrospray ionization conditions; abundant negative ions are formed. | ||
4 Identifying DBPs with LC-MS
Another way to analyze hydrophilic DBPs in drinking water is with LC-MS. Electrospray ionization (ESI) and atmospheric pressure chemical ionization (APCI) are currently the most effective ionization techniques being used with LC-MS, permitting the lowest detection limits. LC-MS involves the introduction of a solution (usually aqueous, typically in amounts of 5–50 µL) onto a chromatographic column, which separates the individual components. As they elute from the column, these compounds enter the mass spectrometer interface, where they are desolvated, ionized and introduced into the mass spectrometer for analysis. Thus, LC-MS differs from GC-MS in that it does not require the analyte to be volatile, which enables the analysis of non-volatile and ionic compounds. LC-MS also allows for the direct analysis of water samples, which often removes the need to extract the analytes into an organic solvent and enables the direct analysis of highly polar, hydrophilic DBPs that are difficult or impossible to extract from water. A more complete discussion of LC-MS can be found in the reference book by Siuzdak.85 Ideally, DBPs should be able to be directly analyzed and identified by LC-MS with the original water sample, but this has been problematic. Low molecular weight (<200 Da) DBPs are virtually impossible to distinguish from the high chemical background inherent in the low molecular weight region of the LC-MS chromatogram. The chemical background is due to the solvents used with LC-MS (e.g. water, acetonitrile, methanol). The chemical background is made up of protonated molecular ions (in the positive ion mode) of these solvents, together with a number of dimers, trimers and adducts of these solvents with sodium and other ions present. In many cases, targeted analytes can be measured by LC-MS, given known masses and retention times.86 However, because chromatographic peaks in this low mass region do not generally rise above the baseline of the chemical background, they are not readily distinguished. This makes unknown identifications extremely difficult for the direct analysis of water. Also, without prior concentration, the low levels of DBPs further complicate the situation.As a result, the most successful studies to date involving the identification of unknown DBPs by LC-MS have also employed derivatization. Derivatization has increased the effectiveness of LC-MS by allowing extraction and preconcentration of the polar compounds, by increasing the molecular weight to a region above the chemical background and by imparting a readily ionizable group to the molecule, thus increasing the sensitivity for detection by MS. An example of this is the use of dinitrophenylhydrazine (DNPH) for derivatizing highly polar carbonyl DBPs (Fig. 6, bottom). This derivatization allows highly polar DBPs from ozonated drinking water to be preconcentrated (which improves their detection); it imparts significant molecular weight to the molecule to boost molecular weights above 200 Da (and out of much of the high chemical background region), and gives the nearly unionizable aldehydes and ketones an ionizable group that permits sensitive detection by negative ion LC-ESI-MS.49
Another derivatization technique that is proving to be promising for utilization with LC-MS is the use of 4-dimethylamine-6-(4-methoxy-1-naphthyl)-1,3,5-triazine-2-hydrazine (DMNTH).87 Like DNPH derivatization, this derivatization method was initially developed for the analysis of carbonyl compounds in air. However, it has been recently shown to be amenable to the analysis of carbonyl compounds in water. Due to the design of this reagent for ultimate sensitivity (by UV and fluorescence, as well as by LC-MS),88–90 it is possible to take as little as 0.5 mL of a drinking water sample, derivatize and inject directly, without preconcentration.87 DMNTH has recently been shown to be effective for detecting and identifying highly polar ozone DBPs, such as pyruvic acid, glyoxylic acid and 5-ketohexanal, in drinking water.87
5 After discovery
Once the DBPs are discovered and identified, optimized analytical methods can be developed for their quantification in drinking water. For these quantitative analyses, GC or LC can often be used without MS, as the retention times are known, and chemical standards would be available for matching. GC with electron capture detection (ECD) is typically used for analyzing halogenated DBPs, as it provides improved detection for halogenated compounds. When MS is used as a quantitative method, often selected ion monitoring is employed (where one or more mass spectral ions are monitored, rather than scanning the entire mass range). This often offers improvements in detection of 1000-fold. Therefore, methods can be refined and optimized for future measurements once the DBPs are known. However, until they are known, there is still a tremendous need for traditional GC-MS and LC-MS, with extensive interpretation of the spectra by the analyst.6 A look to the future
What next? With a significant percentage of DBPs still unaccounted for (>50% of the TOX from chlorinated drinking water, >60% of the AOC from ozonated drinking water, and less known about other disinfectants), and increased public concern over the potential carcinogenicity and reproductive/developmental toxicity to humans, innovative ideas and further work are needed to ensure that the public is adequately protected. An international workshop, sponsored by the International Life Sciences Institute (ILSI, Washington, DC), in 1998, addressed this very issue. The workshop was entitled, ‘Identification of New and Uncharacterized Disinfection By-products in Drinking Water’, and had the primary goal of developing an analytical strategy for the identification, quantification and prioritization of the full range of potential DBPs in drinking water. The results from this workshop have been summarized in a report91 and also by Weinberg92 in an Analytical Chemistry article entitled, ‘Disinfection by-products in drinking water: the analytical challenge’. The workshop brought together scientists with expertise in toxicology, analytical chemistry, physical chemistry, organic and inorganic chemistry, analytical instrumentation, drinking water chemistry and engineering and other areas of environmental chemistry. Many non-drinking water scientists with creative analytical chemistry and instrumentation ideas were brought in to bring to bear new ideas for uncovering the missing DBPs. At this workshop, an expert panel predicted the types of compounds that might be missing. Examples of such compounds include bromoamines, nitrosoamines, chloroketamines, chloroaldamines, polycarboxylic acids and polyamines, organic peroxides and reaction products of disinfectants with carbohydrates or alkylphenol ethoxylates (used as surfactants).91 Experts then presented isolation and concentration techniques and analytical methods (including derivatizations and other analytical instrumental techniques) that could possibly be used to identify some of the unidentified fractions of DBPs.The general consensus was that there are a lot of existing and new techniques that are potentially applicable for the isolation and identification of classes of DBPs that are difficult to characterize (e.g. the polar and non-volatile fractions). These existing and new techniques should be focused upon likely ‘unknown’ DBPs using predictive oxidation chemistry and the likelihood of toxicity. New toxicity screening assays on carcinogenic, reproductive and receptor-mediated effects might also be useful to assist with prioritizing which fractions of DBPs to address first.
Existing techniques discussed included GC-atomic emission detection (AED), which could provide elemental information for chromatographically resolved compounds,93 and combinations of MS and IR techniques (GC coupled to high and low resolution EI-MS, CI-MS and IR), which have been successful in uncovering a number of previously unidentified DBPs.40,41,44–48 New techniques that have not been widely applied to drinking water DBPs included LC coupled to ESI-MS (particularly for identifying polar and higher molecular weight DBPs), membrane introduction mass spectrometry (MIMS) (for continuous in situ water analysis), membrane preconcentration-capillary electrophoresis MS, high volume injection GC-MS (to provide increased sensitivity to GC-MS) and MS-MS. New derivatization techniques discussed included the use of DNPH, chloroformates,94 ferrocenes95 and pentafluorobenzyl alcohol.96–98 Existing derivatization techniques, such as methylation (to indicate the presence of carboxylic acids), acylation with acetic anhydride (to indicate the presence of a phenolic hydroxyl group) and reaction with hydrobromic acid (to determine the presence of an ether functional group), were also discussed. In particular, acylation and reaction with hydrobromic acid have been underutilized for identifying unknown DBPs. Concentration techniques recommended included the on-line coupling of solid phase extraction (SPE) to LC-MS and to GC-MS, and solid phase microextraction (SPME), supercritical fluid extraction (SFE) and semipermeable membrane devices (SPMDs) (for hydrophobic DBPs).
A new analytical instrument, that had not been developed at the time of the ILSI workshop, has recently been shown to provide sensitive analyses for haloacetic acids, bromate and other compounds of environmental concern. This instrument is called a high field asymmetric waveform ion mobility spectrometer (FAIMS).99,100 It is interfaced to ESI-MS and works by separating gas phase ions at atmospheric pressure and room temperature. FAIMS has been shown to effectively reduce background ions that are inherent with ESI-MS, allowing lower detection limits, particularly for compounds below 300 Da. Preconcentration is not required; water samples are diluted with methanol to enable an efficient ESI process and then injected directly. Because no chromatographic columns are involved (ions are separated by the tuning of a voltage on the FAIMS device to allow transmission of a selected ion), analysis times are shortened and the cost per sample is low. Currently, it is being used to analyze target analytes in water; however, its use may be expanded to the identification of unknowns. Another analytical method not mentioned at the ILSI workshop, matrix-assisted laser desorption ionization (MALDI)-MS, has been shown to be successful for analyzing extremely high molecular weight material, including large proteins and microorganisms.85,101–104 This technique, as well as ESI-MS, may prove to be useful in providing information on higher molecular weight, non-volatile DBPs that are not amenable to traditional GC-MS techniques. However, this will be an arduous task, as it is often difficult to identify compounds with molecular weights <300 Da. The number of possible atom combinations and isomers increases dramatically with increasing molecular weight, making this a tremendous challenge.
Before a large effort is made to identify the high molecular weight DBP fraction, it would be useful to determine whether there is a ‘molecular weight cut-off’ in toxicity. Toxicologists, in general, often use a molecular weight of 5000 Da as an upper bound, above which chemicals would not be expected to be absorbed in the body (and therefore would not be toxic).91 It would be useful for researchers to conduct a study to test this hypothesis. If a ‘molecular weight cut-off’ could be determined, this could be used to focus future DBP identification work. It is possible, however, that this high molecular weight material could be broken down in the digestive tract to smaller, absorbable chemicals. Thus, this should be considered before DBPs above a predetermined ‘molecular weight cut-off’ are dismissed as toxicologically unimportant.
Another idea would be to focus on the brominated (and possibly iodinated) fraction of DBPs as a priority. This was suggested at this workshop, as well as at an international workshop on ‘Exposure Assessment for Disinfection By-Products in Epidemiologic Studies’, held in Ottawa, Canada, in 2000.105 The brominated DBPs are generally more toxic (carcinogenic) than their chlorinated analogs. Experiments and identification efforts could be focused on this class of DBP. Labeling studies, such as using 36Cl-labeled chlorine or chloramine, may also be useful for locating and characterizing the higher molecular weight TOX. Minear's group at the University of Illinois have recently begun such an effort by reacting NOM with 36Cl-labeled chlorine, fractionating with ultrafiltration/size exclusion chromatography and studying the DBP fractions with ESI-MS and MS-MS.106
Finally, there is a need for additional fundamental research leading to better characterization of NOM and its reactivity (mechanisms and kinetics) with disinfectants. If this NOM, which serves as the precursor to DBPs, were better understood, it might be possible to predict the formation of particular DBPs in different source waters, and ultimately control their formation.
Once DBPs are identified, occurrence data (quantitative, at many sites) are needed to determine levels of DBPs that people are exposed to through their drinking water. Such an effort is currently in progress. A nationwide DBP occurrence study is underway involving the quantification of ‘high priority DBPs’ in waters across the USA.107, 108 These ‘high priority DBPs’ were selected for study from DBPs that had been reported in the literature to have predicted adverse health effects. Analytical methods were developed for their quantification, and they are currently being measured across the USA to determine how often they occur, under what source water and treatment conditions, and their quantitative levels in drinking water at the drinking water plant and throughout the distribution system. Results from this large effort will help to fill in missing ‘pieces of the pie’ (Fig. 2) and to prioritize new DBPs for future health effects studies.
7 Conclusions
GC-MS has played a pivotal role in the discovery of DBPs in drinking water. Even with all of the work that has taken place over the last 25 years, it is likely that GC-MS will continue to reveal the presence of new DBPs. LC-MS is emerging as a useful analytical tool for identifying highly polar, hydrophilic DBPs, and may be useful for providing information on the higher molecular weight, non-volatile fraction of DBPs. As new analytical techniques are developed, it is hoped that missing fractions of DBPs will be revealed and that we will learn more about the NOM that serves as the precursor to these DBPs. Once DBPs are identified and quantified in waters, health effects studies can be conducted to determine whether any pose a risk to human health and should be controlled. The end goal of this research is to minimize the risk posed by DBPs in drinking water.Acknowledgements
This paper has been reviewed in accordance with the US EPA's peer and administrative review policies and approved for publication. Mention of trade names or commercial products does not constitute endorsement or recommendation for use by the US EPA.References
- S. D. Richardson, in The Encyclopedia of Environmental Analysis and Remediation, ed. R. A. Meyers, Wiley, New York, 1998, pp. 1398–1421 Search PubMed.
- J. J. Rook, Environ. Sci. Technol., 1977, 11, 478 CAS.
- R. F. Christman, D. L. Norwood, D. S. Millington and J. D. Johnson, Environ. Sci. Technol., 1983, 17, 625 CAS.
- D. L. Norwood, J. D. Johnson, R. F. Christman and D. L. Millington, in Water Chlorination: Environmental Impact and Health Effects, ed. R. L. Jolley, W. A. Brungs, J. A. Cotruvo, R. B. Cumming, J. S. Mattice and V. A. Jacobs, Ann Arbor Science, Ann Arbor, MI, 1983, vol. 4, pp. 191–200 Search PubMed.
- US Geological Survey Staff, Humic Substances in the Suwannee River, Georgia; Interactions, Properties, and Proposed Structures, Open File Report 87-557, US Geological Survey, Denver, CO, 1989 Search PubMed.
- C. J. Hwang, S. W. Krasner, M. J. Sclimenti, G. L. Amy, E. Dickenson, A. Bruchet, C. Prompsy, G. Filippi, J.-P. Croué, D. Violleau and J. A. Leenheer, Polar NOM: Characterization, DBPs, Treatment, Report, AWWA Research Foundation, Denver, CO, 2001 Search PubMed.
- R. J. Bull and F. C. Kopfler, Health Effects of Disinfectants and Disinfection By-products, Report, AWWA Research Foundation, Denver, CO, 1991 Search PubMed.
- F. B. Daniel, A. B. DeAngelo, J. A. Stober, G. Olson and N. P. Page, Fundam. Appl. Toxicol., 1992, 19, 159 CrossRef CAS.
- A. B. DeAngelo, F. B. Daniel, B. M. Most and G. R. Olson, Toxicology, 1996, 114, 207 CrossRef CAS.
- A. B. DeAngelo, M. H. George and D. E. House, J. Toxicol. Environ. Health, 1999, 58, 485 CrossRef CAS.
- A. B. DeAngelo, M. H. George, S. R. Kilburn, T. M. Moore and D. C. Wolf, Toxicol. Pathol., 1998, 26, 587 CAS.
- M. H. George, T. M. Moore, S. R. Kilburn, G. R. Olson and A. B. DeAngelo, Toxicol. Pathol., 2000, 28(4), 610 CAS.
- E. S. Hunter, III, E. H. Rogers, J. E. Schmid and A. Richard, Teratology, 1996, 4, 57 CrossRef.
- M. V. Templin, A. A. Constan, D. C. Wolf, B. A. Wong and B. E. Butterworth, Carcinogenesis, 1998, 19, 187 CrossRef CAS.
- M. V. Templin, J. L. Larson, B. E. Butterworth, K. C. Jamison, J. R. Leninger, S. Mery, K. T. Morgan, B. A. Wong and D. C. Wolf, Fundam. Appl. Toxicol., 1996, 23, 109 CrossRef.
- Y. Kurokawa, S. Aoki, Y. Matsushima, N. Takamura, T. Imazawa and Y. Hayashi, J. Natl. Cancer Inst., 1986, 77, 977 CAS.
- H. Komulainen, V.-M. Kosma, S.-L. Vaittinen, T. Vartiainen, E. Kaliste-Korhonen, S. Lotjonen, R. K. Tuominen and J. Tuomisto, J. Natl. Cancer Inst., 1997, 89, 848 CrossRef CAS.
- K. Waller, S. H. Swan, G. DeLorenze and B. Hopkins, Epidemiology, 1998, 9(2), 134 CrossRef CAS.
- S. H. Swan, K. Waller, B. Hopkins, G. Windham, L. Fenster, C. Shaefer and R. R. Neutra, Epidemiology, 1998, 9(2), 136.
- M. D. Gallagher, J. R. Nuckols, L. Stallones and D. A. Savitz, Epidemiology, 1998, 9, 484 CrossRef CAS.
- J. B. Klotz and L. A. Pyrch, Epidemiology, 1999, 10, 383 CrossRef CAS.
- M. D. Kramer, C. F. Lynch, P. Isacson and J. W. Hanson, Epidemiology, 1992, 5, 407.
- F. J. Bove, M. C. Fulcomer, J. B. Klotz, J. Esmarat, E. M. Dufficy and J. E. Savrin, Am. J. Epidemiol., 1995, 141, 850 CAS.
- K. P. Cantor, Cancer Causes Control, 1997, 8, 292 CrossRef CAS.
- K. P. Cantor, C. F. Lynch and M. E. Hildesheim, Epidemiology, 1998, 9, 21 CrossRef CAS.
- D. M. Freedman, K. P. Cantor and N. L. Lee, Cancer Causes Control, 1997, 8, 738 CrossRef CAS.
- W. D. King and L. D. Marrett, Cancer Causes Control, 1996, 7, 596 CrossRef CAS.
- M. A. McGeehin, J. S. Reif, J. C. Becher and E. Mangione, Am. J. Epidemiol., 1993, 138, 492 CAS.
- R. D. Morris, A. M. Audet, I. F. Angellio, T. C. Chalmers and F. Mosteller, Am. J. Public Health, 1992, 82, 965 Search PubMed.
- National Interim Primary Drinking Water Regulations, Fed. Registr., 1979, 44, 68624 Search PubMed.
- National Primary Drinking Water Regulations: Disinfectants and Disinfection By-products; Final Rule, Fed. Registr., 1998, 63(241), 69389 Search PubMed.
- W. L. Budde, Analytical Mass Spectrometry: Strategies for Environmental and Related Applications, American Chemical Society and Oxford University Press, Washington DC, 2001 Search PubMed.
- F. W. McLafferty and F. Tureček, Interpretation of Mass Spectra, University Science Books, Sausalito, CA, 4th edn., 1993 Search PubMed.
- R. M. Silverstein, G. C. Bassler and T. C. Morrill, Spectrometric Identification of Organic Compounds, Wiley, New York, 4th edn., 1981 Search PubMed.
- J. J. Rook, Water Treat. Exam., 1974, 23(2), 234 Search PubMed.
- F. C. Kopfler, R. G. Melton, R. D. Lingg and W. E. Coleman, in Identification and Analysis of Organic Pollutants in Water, ed. L. H. Keith, Ann Arbor Science, Ann Arbor, MI, 1976, pp. 87–104 Search PubMed.
- National Cancer Institute, National Cancer Institute Report on Carcinogenesis Bioassay of Chloroform, Carcinogenesis Program, Division of Cancer Cause and Prevention, Bethesda, MD, 1976 Search PubMed.
- S. W. Krasner, Metropolitan Water District of Southern California, personal communication, 1998.
- S. W. Krasner, B. M. Coffey, P. A. Hacker, C. J. Hwang, C.-Y. Kuo, A. A. Mofidi and M. J. Sclimenti, The Effects of Ozonation and Biofiltration on NOM, Program Summary, Natural Organic Matter Workshop, International Water Distribution Association and Laboratory of Water and Pollution Chemistry, Poitiers, 1996, pp. 14-1–14-4 Search PubMed.
- S. D. Richardson, A. D. Thruston, Jr, T. V. Caughran, P. H. Chen, T. W. Collette, T. L. Floyd, K. M. Schenck, B. W. Lykins, Jr., G.-R. Sun and G. Majetich, Environ. Sci. Technol., 1999, 33, 3368 CrossRef CAS.
- S. D. Richardson, A. D. Thruston, Jr., T. V. Caughran, P. H. Chen, T. W. Collette, T. L. Floyd, K. M. Schenck, B. W. Lykins, Jr., G.-R. Sun and G. Majetich, Environ. Sci. Technol., 1999, 33, 3378 CrossRef CAS.
- M. J. Sclimenti, S. W. Krasner, W. H. Glaze and H. S. Weinberg, Proceedings of the Water Quality Technology Conference, Denver, CO, 1990, AWWA, Denver, CO, 1990 Search PubMed.
- W. H. Glaze, M. Koga and D. Cancilla, Environ. Sci. Technol., 1989, 23(7), 838 CAS.
- J. E. Cavanagh, H. S. Weinberg, A. Gold, R. Sangaiah, D. Marbury, W. H. Glaze, T. W. Collette, S. D. Richardson and A. D. Thruston, Jr., Environ. Sci. Technol., 1992, 26, 1658 CAS.
- S. D. Richardson, A. D. Thruston, Jr., T. W. Collette, K. S. Patterson, B. W. Lykins, Jr., G. Majetich and Y. Zhang, Environ. Sci. Technol., 1994, 28, 592 CAS.
- T. W. Collette, S. D. Richardson and A. D. Thruston, Jr., Appl. Spectrosc., 1994, 48(10), 1181 CAS.
- S. D. Richardson, A. D. Thruston, Jr., T. W. Collette, K. S. Patterson, B. W. Lykins, Jr. and J. C. Ireland, Environ. Sci. Technol., 1996, 30(11), 3327 CrossRef CAS.
- S. D. Richardson, A. D. Thruston, Jr., T. V. Caughran, P. H. Chen, T. W. Collette, K. M. Schenck, B. W. Lykins, Jr., C. Rav-Acha and V. Glezer, Water, Air, Soil Pollut., 2000, 123(1), 95 CrossRef CAS.
- S. D. Richardson, T. V. Caughran, T. Poiger, Y. Guo and F. G. Crumley, Ozone Sci. Eng., 2000, 22(6), 653 Search PubMed.
- W. H. Glaze and H. S. Weinberg, Identification and Occurrence of Ozonation By-Products in Drinking Water, Report, AWWA Research Foundation, Denver, CO, 1993 Search PubMed.
- W. H. Glaze, Environ. Health Perspect., 1986, 69, 151 CAS.
- W. R. Haag and J. Hoigne, Environ. Sci. Technol., 1983, 17, 261 CAS.
- S. D. Killops, Water Res., 1986, 20, 153 CrossRef CAS.
- W. E. Coleman, J. W. Munch, H. P. Ringhand, W. H. Kaylor and D. E. Mitchell, Ozone Sci. Eng., 1992, 14, 51 Search PubMed.
- J. Lawrence, H. Tosine, F. I. Onuska and M. E. Comba, Ozone Sci. Eng., 1980, 2, 55 Search PubMed.
- L. J. Anderson, J. D. Johnson and R. F. Christman, Org. Geochem., 1985, 8, 65 CrossRef CAS.
- R. M. LeLacheur, L. B. Sonnenberg, P. C. Singer, R. F. Christman and M. J. Charles, Environ. Sci. Technol., 1993, 27, 2745 CAS.
- I. N. Najm and S. W. Krasner, J. Am. Water Works Assoc., 1995, 87, 106 Search PubMed.
- S. W. Krasner, J. T. Gramith, E. G. Means, N. L. Patania, I. N. Najm and E. M. Aieta, Proceedings of the Water Quality Technology Conference, Denver, CO, 1991,AWWA, Denver, CO, 1991 Search PubMed.
- C. A. Colclough, J. D. Johnson, R. F. Christman and D. S. Millington, in Water Chlorination: Environmental Impact and Health Effects, ed. R. L. Jolley, Ann Arbor Science, Ann Arbor, MI, 1983, vol. 4, pp. 219–229. Search PubMed.
- A. A. Stevens, Environ. Health Perspect., 1982, 46, 101 CAS.
- P. Backlund, L. Kronberg and L. Tikkanen, Chemosphere, 1988, 17, 1329 CrossRef CAS.
- J. G. Jacangelo, N. L. Patania, K. M. Reagan, E. M. Aieta, S. W. Krasner and M. J. McGuire, J. Am. Water Works Assoc., 1989, 81, 74 Search PubMed.
- R. Kanniganti, J. D. Johnson, L. M. Ball and M. J. Charles, Environ. Sci. Technol., 1992, 26(10), 1998 CAS.
- B. Kundu, S. H. Warren, D. M. DeMarini, S. D. Richardson, E. D. Wagner and M. J. Plewa, presented at the Environmental Mutagen Society Conference, San Diego, CA, 2001 Search PubMed.
- Y. Kargalioglu, E. D. Wagner, S. D. Richardson, R. A. Minear and M. J. Plewa, Environ. Mol. Mutagenesis, 2000, 35, Suppl. 31 Search PubMed.
- L. Kronberg, B. Holmbom, M. Reunanen and L. Tikkanen, Environ. Sci. Technol., 1988, 22, 1097 CAS.
- B. R. Holmbom, R. H. Voss, R. D. Mortimer and A. Wong, Tappi, 1981, 64(3), 172 Search PubMed.
- B. R. Holmbom, R. H. Voss, R. D. Mortimer and A. Wong, Environ. Sci. Technol., 1984, 18, 333 CAS.
- J. Hemming, B. Holmbom, M. Reunanen and L. Kronberg, Chemosphere, 1986, 15, 549 CrossRef.
- L. Kronberg and T. Vartiainen, Mutat. Res., 1988, 206, 177 CAS.
- H. Horth, M. Fielding, H. James, M. Thomas, T. Gibson and P. Wilcox, in Water Chlorination: Chemistry, Environmental Impact and Health Effects, eds. R. L. Jolley, L. W. Condie, J. O. Johnson, S. Katz, R. A. Minear, J. S. Mattice and A. Jacobs, Lewis Publishers, Chelsea, MI, 1990, vol. 6, pp. 107–124 Search PubMed.
- J. R. Meier, R. B. Knohl, W. E. Coleman, H. P. Ringhand, J. W. Munch, W. H. Kaylor, R. P. Streicher and F. C. Kopfler, Mutat. Res., 1987, 189, 363 CAS.
- L. Kronberg, R. F. Christman, R. Singh and S. M. Ball, Environ. Sci. Technol., 1991, 25, 99 CAS.
- H. Horth, M. Fielding, C. P. James, H. A. James and R. D. Gwilliam, Identification of Mutagens in Drinking Water, Final report to the Department of the Environment, DoE 2489-M(P), Wrc plc, Medmenham, 1991 Search PubMed.
- N. Suzuki and J. Nakanishi, Chemosphere, 1995, 30(8), 1557 CrossRef CAS.
- D. Khiari, S. W. Krasner, C. J. Hwang, R. Chinn and S. E. Barrett, Proceedings of the Water Quality Technology Conference, Denver, CO, 1996, AWWA, Denver, CO, 1996 Search PubMed.
- Y. Inoue, T. Sakai, H. Kumagai and Y. Hanaoka, Anal. Chim. Acta, 1997, 346(3), 299 CrossRef CAS.
- H. S. Weinberg and H. Yamada, Anal. Chem., 1998, 70(1), 1 CrossRef CAS.
- J. T. Creed and C. A. Brockhoff, Anal. Chem., 1999, 71(3), 722 CrossRef CAS.
- EPA Method 321.8, Determination of Bromate in Drinking Water by Ion Chromatography Inductively Coupled Plasma-Mass Spectrometry, US EPA, Office of Research and Development, National Exposure Research Laboratory, Cincinnati, OH, 1998 Search PubMed.
- M. Nowak and A. Seubert, Anal. Chim. Acta, 1998, 359(1–2), 193 CrossRef CAS.
- H. Yamada and I. Somiya, Ozone Sci. Eng., 1989, 11(2), 127 Search PubMed.
- Y. Xie and D. A. Reckhow, J. Mass Spectrom., 1997, 32, 99 CrossRef CAS.
- G. Siuzdak, Mass Spectrometry for Biotechnology, Academic Press, New York, 1996 Search PubMed.
- M. L. Magnuson and C. A. Kelty, Anal. Chem., 2000, 72(10), 2308 CrossRef CAS.
- S. D. Richardson and U. Karst, Preprints of Extended Abstracts, American Chemical Society National Meeting, Division of Environmental Chemistry, San Diego, CA, 2001 Search PubMed.
- C. Kempter, W. Pötter, N. Binding, H. Kläning, U. Witting and U. Karst, Anal. Chim. Acta, 2000, 410, 47 CrossRef CAS.
- C. Kempter and U. Karst, Analyst, 2000, 125, 433 RSC.
- C. Kempter, G. Zurek and U. Karst, J. Environ. Monit., 1999, 1, 307 RSC.
- Identification of New and Uncharacterized Disinfection By-products in Drinking Water, Workshop Report, International Life Sciences Institute, Washington DC, 1999 Search PubMed.
- H. Weinberg, Anal. Chem., 1999, 71(23), 801A CrossRef CAS.
- B. D. Quimby and J. J. Sullivan, Anal. Chem., 1990, 62, 1027 CrossRef CAS.
- V. Maurino, C. Minero, E. Pelizzetti, S. Angelino and M. Vincenti, J. Am. Soc. Mass Spectrom., 1999, 10(12), 1328 CrossRef CAS.
- J. M. E. Quirke, C. L. Adams and G. J. van Berkel, Anal. Chem., 1994, 66, 2096 CrossRef.
- R. S. Spaulding, P. Frazey, X. Rao and M. J. Charles, Anal. Chem., 1999, 71, 3420 CrossRef CAS.
- C. J. Chien, M. J. Charles, K. G. Sexton and H. E. Jeffries, Environ. Sci. Technol., 1998, 32(2), 299 Search PubMed.
- P. Frazey, X. Rao, R. Spaulding, B. Beld and M. J. Charles, Int. J. Mass Spectrom., 1999, 190–191, 343 CrossRef.
- D. A. Barnett, R. Guevremont and R. W. Purves, Appl. Spectrosc., 1999, 53(11), 1367 CAS.
- B. Ells, D. A. Barnett, K. L. Froese, R. W. Purves, S. Hrudey and R. Guevremont, Anal. Chem., 1999, 71(20), 4747 CrossRef CAS.
- G. Siuzdak, J. Mass Spectrom., 1998, 33(3), 203 CrossRef CAS.
- R. D. Holland, J. G. Wilkes, F. Rafii, J. B. Sutherland, C. C. Persons, K. J. Voorhees and J. O. Lay, Rapid Commun. Mass Spectrom., 1996, 10(10), 1227 CrossRef CAS.
- M. A. Claydon, S. N. Davey, V. Edwards-Jones and D. B. Gordon, Nature Biotechnol., 1996, 14(11), 1584 CAS.
- T. Krishnamurthy, P. L. Ross and U. Rajamani, Rapid Commun. Mass Spectrom., 1996, 10, 883 CrossRef CAS.
- T. E. Arbuckle, S. E. Hrudey, S. W. Krasner, J. R. Nuckols, S. D. Richardson, P. Singer, P. Mendola, L. Dodds, C. Weisel, D. L. Ashley, K. L. Froese, R. A. Pegram, I. R. Schultz, J. Reif, A. M. Bachand, F. M. Benoit, M. Lynberg, C. Poole and K. Waller, Environ. Health Perspect., in press. Search PubMed.
- X. Zhang and R. A. Minear, Preprints of Extended Abstracts, American Chemical Society National Meeting, Division of Environmental Chemistry, Chicago, IL, 2001 Search PubMed.
- S. W. Krasner, S. Pastor, R. Chinn, M. J. Sclimenti, H. S. Weinbergand S. D. Richardson, Proceedings of the Water Quality Technology Conference, Nashville, TN, 2001, AWWA, Denver, CO, 2001 Search PubMed.
- H. S. Weinberg, S. W. Krasner, and S. D. Richardson, Proceedings of the Water Quality Technology Conference, Nashville, TN, 2001, AWWA, Denver, CO, 2001 Search PubMed.
| This journal is © The Royal Society of Chemistry 2002 |