Direct comparison of the electrocatalytic oxidation of hydrogen by an enzyme and a platinum catalyst†
Anne K.
Jones
a,
Emma
Sillery
a,
Simon P. J.
Albracht
b and
Fraser A.
Armstrong
*a
aDepartment of Chemistry, Oxford University, Oxford, UK OX1 3QR. E-mail: fraser.armstrong@chem.ox.ac.uk
bSwammerdam Institute for Life Sciences, Biochemistry, University of Amsterdam, Plantage Muidergracht 12, NL-1018 TV, Amsterdam, The Netherlands
First published on 21st March 2002
Abstract
It is shown that for molecules of Allochromatium vinosum [NiFe]-hydrogenase adsorbed on a pyrolytic graphite electrode the nickel–iron active site catalyzes hydrogen oxidation at a diffusion-controlled rate matching that achieved by platinum.
Hydrogen oxidation is one of the most vital reactions known, since hydrogen is both an ancient biological fuel1 and also the fuel of the future.2 Limitations of conventional fuel cells have led to attempts to develop biofuel cells, in which the electrocatalyst at either the cathode or anode, or both, is an organism or enzyme.3–6 However, the aptness of biocatalysts for such applications is not established since there have been no experiments conducted to compare their activities directly with those of industrial catalysts. The biological and industrial catalysts differ structurally and mechanistically. [NiFe]-hydrogenases, biological hydrogen oxidation catalysts, are large molecules (98 kDa) that contain a buried bimetallic active site composed of only the base metals nickel and iron, in a sulfur- and carbon-rich coordination environment.7,8 The active site is connected to the surface via a series of Fe–S clusters that transfer electrons through the protein matrix. By contrast, fuel cells typically employ platinum, a precious metal. We now report a direct comparison between the hydrogen oxidation activity of Allochromatium vinosum [NiFe]-hydrogenase (AvH2ase) adsorbed at a pyrolytic graphite ‘edge’ (PGE) electrode and that of platinum deposited on an identical PGE (or gold) electrode. To approach operating conditions for a fuel cell, the experiments have been carried out under 1 atm hydrogen at 45 °C.
Fig. 1 shows hydrogen oxidation current densities (current/electrode area) obtained for an AvH2ase film formed on a PGE electrode, alongside those obtained for the same electrode surface (or gold) coated with platinum.10 These are measured at a potential where the current is virtually independent of potential (see below). The current (i) is directly proportional to turnover rate, and follows the Levich equation, i = 0.62nFAD2/3ω1/2ν−1/6C; where n is the number of electrons in the reaction, F the Faraday constant, A the electrode area, D the diffusion coefficient, ω the electrode rotation rate, ν the kinematic viscosity of the solution and C the concentration of hydrogen. This equation predicts that the current due to a diffusion-controlled process depends linearly on the square root of the rotation rate. The results show that hydrogen oxidation rates by platinum and the active site of AvH2ase are indistinguishable and limited by mass transport of hydrogen to the electrode surface (this is also true for 0.01 and 0.10 atm hydrogen; see ESI†); the average diffusion coefficient for hydrogen determined from these results, 4.1 × 10−5 cm2 s−1 at 45 °C, accords well with the literature value of 3.83 × 10−5 cm2 s−1 at 23 °C.11 The electroactive coverage (Γ) of enzyme on the electrode surface was maximally 3 pmol cm−2. This value was obtained after introducing CO, whereupon the CO-inhibited enzyme displays faint signals due to the oxidation/reduction of the Fe–S clusters.12 A lower limit of 6000 s−1 for the turnover number, kcat, for enzymatic hydrogen oxidation can then be calculated using kcat = i/2FAΓ.
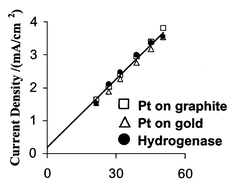 | ||
| Fig. 1 Levich plots for hydrogen oxidation currents from hydrogenase and platinum at +0.242 V vs. SHE, 45 °C and pH 7 under 1 atm hydrogen. The line of best fit is from the hydrogenase data. Platinum currents are from chronoamperometric experiments in which the potential was held at +0.242 V vs. SHE and the rotation rate varied. Hydrogenase currents are from cyclic voltammetry experiments at 1 V s−1, since maintaining the film at an oxidative potential over extended periods inactivates the enzyme.9 | ||
Four reasons, derived from the large size of the enzyme, might cause hydrogen oxidation currents from AvH2ase to be lower than those from platinum. These are impeded accessibility of hydrogen to the buried active site, long-range intramolecular proton transfer from the active site, the relatively low density of catalytic sites able to fit on the electrode and the requirement for electron transfer to occur over a large distance.13 Structural analysis suggests that substrate access and product removal are facilitated by protein channels.14 Since the density of catalytic sites on the electrode is likely to be much lower for the enzyme than for platinum, each hydrogenase active site must turn over faster than a corresponding platinum atom to attain the same overall current.
The important difference between Pt and AvH2ase is clear from Fig. 2. High rates of enzyme turnover are achieved at a cost, i.e. the maximal rate under 1 atm hydrogen is not attained until a significant driving force (+0.2 V vs. SHE) is applied (Fig. 2(b)); whereas with platinum the same maximum is reached by −0.4 V vs. SHE (Fig. 2(a)). At low hydrogen concentrations, i.e. when the demand for a high rate of electron removal is relaxed, the hydrogen oxidation currents at all potentials are similar for both catalysts (Fig. 2(c), platinum data not shown). While the Pt catalyst can exchange electrons directly with the electrode, the [NiFe] active site of the enzyme is too buried within the protein to be in direct electronic connection with the electrode,13 and electron transfer (ET) depends upon the nature of the electrode/enzyme interface and intramolecular mediation by the Fe–S clusters. Either of these can provide a resistance to the electron flow, and they represent important focuses for technological development.
 | ||
| Fig. 2 Potential dependence of hydrogen oxidation currents for platinized gold and hydrogenase/PGE rotating-disk electrodes at pH 7, 45 °C, rotation speed 2500 rpm. (a) Platinized gold; 1 atm hydrogen, 1 V s−1. (b) Hydrogenase; 1 atm hydrogen, 1 V s−1. (c) Hydrogenase; 0.1 atm hydrogen, 0.1 V s−1. | ||
Sustainable activity is another important consideration in catalyst evaluation. One aspect is the well-known oxidative inactivation of the enzyme;9 this process should become more problematic as the potential is made more oxidizing to drive the reaction. However, this inactivation will be countered increasingly as the hydrogen concentration is raised. A more significant aspect is the irreversible poisoning of platinum by carbon monoxide.15 Carbon monoxide also binds to and inhibits hydrogenase,16 but in this case it competes with hydrogen. This means that while exposure to CO causes the immediate poisoning of platinum, the AvH2ase film quickly recovers its activity simply upon removal of carbon monoxide (see ESI†).
In conclusion, the [NiFe] site in molecules of AvH2ase adsorbed at a PGE electrode can oxidize hydrogen at rates comparable to electrodeposited platinum and with less susceptibility to CO poisoning. The extremely high activity of [NiFe]-hydrogenases has, until now, been little realized. Thus it is important that our results confirm that a biological catalyst containing only the base metals nickel and iron coordinated by electron-rich sulfur ligands can perform the task hitherto ascribed at ambient temperatures only to precious metals. The implications are numerous. First, there can be no doubt that it must be possible to synthesize functional biomimetic complexes from cheap metals for use as hydrogen oxidation catalysts that compare favorably with platinum catalysts.17 Second, the relatively unexplored realm of enzyme-based energy production coupled with the ability to optimize desirable properties of the enzyme (such as improving the ET rate) by genetic engineering should prompt greater development of biological-based energy. With the unlimited ability to manufacture enzymes cheaply, this makes hydrogenases particularly attractive candidates for use as oxidation catalysts in fuel cells.
This research was supported by the EPSRC, BBSRC (43/B10492, 43/B11675) and the Netherlands Organization for Scientific Research (NWO) division for Chemical Science (CW). Mr W. Roseboom is acknowledged for his help with enzyme preparation. A. K. J. thanks the Rhodes Trust and the National Science Foundation for their support.
Notes and references
- P. M. Vignais, B. Billoud and J. Meyer, FEMS Microbiol. Rev., 2001, 25, 455–501 CrossRef CAS.
- Hydrogen as a Fuel: Learning from Nature, ed. R. Cammack, M. Frey and R. Robson, Taylor and Francis, New York and London, 2001 Search PubMed.
- G. T. R. Palmore and H.-H. Kim, J. Electroanal. Chem., 1999, 464, 110–117 CrossRef CAS.
- E. Katz, I. Willner and A. B. Kotlyar, J. Electroanal. Chem., 1999, 479, 64–68 CrossRef CAS.
- S. Tsujimura, M. Fujita, H. Tatsumi, K. Kano and T. Ikeda, Phys. Chem. Chem. Phys., 2001, 3, 1331–1335 RSC.
- T. Chen, S. C. Barton, G. Binyamin, Z. Gao, Y. C. Zhang, H.-H. Kim and A. Heller, J. Am. Chem. Soc., 2001, 123, 8630–8631 CrossRef CAS.
- R. P. Happe, W. Roseboom, A. J. Pierik, S. P. J. Albracht and K. A. Bagley, Nature, 1997, 385, 126–126 CrossRef CAS.
- A. Volbeda, E. Garcia, C. Piras, A. L. de Lacey, V. M. Fernandez, E. C. Hatchikian, M. Frey and J. C. Fontecilla-Camps, J. Am. Chem. Soc., 1996, 118, 12989–12996 CrossRef CAS.
- J. M. C. C. Coremans, J. W. Van der Zwaan and S. P. J. Albracht, Biochim. Biophys. Acta, 1992, 1119, 157–168 CAS.
- Gold (99.9985%, Alfa, UK) rotating disk electrodes were manufactured analogously to graphite electrodes (A. Sucheta, R. Cammack, J. Weiner and F. A. Armstrong, Biochemistry, 1993, 32, 5455–5465) and cleaned as described previously (L. J. C. Jeuken and F. A. Armstrong, J. Phys. Chem. B, 2001, 105, 5271–5282). Clean Pt surfaces were electrodeposited onto gold or PGE from 5 mM hydrogen hexachloroplatinate(IV) hydrate (Aldrich) (S. F. White, A. P. F. Turner, R. D. Schmid, U. Bilitewski and J. Bradley, Electroanalysis, 1994, 6, 625–632). Electrodepositing was continued until a stable cyclic voltammogram was recorded. Purification of AvH2ase, preparation of films at PGE and electrochemistry were carried out as reported before (ref. 12). All hydrogenase experiments were undertaken at 45 °C, pH 7 in mixed buffer as described previously (ref. 12). Platinum experiments were also undertaken at pH 7, 45 °C in a chloride-free 0.1 M phosphate buffer. Experiments in 1% or 10% hydrogen/nitrogen mixtures were conducted by replacing the glovebox atmosphere. Experiments with 1 atm H2 were conducted by bubbling the gas directly into the electrochemical cell. All current densities are calculated based on the geometric area (3.1 mm2) of the electrode. Diffusion coefficients were obtained with hydrogen concentrations taken from Handbook of Chemistry and Physics, ed. D. R. Lide, CRC Press, Inc.. London, 77th edn., 1996.
- R. N. Adams, Electrochemistry at Solid Electrodes, Marcel Dekker, New York, Inc., 1969 Search PubMed.
- H. R. Pershad, J. L. C. Duff, H. A. Heering, E. C. Duin, S. P. J. Albracht and F. A. Armstrong, Biochemistry, 1999, 38, 8992–8999 CrossRef CAS.
- C. C. Page, C. C. Moser, X. X. Chen and P. L. Dutton, Nature, 1999, 402, 47–52 CrossRef CAS.
- Y. Montet, P. Amara, A. Volbeda, X. Vernede, E. C. Hatchikian, M. J. Field, M. Frey and J. C. Fontecilla-Camps, Nat. Struct. Biol., 1997, 4, 523–526 CrossRef CAS.
- N. M. Markovic, C. A. Lucas, B. N. Grgur and P. N. Ross, J. Phys. Chem. B, 1999, 103, 9616–9623 CrossRef CAS.
- J. W. van der Zwaan, S. P. J. Albracht, R. D. Fontijn and Y. B. M. Roelofs, Biochim. Biophys. Acta, 1986, 872, 208–215 CAS.
- A. C. Marr, D. J. E. Spencer and M. Schröder, Coord. Chem. Rev., 2001, 219–221, 1055–1074 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Levich plots at 1% and 10% hydrogen, and a comparison of the effect of carbon monoxide on oxidation currents obtained at platinum and enzyme-modified electrodes. See http://www.rsc.org/suppdata/cc/b2/b201337a/ |
| This journal is © The Royal Society of Chemistry 2002 |