
Advanced electrospun nanomaterials for highly efficient electrocatalysis
Xiaofeng
Lu
 *a,
Meixuan
Li
a,
Huiyuan
Wang
*bc and
Ce
Wang
*a
*a,
Meixuan
Li
a,
Huiyuan
Wang
*bc and
Ce
Wang
*a
aAlan G. MacDiarmid Institute, College of Chemistry, Jilin University, Changchun, 130012, P. R. China. E-mail: xflu@jlu.edu.cn; cwang@jlu.edu.cn; Fax: +86-431-85168292; Tel: +86-431-85168292
bKey Laboratory of Automobile Materials of Ministry of Education & School of Materials Science and Engineering, Nanling Campus, Jilin University, No. 5988 Renmin Street, Changchun 130025, P. R. China. E-mail: wanghuiyuan@jlu.edu.cn
cInternational Center of Future Science, Jilin University, Changchun, 130012, P. R. China
First published on 30th August 2019
Abstract
In recent years, electrospun nanomaterials have been regarded as efficient electrocatalysts for energy storage and conversion due to their large surface area, unique chemical structure, easily tunable composition, and excellent electron and mass transportation properties. In this review, we will discuss the recent progress regarding the electrochemical catalytic reactions based on advanced electrospun nanomaterials, including the hydrogen evolution reaction (HER), oxygen evolution reaction (OER), oxygen reduction reaction (ORR), carbon dioxide reduction reaction (CO2RR), nitrogen reduction reaction (NRR), and small molecule oxidation reaction. After a brief description of the electrospinning technique and electrospun nanomaterials, we will focus on the relationship among the morphology, composition, and architecture of the electrospun nanomaterial-based electrocatalysts and their electrocatalytic performance. In addition, the remaining challenges and perspectives for further advances have been addressed. With the fast development of the electrocatalytic energy conversion devices, the research on the advanced electrospun nanomaterials in this field is anticipated to be significantly attractive and exciting in the near future.
 Xiaofeng Lu | Xiaofeng Lu received his B.S. degree in 2003 and Ph.D. degree in 2007 both from the College of Chemistry, Jilin University. After that he worked as a postdoctoral fellow at Washington University in St Louis until 2008. He is now a professor of Alan G. MacDiarmid Institute, College of Chemistry, Jilin University. His current research focuses on the fabrication of multifunctional 1D nanomaterials for applications in catalysis and energy devices. |
 Meixuan Li | Meixuan Li received her B.S. degree in 2015 from the Department of Food Quality and Safety, Jilin University. She is currently a PhD candidate under the supervision of Prof. Xiaofeng Lu in the College of Chemistry, Jilin University. Her research interests focus on the preparation of electrospun nanomaterials as electrochemical catalysts for the hydrogen evolution, oxygen evolution, and oxygen reduction reactions. |
 Huiyuan Wang | Huiyuan Wang received his B.S. degree from the Jilin University of Technology in 1998 and Ph.D. degree in Materials Processing Engineering from Jilin University in 2004. He is currently a professor of the School of Materials Science and Engineering, Jilin University. His research mainly focuses on design and fabrication of novel energy materials and advanced metallic materials. |
 Ce Wang | Ce Wang received her B.S. degree from the Department of Chemistry in Jilin University in 1982 and Ph.D. degree at the Technische Universität Wien, Austria in 1995. After that she worked as a postdoctoral fellow at Drexel University until 1997. She is currently a professor of Alan G. MacDiarmid Institute, College of Chemistry, Jilin University. Her current research interests include the synthesis of electrospun materials for electronics, sensors, environment, and energy applications. |
1. Introduction
To solve the energy shortage, environmental pollution and climate change issues, it is essential to develop clean and sustainable energy to reduce the consumption of fossil fuels. At present, a large number of efficient advanced technologies, such as fuel cells, metal–air batteries, water electrolysis, and small molecule to fuel conversion, have been explored to be used as efficient energy conversion devices.1–4 These typical devices are usually involved in a series of electrochemical reactions, including the hydrogen evolution reaction (HER), oxygen evolution reaction (OER), oxygen reduction reaction (ORR), carbon dioxide reduction reaction (CO2RR), nitrogen reduction reaction (NRR), small molecule (methanol, ethanol, formic acid, and urea) oxidation reaction, etc.5–15 Generally, to obtain high-performance energy conversion devices, the fabrication of a highly efficient electrocatalyst is required, which can lower the electrochemical reaction potential and increase its rate. Generally, the electrocatalyst is often used to modify the electrode to facilitate the electron transport between the electrode and reactants or intermediates.Among a large variety of electrocatalysts, noble metal-based materials are the most efficient ones, for example, Pt-based nanomaterials for the HER and ORR, RuO2 and IrO2 for the OER, and Au-based nanomaterials for the CO2RR and NRR.16 Although the noble metal-based electrocatalysts show excellent catalytic activity for these typical electrochemical reactions, the high cost, scarce reserves, and poor long-term stability extremely restrict their large scale commercialization in sustainable energy technologies. Therefore, noble metal-free nanocatalysts have aroused increasing attention and led to great advances in this fascinating field. Typical noble metal-free nanocatalysts mainly include metal oxides, hydroxides/oxyhydroxides, chalcogenides, carbides, nitrides, phosphides, doped carbon materials, metal–organic frameworks (MOFs), functional hybrids, etc. By tuning the types of the electrocatalysts and their compositions, outstanding electrocatalytic performance for the HER, OER, ORR, CO2RR, NRR, and small molecule oxidation reaction could be achieved.
To promote the electrochemical performance of the energy conversion devices, the architecture design is very important. Over the last few decades, zero-dimensional (0D) quantum dots, one-dimensional (1D) nanofibers and nanotubes, two-dimensional (2D) nanosheets, and three-dimensional (3D) microstructures have been broadly discovered to show brilliant electrocatalytic activity and stability. For example, phosphorene quantum dots functionalized with nitrogen-containing groups (FPQDs) that were synthesized from an electrochemical exfoliation process showed excellent OER activity with an overpotential of 1.66 V at a current density of 10 mA cm−2.17 The FPQD electrocatalyst also exhibited long-term stability with no obvious current decay within 10 h. 1D nanostructured marokite CaMn2O4 nanorods in a post-spinel phase have also been fabricated via a solvothermal strategy, which showed outstanding ORR catalytic performance including favorable specific and mass activities, a low Tafel slope and high stability under alkaline conditions.18 Xie and co-worker reported that 2D NiSe2 nanosheets with structural distortions showed superior OER activity with a low η of 0.33 V to reach a current density of 10 mA cm−2, which is much better than those of a bulk NiSe2 sample and the benchmark RuO2 material.19 Recently, a 3D graphene aerogel/Ni/MnO material has been explored as an efficient bifunctional electrocatalyst for both the ORR and OER.20 Then a Zn–air battery based on the bifunctional catalyst exhibited a superior specific capacity, power density and cycling stability compared with a Pt/C//RuO2 catalyst.
Among the four typical chemical architectures mentioned, 1D nanomaterials possessing a fiber- or tube-like structure with a diameter ranging from several to several hundreds of nanometers have attracted great attention due to their unique physical and chemical structures.21,22 The 1D nanomaterials showed the advantages of small dimensions, large surface areas, and high aspect ratios, enabling excellent electron and mass transfer along one controllable direction.23 Therefore, the 1D nanomaterials have been widely developed to be efficient electrocatalysts for energy conversion and storage. Until now, a large number of methods, such as a hard and sacrificial template approach, hydrothermal and solvothermal reactions, chemical vapor deposition, self-assembly, the electrospinning technique etc., have been explored to generate 1D nanomaterials. Among these methods, electrospinning is one of the most facile and universal ways to produce fiber or tube-like nanomaterials with tunable compositions and controllable architectures. Over the past twenty years, many types of 1D nanomaterials from polymers, ceramics, metals, and their hybrids have been fabricated via the electrospinning technique, and have shown a large variety of applications in the fields of catalysis, electronic devices, filters, environmental science, biomedical engineering, and energy devices.24–33
To date, although a large number of reviews have been published on the subject of highly efficient electrocatalysis based on various types of functional nanomaterials,5–15 there are few comprehensive reviews focused on the application of electrospun nanomaterials for this subject yet. More importantly, more and more studies on electrospun nanomaterials for electrocatalytic applications have emerged in the last few years. In this review, the recent advances of electrospun nanomaterials in electrocatalytic reactions including the HER, OER, ORR, CO2RR, NRR, and small molecule oxidation process have been summarized (Fig. 1). In particular, the correlation between the architectures of the electrospun nanomaterials and their electrocatalytic performance is highlighted. We first provide an introduction of the fundamental electrospinning technique and electrospun nanofibers, and then move to a detailed discussion on the HER, OER, ORR, CO2RR, NRR, and small molecule oxidation performance of various types of electrospun nanomaterials in acidic, neutral and alkaline electrolytes. Finally, the remaining challenges and perspectives related to the fabrication of electrospun nanomaterial-based architectures toward the advances of high-performance electrocatalysts will be addressed.
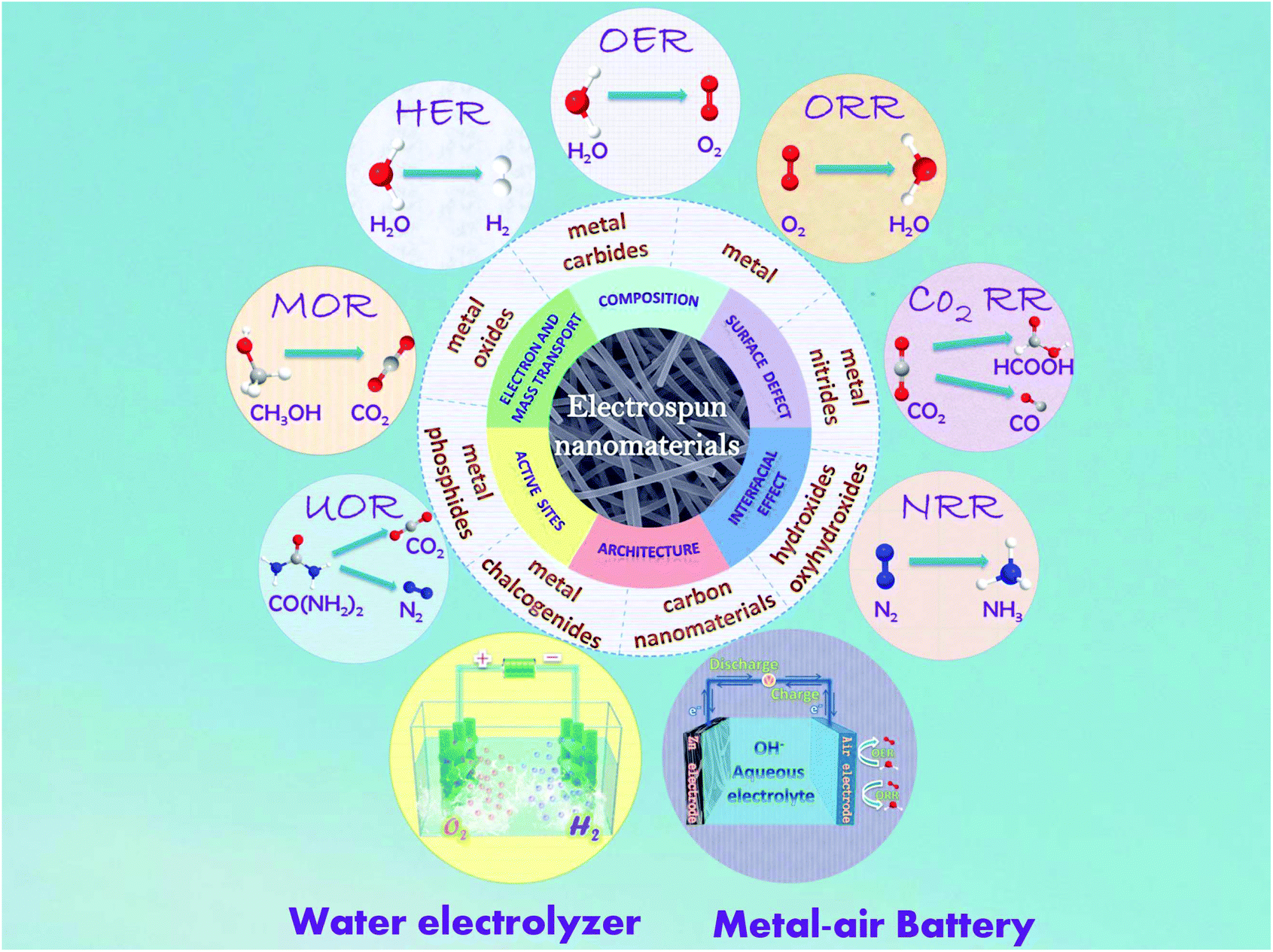 | ||
| Fig. 1 Schematic illustration of the electrospun nanomaterials for their applications in electrocatalysis. | ||
2. Fundamentals of the electrospinning technique and electrospun nanomaterials
2.1 Principle of the electrospinning process
The electrospinning technique has descended from the electrospray technique, which has been studied in detail employing the destabilization of a liquid jet as a model with an increase in electric charge.34 The electrospray approach has been widely applied to prepare aerosols. Based on this principle, polymeric particles could also be obtained if a certain polymer was added into the electrospray solution. When the concentration of the polymer is increased to be sufficiently large, polymeric nanofibers will be formed, which is called the electrospinning process.35 With the quick development of nanoscience and nanotechnology, the electrospinning technique has become a hot topic of research after the 1990s owing to its strong ability to produce a large variety of 1D nanostructured materials. By the introduction of post-treatment processes, not only polymeric but also ceramic, metallic and hybrid 1D nanomaterials could be fabricated via the electrospinning technique.36–39 The electrospinning equipment is very simple, and mainly consists of three parts: a high-voltage source, a spinneret and a collector device. The high-voltage supply could come from either direct current or alternating current sources, and provide a voltage of up to tens of kilovolts between the spinneret and the collector device. During the electrospinning process, a strong electrostatic force from the high-voltage electrostatic-field is formed, which can keep the force balance with the surface tension of the polymer solution droplet, deforming into a Taylor cone at the needle tip.40 When the intensity of the electrostatic-field increases to a critical value, the force balance is broken and a charged jet ejects from the tip of the Taylor cone. After the evaporation of the solvent or melt solidification, solid polymeric nanofibers will be formed on the collector device.A metal nozzle spinneret with a chamfered or flat tip is usually used to stabilize the Taylor cone and produce the electrospinning jet. Compared with the chamfered tip, little oscillation of the jet and even multiple jets could be generated from the flat tip at a higher feed rate. The diameter of the nozzle has an influence on the size of the polymeric nanofibers.41 Generally, a smaller diameter of the nozzle produces thinner nanofibers. To increase the yield of the nanofibers, especially for mass production, a multi-nozzle spinneret has also been used for electrospinning.42 However, the nearby multi-nozzle spinnerets might show a significant influence on the jets, producing a large distribution of the diameters of the nanofibers. On the other hand, a typical electrospinning device without any spinnerets has also been reported, which is named a nozzleless electrospinning device.43 For instance, Chase and co-workers introduced a polyethylene porous tube in which was installed a metal electrode and supplied with nitrogen gas on one side of the tube.44 During the electrospinning process, the jets were produced from the droplets as a Taylor cone at the holes under a high electric field. Finally, the generated polymer nanofibers were formed on the surface of the collector. The collector is also a necessary part of the electrospinning setup, which usually refers to conductive or less conductive substrates such as metal foil, silicon wafer, non-woven fabrics, and paper. The conductivity of the collector can influence the density of the nanofibers on its surface because of the difference of the charges on the fibers to be conducted to ground.
2.2 Electrospun nanomaterials
One of the most significant advantages of the electrospinning technique is its ability to produce 1D nanomaterials with tunable compositions. Until now, a large number of electrospun nanofibers or nanotubes have been prepared through the electrospinning technique or a combination with a post-treatment process.45–49 In addition to the compositions, the architecture of the electrospun nanomaterials could also be tunable by regulating the electrospinning parameters such as the solution viscosity and conductivity, kinds of solvents and their volatility, applied voltage, tip-to-collector distance, feeding rate, humidity, temperature, etc. The solution viscosity which is usually determined from the molecular weight of the polymer and its concentration in the solution plays a key role in regulating the diameter of the as-electrospun nanofibers.50 Generally, a lower solution viscosity will produce smaller diameters of the nanofibers. However, more and more beads will be formed when the solution viscosity reduces to a critical value. In addition, the diameter of the electrospun nanofibers is also correlated with the applied voltage, tip-to-collector distance and feeding rate.51–53 A higher applied voltage, longer tip-to-collector distance and slower feeding rate may produce nanofibers with a smaller diameter.Furthermore, the architecture of the electrospun nanofibers can be regulated by adjusting the spinneret and the collector. For example, a coaxial spinneret has been broadly used to generate core–shell structured nanofibers and nanotubes.54–56 Sun and co-workers demonstrated the fabrication of core–shell structured polysulfone (PSU)/poly(ethylene oxide) (PEO) nanofibers through such a coaxial electrospinning approach.54 Similarly, core–shell structured PCL-r-gelatin bi-component nanofibers have also been prepared through electrospinning of the precursors of gelatin/2,2,2-trifluoroethanol (TFE) and PCL/TFE (Fig. 2a and b).55 Furthermore, the coaxial electrospinning technique can also be applied for the preparation of hollow nanotubes.57 For instance, Xia and co-workers prepared poly(vinyl pyrrolidone) (PVP)/TiO2 hollow nanotubes by using the coaxial electrospinning process with Ti(OiPr)4 and PVP in ethanol solution as the sheath liquid and mineral oil as the core liquid, and subsequently removed the mineral oil by extraction with octane.57 Under a calcination process, the PVP/TiO2 nanotubes could be converted to anatase nanotubes (Fig. 2c and d). Furthermore, hierarchical multichannel microtubes could also be prepared via a modified multifluidic compound jet electrospinning technique (Fig. 2e and f).58
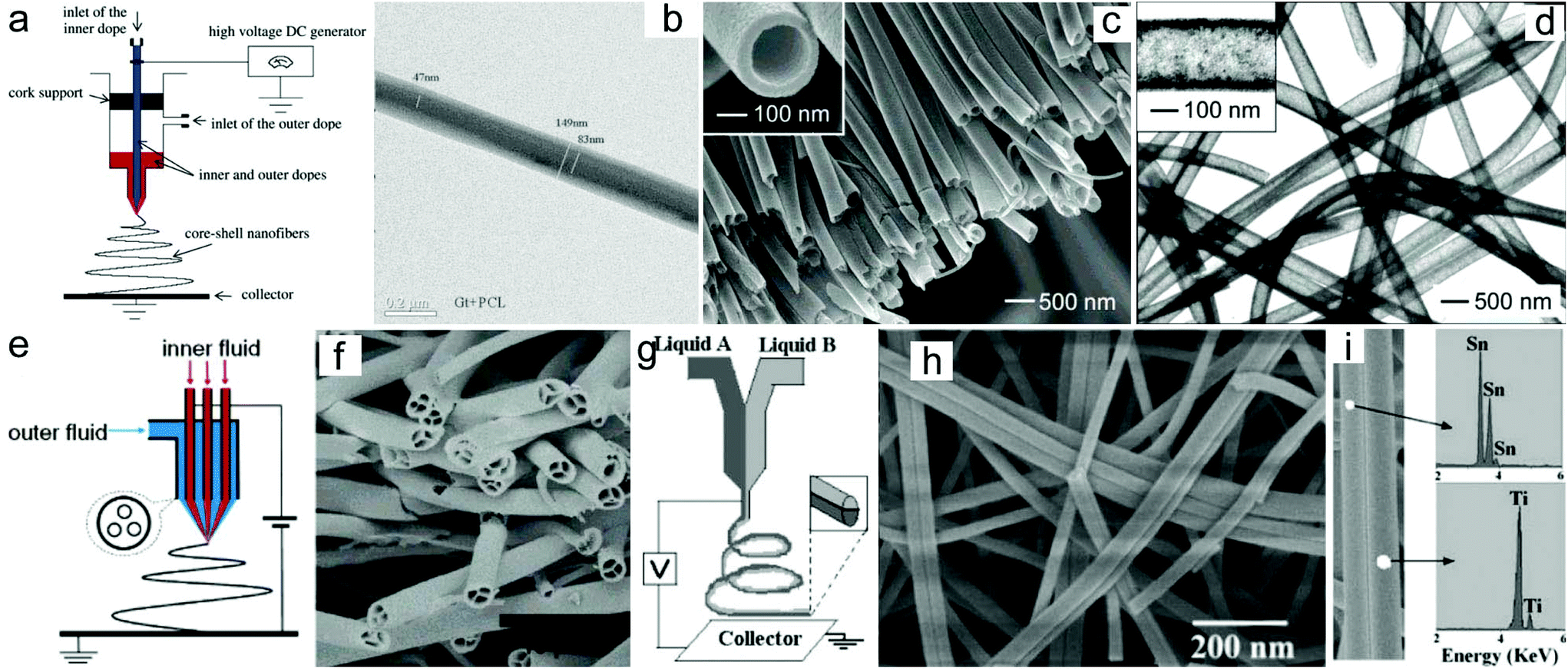 | ||
| Fig. 2 Electrospun nanomaterials with designed architectures: (a) schematic drawing of the electrospinning setup to prepare core/shell structured nanofibers. (b) TEM image of core–shell structured PCL-r-gelatin biocomponent nanofibers. Reprinted with permission from ref. 55. Copyright 2004, American Chemical Society. (c) SEM and (d) TEM images of the hollow TiO2 nanotubes. Reprinted with permission from ref. 57. Copyright 2004, American Chemical Society. (e) Schematic drawing of the electrospinning setup to prepare multichannel nanotubes. (f) SEM image of the TiO2 multichannel nanotubes. Reprinted with permission from ref. 58. Copyright 2007, American Chemical Society. (g) Schematic drawing of the electrospinning setup to prepare Janus nanofibers. (h) SEM image of the TiO2/SnO2 Janus nanofibers. (i) EDX analysis of the prepared TiO2/SnO2 Janus nanofibers. Reprinted with permission from ref. 61. Copyright 2007, American Chemical Society. | ||
In addition to the core–shell structured nanofibers, Janus nanostructures with various exposed surfaces on their two opposite sides could be fabricated via a side-by-side electrospinning technique. This process has been achieved by using side-by-side spinnerets. Through such a strategy, a series of polymer/polymer nanofibers such as poly(vinyl chloride)/segmented polyurethane (PU), poly(vinyl chloride)–poly(vinylidene fluoride) and polyacrylonitrile (PAN)/PU nanofibers have been prepared. In addition to polymer/polymer nanofibers, ceramic/ceramic nanofibers such as TiO2/SnO2 nanofibers with a Janus structure have also been prepared (Fig. 2g–i).59–61 Recently, Dong and co-workers prepared multifunctional Janus nanoribbon arrays through a modified side-by-side electrospinning technique.62 The synthesized Janus nanoribbons exhibited not only magnetism and photoluminescence properties but also anisotropic electrical conductivity, and have potential applications in subminiature electronic equipment.
Generally, electrospun nanofibers with a porous structure are very important for their applications in sensors, catalysis, and energy devices because of their large surface area and unique mass transport. During the electrospinning process, the porous structure can be achieved via a phase separation method. A typical example is that of porous poly-L-lactide (PLLA) nanofibers which have been fabricated from an electrospinning process of PLLA solution in dichloromethane.63 Owing to the phase separation during the dichloromethane evaporation, the PLLA-rich phase solidifies to form a matrix while the dichloromethane-rich phase creates pores. In addition, the porous carbon nanofibers (CNFs) have also been reported from physical or chemical activation processes with some special reagents.64,65 Oxidizing gases such as steam, air and carbon dioxide are usually used for physical activation of CNFs, while potassium hydroxide, phosphoric acid, and sodium hydroxide are typical chemical activating agents. By using 20 wt% KOH as the activating agent, the CNFs showed an increase in the BET surface area from 524 to 1317 m2 g−1 and the percentage of mesopores and macropores.65 The ceramic nanofibers with a porous structure could be prepared by a selective calcination process. For instance, porous TiO2 nanofibers have been fabricated through a microemulsion electrospinning technique followed by the calcination process.66 The pores are produced from the vacancies with the removal of the oil droplets.
3. Application of electrospun nanomaterials for highly efficient electrocatalysis
3.1 Application of electrospun nanomaterials towards the HER
The HER is a typical half-reaction during the water splitting process to generate hydrogen gas.67 It is generally considered that the HER process mainly includes a hydrogen adsorption process on the surface of the electrocatalyst via a charge transfer or the Volmer reaction (H3O+ + e− + * → Had + H2O in acidic solution and H2O + e− + * → Had + OH− in alkaline solution) and a molecular hydrogen production process via a Tafel reaction (Had + Had → H2) or a Heyrovsky reaction (H3O+ + Had + e− → H2 + H2O + * in acidic solution and H2O + Had + e− → H2 + OH− + * in alkaline solution) (Fig. 3).68,69 Therefore, both the adsorption and production processes should be considered for the design of an efficient electrocatalyst for the HER. Based on the Sabatier theory, the moderate adsorption of Had on the active sites is beneficial for the HER activity. Among a large variety of electrocatalysts, Pt is regarded as the most efficient one for the HER because of the appropriate adsorption ability of Had on the Pt surface.69,70 To develop low-cost HER catalysts, a series of non-noble metal-based materials have emerged in the past few years, including non-noble metals, metal oxides, metal sulfides, metal nitrides, metal phosphides, metal carbides, metal borides, their hybrids, etc.71–76 | ||
| Fig. 3 The HER mechanism on the surface of an electrocatalyst in acidic medium. Reprinted with permission from ref. 69. Copyright 2014, Royal Society of Chemistry. | ||
Among various types of non-noble metals, Ni, Co and Cu have been considered as good electrocatalysts for the HER because of their relatively high catalytic activity and low cost.77–81 However, to prevent the aggregation of Ni and Co nanoparticles which can reduce their electrocatalytic activity, they are usually deposited on the carbon matrix. For example, Ni-CNFs have been prepared through electrospinning a precursor of Ni2+/poly(amic acid) and a subsequent carbonization process as efficient HER catalysts.77 The prepared sample showed that Ni nanoparticles were uniformly distributed on the CNF surface, enabling strong interactions and available charge transfer between Ni and CNFs. Thus the Ni-CNFs with 8% Ni displayed a low overpotential of −0.17 V and a Tafel slope of 105 mV dec−1 in a 2 M KOH electrolyte. To further enhance the HER activity of non-noble metal nanoparticles, the fabrication of a metal alloy is one of the most efficient routes. Tang, Li, Xu and co-workers prepared a novel hierarchical architecture of Ni3Fe nanoalloy-encapsulated carbon nanotubes decorated on N-doped CNFs through an electrospinning strategy for the HER (Fig. 4a–c).78 The catalyst showed a low overpotential of 72 mV at a current density of 10 mA cm−2 in an alkaline electrolyte and a Tafel slope of 98 mV dec−1 (Fig. 4d–f). The superior HER performance could be the result of abundant Ni3Fe catalytic sites and the efficient electron transport and mass diffusion of fibrous nanostructures, which is reflected by the large double-layer capacitance (Cdl) value (16.3 mF cm−2) of the catalyst (Fig. 4g). The prepared catalyst also showed an outstanding long-term stability, with unchanged LSV curves after 1000 cycles and no evident current density decay after 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s chronoamperometry measurement (Fig. 4h and i). In addition, the fabrication of non-noble metal/noble metal and noble metal/noble metal hybrids is also proved to be an efficient route to construct HER electrocatalysts.82–85 For example, we have shown that the addition of Ni in the Pt/CNFs could enhance the HER activities in a wide pH range from acidic to alkaline solutions.82 The superior HER performance could be related to the synergistic effect between the two types of metals.
000 s chronoamperometry measurement (Fig. 4h and i). In addition, the fabrication of non-noble metal/noble metal and noble metal/noble metal hybrids is also proved to be an efficient route to construct HER electrocatalysts.82–85 For example, we have shown that the addition of Ni in the Pt/CNFs could enhance the HER activities in a wide pH range from acidic to alkaline solutions.82 The superior HER performance could be related to the synergistic effect between the two types of metals.
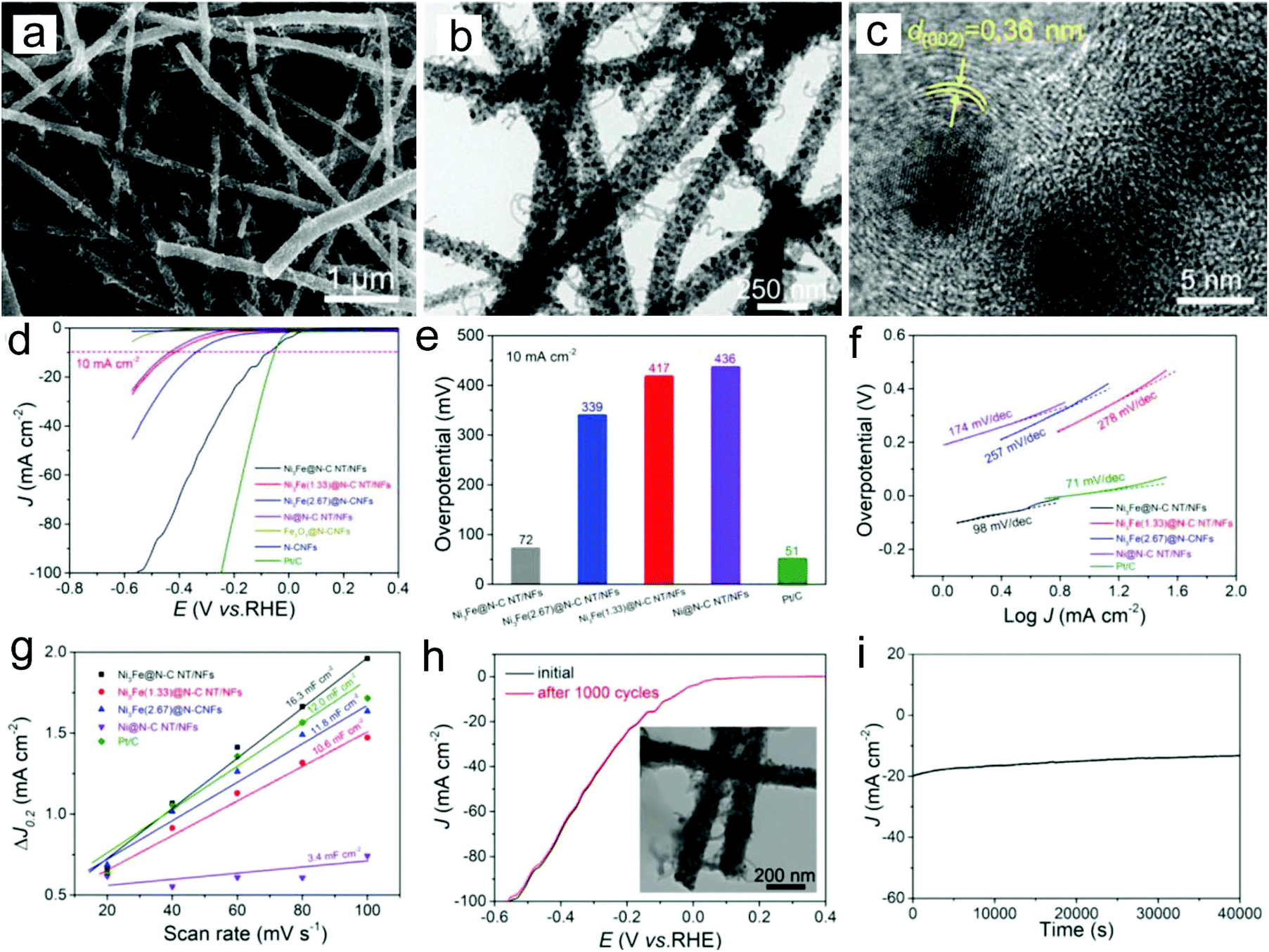 | ||
| Fig. 4 (a) SEM, (b) TEM and (c) HRTEM images of the prepared Ni3Fe nanoalloy-encapsulated carbon nanotubes decorated on N-doped CNFs. (d) LSC curves, (e) overpotentials, (f) Tafel slopes, (g) capacitive current versus scan rates, (h) long-term stability, and (i) i–t curve at an overpotential of 200 mV for the prepared Ni3Fe nanoalloy-encapsulated carbon nanotubes decorated on N-doped CNFs. The inset in Fig. 4h shows the TEM image of the catalyst after the stability test. Reprinted with permission from ref. 78. Copyright 2018, John Wiley and Sons. | ||
Metal oxides are another type of electrocatalyst towards the HER.86–94 It has been reported that the HER rates of NiO and Co3O4 electrospun nanofibers examined by four electrode CV approaches and a two-phase reaction were much higher than those of bulk samples, demonstrating the improved surface to volume ratio of the fibrous structures.86 Recently, WO3−x nanoplates have attracted much attention for their use in HER catalysts. For example, Du and co-workers prepared WO3−x nanoplates on CNFs via electrospinning and carbonization processes for HER applications.87 It was found that the HER activities of the prepared WO3−x/CNFs increase with the increment of the W content. Owing to the poorly crystalline, large specific surface area, high conductivity and numerous active sites, the optimized WO3−x/CNFs exhibited a low overpotential (180 mV at a current density of 10 mA cm−2), and a Tafel slope of 89 mV per decade in a 0.5 M H2SO4 electrolyte, as well as long-term stability (12 h). Compared with single metal oxides, binary transition metal oxides have also become a hot topic for their applications in electrocatalytic water splitting. For example, IrxRu1−xOy nanofibers have been fabricated via an electrospinning technique, and showed a surprisingly high HER activity in alkaline solution.88 The HER activity of the prepared Ir0.80Ru0.20Oy was superior to those of single IrOy, RuOy, commercial Pt and commercial Ir catalysts. The density functional theory revealed that the outstanding HER performance of the catalyst could result from the suitable hydrogen adsorption ability, which was comparable with that of Pt.
Transition metal chalcogenides including MoxS, CoxS, NixS, WS2, MoSe2, and CoTe2 have been used as good alternatives to Pt for the HER due to their abundant active sites and unique electrochemical properties.95–119 Among these, MoS2 nanomaterials have been mostly studied because of their unique lamellar structure and outstanding catalytic performance.95–108 It has been reported that the edge sites of MoS2 play a key role in its electrocatalytic activity compared with the basal planes. In addition, the conductivity of MoS2 nanomaterials is poor, which usually hindered their electrocatalytic activity. Therefore, the synthesis of MoS2 nanomaterials with abundant exposed edges supported on CNFs with high conductivity is an ideal choice to construct highly efficient HER catalysts. For example, the MoS2/CNFs synthesized by electrospinning and subsequent treatment showed a uniform morphology consisting of MoS2 nanoplates on CNFs, offering abundant exposed edges and strong bonding of MoS2 with conductive CNFs.95 Therefore, the prepared MoS2/CNFs displayed an ultrahigh HER activity in an acidic electrolyte, with overpotentials of 380 and 450 mV to reach densities of 500 and 1000 mA cm−2, which was better than those of most of the previously reported MoS2-based and commercial Pt/C catalysts. Recently, we have demonstrated the preparation of CoS2-C@MoS2 core–shell nanofibers for HER applications. Resulting from the excellent electron transport and synergistic effect, the synthesized CoS2-C@MoS2 core–shell nanofibers with an optimized MoS2 shell thickness delivered an overpotential of 173 mV at a current density of 10 mA cm−2 with a Tafel slope of 61 mV dec−1 in an acidic medium (Fig. 5a and b).104 The catalyst exhibited a large electrochemically active surface area with a Cdl value of 6.32 mF cm−2 (Fig. 5c and d) and excellent electron transfer ability from active sites (Fig. 5e). Furthermore, the prepared CoS2-C@MoS2 core–shell nanofibers delivered a favorable long-term stability with a stable LSV curve after 1000 cycles (Fig. 5f). The catalyst is a promising material for its application in energy-related devices. Similarly, CoS2/CNFs have also been prepared via electrospinning, carbonization and in situ sulfurization processes for the HER, generating an overpotential of 136 mV to reach a current density of 10 mA cm−2 and remarkable stability to maintain the catalytic activity for 20 h.109 In addition to the metal sulfides, metal selenides have also been reported to be efficient HER catalysts. For instance, MoSe2 decorated porous CNFs (MoSe2/CNFs) have been fabricated via pyrolysis of polydopamine coated polystyrene fibers and subsequent hydrothermal reaction.110 By using the MoSe2/CNFs as the electrocatalyst for the HER, a small onset potential of 70 mV, a Tafel slope of 65 mV dec−1 and long-term stability (no deterioration after 1000 cycles) were achieved.
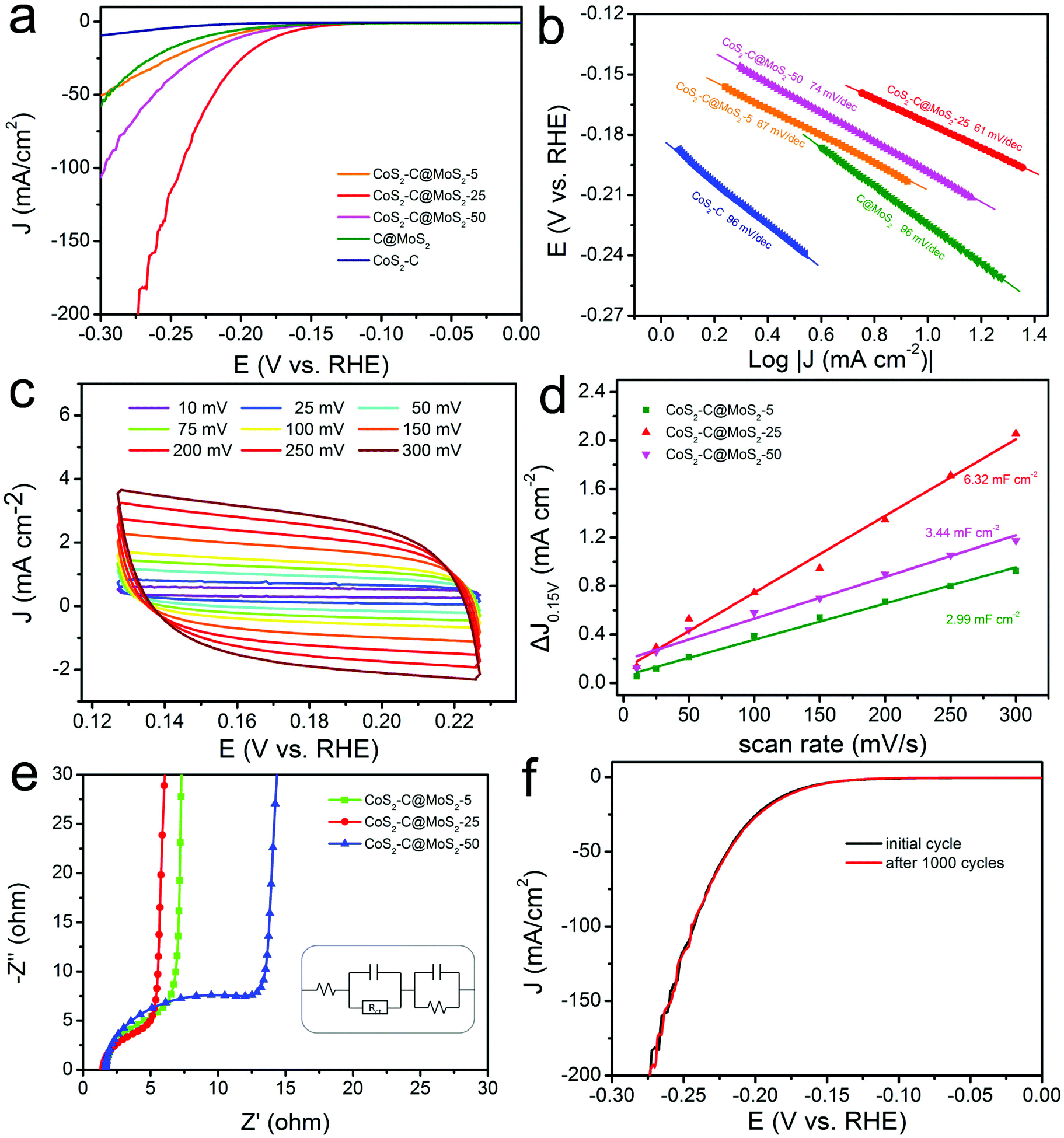 | ||
| Fig. 5 Analysis of the HER performance of CoS2-C@MoS2 samples. (a) LSC curves and (b) Tafel slopes of CoS2-C@MoS2-25 and control samples. (c) CV curves of the prepared CoS2-C@MoS2-25 catalyst. (d) Capacitive current versus scan rates of the CoS2-C@MoS2-25 and control samples, showing their double-layer capacitance values. (e) Nyquist plots of the different samples. (f) LSV curves of the CoS2-C@MoS2-25 catalyst before and after 1000 CV cycles. Reprinted with permission from ref. 104. Copyright 2019, American Chemical Society. | ||
Transition metal phosphides are significantly studied because of their optimized hydrogen adsorption ability, which is located near the top of the HER volcano.120,121 On the other hand, one-dimensional nanostructures, especially hollow nanotubes, have aroused increasing attention for energy conversion applications because of their large specific surface area and fast charge/mass transport properties.122–127 Therefore, transition phosphide nanotubes are ideal candidates as HER electrocatalysts. Miao, Liu and co-workers prepared CoP nanotubes as HER catalysts through electrospinning, thermal stabilization and phosphorization reaction.122 As a result, the prepared CoP nanotubes showed a low overpotential of 152 mV at a current density of 10 mA cm−2 and a low Tafel slope of 50 mV dec−1 in acidic solution. Furthermore, the CoP nanotube catalyst displayed excellent cycling stability, and did not show much increase of the overpotential over 24 h operation. The enhanced HER activity of the CoP nanotubes compared with those of the control samples results from their unique hollow tubular structure for efficient mass diffusion and electron transfer properties. Recently, Ni2P nanoparticles encapsulated in N-doped porous CNFs (Ni2P@NPCNFs) have also been prepared via an electrospinning technique for the HER.123 The introduction of NPCNFs increased the conductivity of the Ni2P@NPCNFs, enabling their remarkable HER performance. It was found that the prepared Ni2P@NPCNFs exhibited a superior HER activity over the whole pH values, showing the overpotentials of 63.2, 104.2 and 185.3 mV to reach a current density of 10 mA cm−2 in acidic, basic and neutral electrolytes, respectively. Furthermore, the polarization curves almost did not change after 3000 cycles, demonstrating their long-term stability. The superior HER performance of the Ni2P@NPCNFs should be attributed to the less interfacial resistance among Ni2P nanoparticles, large surface area, abundant exposed active sites, proven H* adsorption by N-doping, and the synergistic effect between Ni2P and CNFs. Most importantly, this strategy could also be extended to fabricate many other types of transition metal phosphides such as Fe2P@NPCNFs, Co2P@NPCNFs, and Cu3P@NPCNFs, broadening their applications for electrocatalytic water splitting.
Recently, metal carbides have emerged as promising HER electrocatalysts due to their metal-like electronic structure, outstanding H* adsorption ability, high stability and low cost.128–130 In the past few years, a series of transition metal carbides including Mo2C, W2C, and Fe3C have been extensively studied for HER applications. Among these, Mo2C is remarkable because of its high HER activity and long-term stability. Recently, hexagonal-phase Mo2C nanocrystals in CNFs (H-Mo2C/CNFs) have been prepared via an electrospinning and pyrolysis process, which exhibited an outstanding HER activity in both acidic and basic solutions.130 The overpotentials of the H-Mo2C/CNFs were 160 mV and 92 mV in 0.5 M H2SO4 and 1 M KOH to reach a current density of 10 mA cm−2, respectively. Accordingly, the Tafel slopes were 66 mV dec−1 and 63 mV dec−1 in acidic and basic solutions. In addition, long-term stability was achieved in not only acidic but also basic media. The outstanding catalytic performance of the prepared H-Mo2C/CNFs could be explained as follows: (1) abundant active sites are generated through the distribution of small Mo2C nanocrystals in interconnected porous CNFs; (2) the porosity and interconnected architecture of the H-Mo2C/CNFs provide good ion, electron and mass transport pathways; and (3) the porous CNFs enable excellent conductivity of Mo2C and regulate the Mo–H bonding energy, which provides a synergistic effect between Mo2C and CNFs.
3.2 Application of electrospun nanomaterials towards the OER
The OER is another typical half reaction in water splitting to produce oxygen gas, which is involved in a classic reaction as 2H2O → O2 + 4H+ + 4e− in an acidic electrolyte or 4OH− → O2 + 2H2O + 4e− under alkaline conditions.131 Compared with the HER, the OER usually requires a larger overpotential to overcome the kinetic barrier because it is a reaction involving four electrons.131 Over the past few years, a large variety of electrocatalysts have been developed to improve the electrode kinetics. Among those catalysts, RuO2 and IrO2 are the most typical and efficient OER electrocatalysts; however, the high cost and scarce sources restrict their broad applications. In recent years, research studies have been focusing on the fabrication of non-precious catalysts for the OER. For example, transition metals, metal oxides, metal carbides, metal phosphides, metal nitrides, hydroxides/oxyhydroxides, and their hybrids as well as C3N4 nanotubes have been demonstrated to show excellent OER properties.132–157Co, Fe, and Ni-based electrocatalysts have been regarded as good candidates to replace RuO2 and IrO2 catalysts due to their good OER activity and easy availability.133–137 Generally, Co, Fe and Ni nanoparticles and especially their alloy have been reported to show excellent OER activities. For example, NiFe-CNFs have been prepared via electrospinning and a subsequent pyrolysis process for the OER in an alkaline electrolyte.133 It was found that NiFe nanoparticles were homogeneously distributed in CNFs with pores of 3–9 nm diameter, which enhanced the OER activity and durability of the NiFe-CNFs. The prepared 50 wt% NiFe-CNFs exhibited an overpotential value of 270 mV at a current density of 10 mA cm−2, which was only 20 mV higher than that of Ir black. Recently, a ternary FeCoNi alloy embedded in mesoporous CNFs (FeCoNi-CNFs) has also been prepared via a similar procedure for the OER.134 The prepared FeCoNi-CNFs showed an overpotential of 220 mV to reach 10 mA cm−2 and a Tafel slope of 57 mV dec−1, which were much lower than those of a commercial IrO2 catalyst, indicating the high OER activity of the FeCoNi-CNFs. Furthermore, after 2000 CV cycles, the LSV of FeCoNi-CNFs almost coincided, confirming their long-term stability.
Co, Fe, and Ni-based transition metal oxides are the most promising electrocatalysts for the OER due to their low cost, high activity and stability which are even superior to those of RuO2 and IrO2 catalysts.138–150 Among these, spinel oxides have been explored to show excellent OER performance due to their unique chemical structures and rich redox properties. It has been reported that the electrospun CoFe2O4 nanofibers exhibited a better OER activity than the commercial Ir/C (20%) catalyst.138 Recently, oxygen-vacancy-rich CoFe2O4 nanofibers have been reported to show an enhanced OER performance.139 Through heat treatment under a 10% H2/Ar atmosphere, oxygen-vacancy-rich CoFe2O4 (CoFe2 alloys/CoFe2O4) porous hollow nanofibers were prepared, which showed an improved OER performance compared with pure CoFe2O4 nanofibers in a basic medium. The CoFe2 alloys/CoFe2O4 hollow nanofibers showed a low overpotential (300 mV at a current density of 10 mA cm−2), a low Tafel slope (73.34 mV dec−1) and excellent long-term stability (almost no potential change over 12.5 h). The superior OER performance of the CoFe2 alloys/CoFe2O4 nanofibers could be the result of their unique porous structure, large oxygen vacancies, numerous catalytically active sites, and strong coupling between CoFe2 alloys and CoFe2O4 nanofibers, which provide efficient electron transport, mass diffusion and a synergistic catalytic effect. To increase the electrical conductivity of the spinel oxides during the electrocatalytic process, the hybridization of spinel oxides with conductive carbon-based substrates has become another effective strategy. The carbon/spinel oxide hybrid enables excellent charge transfer, thus leading to an improved OER performance. For instance, Tang, Xu and co-workers prepared CoFe2O4 nanoparticles encapsulated in N-doped carbon nanofibers (CoFe2O4@N-CNFs) through a facile electrospinning strategy combined with a controlled calcination process.140 As an OER electrocatalyst, the prepared CoFe2O4@N-CNFs showed an overpotential of 349 mV to reach a current density of 10 mA cm−2, which is comparable with that of the commercial RuO2 catalyst (342 mV). Importantly, the catalyst showed a much better OER activity at a high current density, implying its promising practical applications. For example, the overpotential of the catalyst is around 408 mV at a current density of 30 mA cm−2, which is much lower than that of the RuO2 catalyst (594 mV). Accordingly, the prepared CoFe2O4@N-CNFs presented a current density 2.6 times higher than that of RuO2 at 1.7 V. The Tafel slope of the catalyst was 80 mV dec−1, which is comparable with that of RuO2. The prepared CoFe2O4@N-CNFs also showed outstanding durability, demonstrating a 7.3% current attenuation after 40000 s compared with a 39.8% current loss of the RuO2 catalyst. The superior OER performance benefits from not only the interconnected nanofibrous feature for efficient electron transport and mass diffusion but also the hybridization of CoFe2O4 nanoparticles with N-doped CNFs for more catalytically active sites.
In addition to the spinel oxides, perovskite oxides with a formula of ABO3 have also been developed as OER catalysts due to their excellent OH− adsorption ability and high O2 transfer properties. Recently, hollow perovskite La0.7Sr0.3Co0.25Mn0.75O3 nanofibers (LSCM NFs) have been prepared through an electrospinning technique and a calcination process.151–154 The prepared hollow LSCM NFs consisted of a large amount of dense nanoparticles, showing the advantages of a large surface area, high porosity and ample inner space. On controlling the calcination temperature, the LSCM NFs showed a perfect OER performance, with an overpotential of 340 mV to reach a current density of 10 mA cm−2 and a Tafel slope of 111 mV dec−1. Furthermore, the La0.7Sr0.3Co0.25Mn0.75O3 nanofibers also showed excellent long-term stability, with a current density decrement of only 10% after 10 h, which is superior to that of a commercial IrO2 catalyst. The high-OER performance of the La0.7Sr0.3Co0.25Mn0.75O3 nanofibers implies their promising practical applications.
We have shown that transition metal phosphides are efficient HER catalysts towards water splitting. In fact, the metal phosphides are also regarded as efficient OER catalysts.155,156 For example, FeNiP@N-doped CNFs have been prepared through electrospinning and phosphorization, and showed an excellent OER performance in 1.0 M KOH with a low overpotential (300 mV to reach a current density of 10 mA cm−2), a small Tafel slope (47 mV dec−1), and long-term stability (retention up to 99.3% over 20 h).155 In addition to the metal phosphides, recently, Fe/Ni phosphate nanosheets on the surface of electrospun CNFs (ESC@FNPO) have been developed via a four-step strategy for the OER.156 Firstly, PAN nanofibers were prepared via an electrospinning technique. Then CNFs were obtained through a calcination process. After that, an electrodeposition process was carried out to synthesize Fe/Ni hydroxide nanosheet arrays on CNFs. Finally, ESC@FNPO was produced via a phosphatized process using NaH2PO2 as a P source. The prepared ESC@FNPO has been proved to be an efficient OER catalyst, which showed an overpotential of 280 mV at a current density of 10 mA cm−2, lower than those of the RuO2 catalyst. The superior OER performance is attributed to the superior adsorption of hydroxyls on the surface of catalysts because the phosphates promote the metal oxidation and the distorted local metal geometry. In addition, the overpotential of the prepared ESC@FNPO was also much lower than that of Fe/Ni phosphate nanosheets on the surface of carbon cloth (ESC@CC), demonstrating the important role of the electrospun CNFs owing to their larger surface area and lower charge-transfer resistance.
A transition metal carbide is another promising candidate as an OER catalyst. Architectured Fe3C nanoparticle@nitrogen-enriched carbon nanotube (NCNT)-nitrogen-doped carbon nanofibers (NCNFs) were prepared via electrospinning and subsequent high temperature treatment.157 The prepared Fe3C@NCNTs-NCNFs with a unique heterostructure showed large exposed active sites, outstanding electron transfer properties and a strong synergistic catalytic effect (Fig. 6a and b). Therefore, the catalyst presented a superior OER catalytic activity in 1 M KOH media with an overpotential of 284 mV at a current density of 10 mA cm−2 and a low Tafel slope (56 mV dec−1) (Fig. 6c and d). These values are much lower than those of Fe3C-NCNF, RuO2 and NCNF samples, confirming the excellent OER kinetics of the prepared Fe3C@NCNTs-NCNFs. Furthermore, the catalyst presented an outstanding long-term stability, showing almost the same polarization curve after 5000 cycles as the initial one and only 4.4% degradation of the current density after 15 h testing (Fig. 6e and f). The superior OER performance could be attributed to the highly conductive CNF substrate and the unique encapsulated architecture.
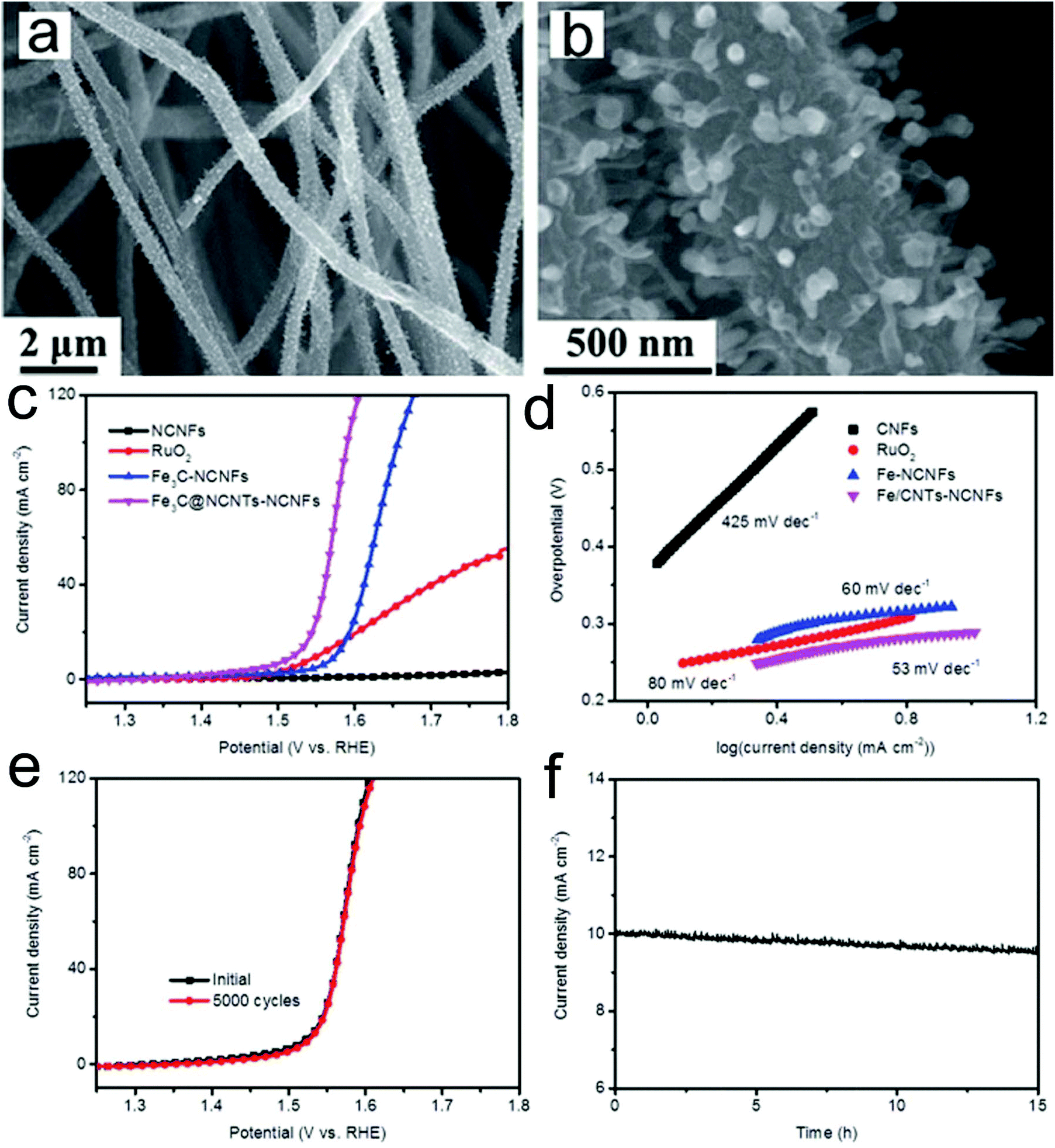 | ||
| Fig. 6 (a and b) SEM images of the prepared Fe3C@NCNTs-NCNFs. (c) Polarization curves and (d) the corresponding Tafel plots of Fe3C@NCNTs-NCNFs and control samples. (e) Stability test of the prepared Fe3C@NCNTs-NCNFs through (e) potential cycling and (f) time dependence of catalytic current density at a static overpotential. Reprinted with permission from ref. 157. Copyright 2017, Royal Society of Chemistry. | ||
3.3 Application of electrospun nanomaterials towards the ORR
The ORR is usually involved in a cathode reaction of fuel cells and metal–air batteries, and plays a key role in modern energy technologies.158,159 As the ORR delivers an intrinsically sluggish kinetics, it is necessary to introduce a high-performance catalyst to accelerate this process. Generally, the ORR process on the surface of an electrocatalyst is mainly divided into two steps.159 First, O2 molecules are diffused and adsorbed on the catalyst surface to form O2* (* stands for sites on the surface of the catalyst). Then, the O2* molecules are reduced to generate H2O or H2O2 through a two- or four-electron transfer process. The four-electron transfer process usually exhibits higher reaction kinetics and efficiency. During the ORR process, the removal of the typical intermediate species such as OOH* and OH* from catalysts is regarded as the rate-determining step (RDS) when the binding strength between the intermediates and catalyst is strong. In contrast, the formation of OOH* from the O2 protonation is the RDS when the binding strength is weak. Therefore, the regulation of the electronic structure of the catalyst to modulate the adsorption energy of the intermediates is an efficient means to achieve high-performance ORR.Pt-based materials are the most efficient commercial catalysts for the ORR due to their moderate binding energies with the intermediates.160–165 However, Pt-based catalysts suffer from the significant disadvantages of high cost, poor operation durability, and low tolerance against a fuel crossover effect. Consequently, non-noble-metal catalysts have been extensively developed as candidates for the ORR in the past few years. To date, a large number of ORR catalysts including heteroatom-doped carbons, metal–nitrogen–carbon (M–N/C), transition metal oxides, metal chalcogenides, carbon carbides, etc. have emerged as efficient catalysts for the ORR. One of the limitations towards enhancing the activity of these catalysts is their lack of active sites. Therefore, it is required to fabricate ORR catalysts with a unique structure and morphology. Electrospun nanomaterials play this role due to their unique 1D structures and directed electron transfer ability.160–230 Owing to the high cost of the Pt-based catalyst, Ag nanoparticles deposited on lignin-derived electrospun CNFs have been used for the ORR in a basic medium, delivering a much higher mass activity (119 mA mg−1) than the commercial Pt/C catalyst (98 mA mg−1).166 Recently, bimetallic FeAg-CNFs have also been developed as efficient ORR catalysts in KOH solution.167 The prepared FeAg-CNFs showed an improved electrocatalytic activity with a better onset potential and half-wave potential than the Pt/C catalyst.
Heteroatom-doped CNFs are the most common non-noble-metal catalysts for the ORR.169–186 For instance, the obtained N-doped CNFs that are prepared by carbonization of electrospun PAN nanofibers under an NH3 atmosphere possess a fiber-like structure and show some protruding graphitic layers with exposed edges.169 Owing to the unique characteristics, the N-doped CNFs delivered an excellent electrocatalytic activity for the ORR. In addition, the carbonization atmosphere and temperature strongly influenced the ORR activity of the N-doped CNFs. The NCNFs obtained in NH3 at 800, 900 and 1000 °C and N2 at 1000 °C showed the onset potentials at −0.23, −0.14, −0.09 and −0.13 V, demonstrating the superior ORR activity of the sample prepared in NH3 at 1000 °C (Fig. 7a and b). The sample with a carbonization temperature of 1000 °C under the NH3 atmosphere exhibited the best performance with the overall number of electrons transferred (n) being 3.96 at −0.40 V and a kinetic-limiting current density (JK) of 21.50 mA cm−2, demonstrating the outstanding electron-transfer kinetics of the ORR. Furthermore, the prepared N-doped CNFs showed favorable tolerance to the crossover effect and excellent electrocatalytic stability. There were few changes of the CV curves in methanol after 1000 cycles and a persisting relative current of 97.7% after 20000 s (Fig. 7c and d). From the X-ray photoelectron spectroscopy (XPS) results (Fig. 7e), it could be concluded that the small diameter, the abundant exposed layer edges, and the increased pyrrolic-N numbers contribute to the high-performance of the ORR. Compared with the single heteroatom-doped carbons, multiple heteroatom co-doped carbon materials have received more and more attention due to their enhanced electrocatalytic activity.185,186 For example, Jia and co-workers prepared morning glory-like N,S-co-doped carbon nanobelts (NSCNBs) through an electrospinning technique for ORR applications.185 The catalyst delivered a peak potential (Ep) of 0.812 V and a reduction current of 4.79 mA cm−2, which were better than those of many previously reported heteroatom co-doped carbon catalysts. In addition, the prepared NSCNBs delivered a half potential (E1/2) of 0.831 V, which was better than that of the commercial Pt/C catalyst (0.812 V). The n value was estimated to be ca. 3.64–3.94, which was also comparable with that of the Pt/C catalyst, indicating the superior ORR activity and a 4e−1 pathway. The Tafel slope of the NSCNBs (37.6 mV dec−1) was also close to that of the Pt/C sample (31.3 mV). Furthermore, the prepared NSCNBs exhibited superior durability compared with the commercial Pt/C catalyst. The high performance of the NSCNBs could be attributed to not only the unique belt-like microstructure but also the synergistic effect of both S and N doping.
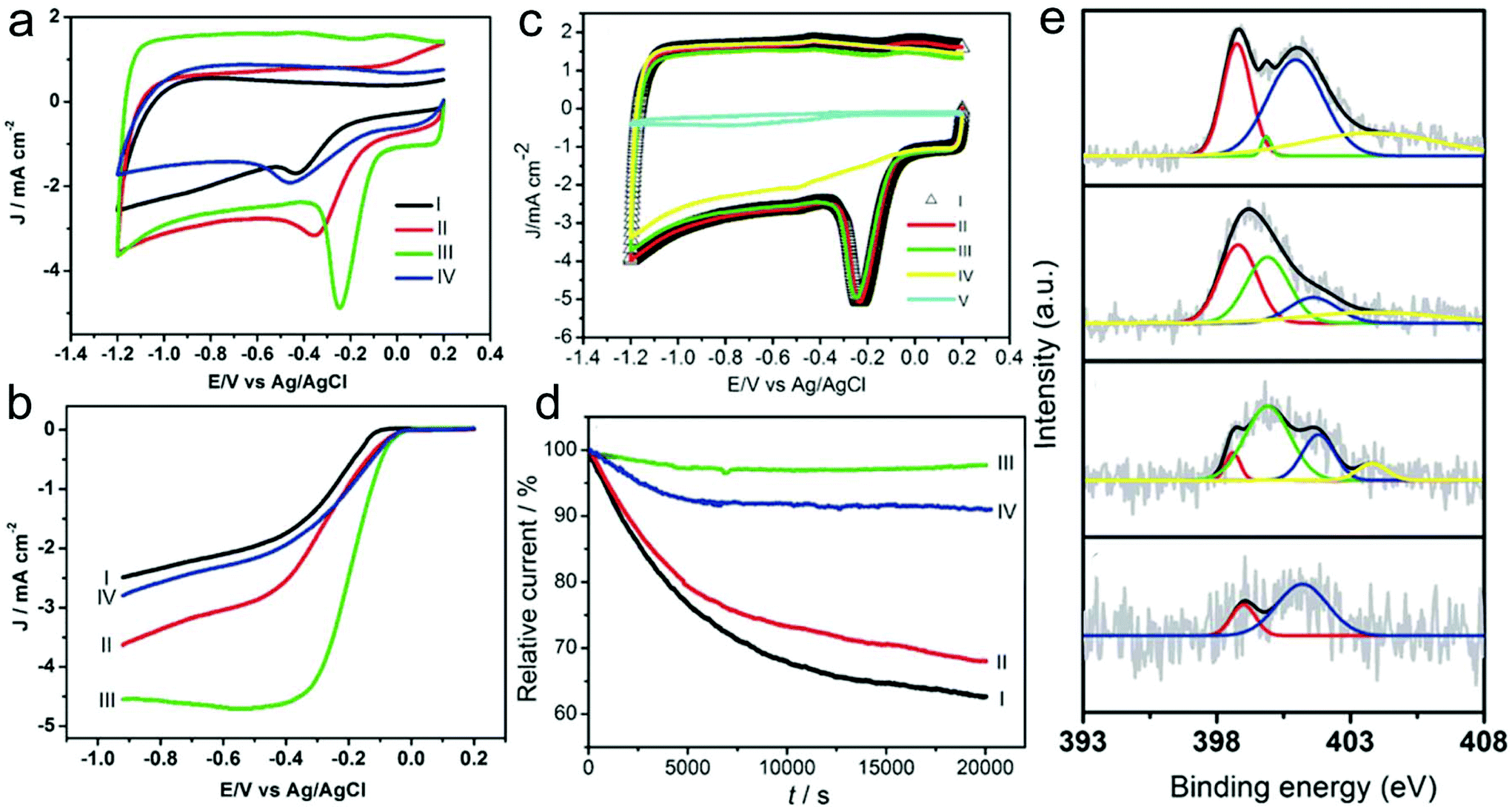 | ||
| Fig. 7 (a) CV curves and (b) RDE voltammograms of the NCNFs in O2-saturated 0.1 M KOH solution. I: NH3, 800 °C; II: NH3, 900 °C; III: NH3, 1000 °C; and IV: N2, 1000 °C; (c) tolerance to the crossover effect of the NCNFs measured from CV curves. I: saturated with O2, II: saturated with O2 containing 3.0 M methanol, III: saturated with O2 containing 3.0 M methanol and after a continuous potential cycling of about 1000 cycles, IV: saturated with N2; and V: saturated with O2, bare GC electrode. (d) I–t chronoamperometric curve of the NCNFs obtained in NH3 at I: 800 °C, II: 900 °C, III: 1000 °C and IV: in N2 at 1000 °C. (e) XPS N 1s curves of the NCNFs: (1) NH3, 800 °C; (2): NH3, 900 °C; (3): NH3, 1000 °C; and (4): N2, 1000 °C. Reprinted with permission from ref. 169. Copyright 2011, Elsevier. | ||
Recently, transition metals such as Fe and Co which enhance ORR activity in carbon materials have also been widely studied.187–218 Metal nanoparticles or metal atoms have been proved to be coordinated with N atoms to provide more catalytically active sites, which efficiently improve the electrocatalytic performance. Liang and co-workers prepared hierarchically porous Fe and N co-doped CNFs (HP-Fe–N/CNFs) through pyrolysis of polypyrrole-decorated polystyrene/FeCl3 fibers for the ORR.187 The prepared Fe–N/CNFs delivered a large surface area of 569.6 m2 g−1 and moderate doping of Fe and N, resulting in outstanding ORR activity. Accordingly, the HP-Fe–N/CNFs exhibited a comparable onset potential (0.93 V) and half-wave potential (0.80 V) to the Pt/C catalyst (0.92 and 0.81 V). Meanwhile, the Tafel slope of the HP-Fe–N/CNFs (72 mV dec−1) was also similar to that of the Pt/C catalyst (68 mV dec−1). Furthermore, the prepared HP-Fe–N/CNFs showed remarkable selectivity, long-term stability and tolerance against methanol. The outstanding ORR performance was ascribed to the efficient mass transfer from the hierarchical porous structure of the HP-Fe–N/CNFs and the abundant active sites from the effective Fe and N doping. Similar to the Fe–N-CNFs, Co–N-CNFs have also been developed as efficient ORR catalysts, which display a comparable ORR performance with the commercial Pt/C catalyst.
Transition metal oxides are another type of efficient ORR catalyst, showing the advantages of low price, good methanol tolerance, and superior stability.219–225 To enhance the electron transport of a metal oxide-based catalyst, the hybridization of a metal oxide with conductive carbon materials is an effective strategy. For instance, Co3O4 nanoparticles embedded in porous N-doped graphitic CNFs have been prepared through electrospinning and a subsequent annealing process for ORR applications.219 During the synthetic process, metal nanoparticles were firstly formed, and they catalyzed graphitization of the polymer nanofibers and converted to Co3O4 nanoparticles via an oxidation process. Therefore, the prepared Co3O4@N-doped graphitic CNFs exhibited a large surface area, multiple pore channels and plenty of active sites, offering them excellent electrocatalytic performance. The catalyst delivered a half-wave potential of 10 mV more positive than Pt/C, demonstrating its superior OER activity in alkaline solutions. Furthermore, the prepared Co3O4@N-doped graphitic CNFs showed better stability and methanol tolerance than Pt. In a word, the high-performance of the ORR was related to the synergistic catalytic effect between Co3O4 and N-doped graphitic CNFs. It is well known that Co3O4 shows magnetic susceptibility because of the spin/spin–orbit coupling-induced magnetic moment. Therefore, the ORR properties were strongly influenced by the magnetic field. With the assistance of a 1.32 mT magnetic field, a 3.92-electron pathway was attained.
A transition metal carbide is an efficient non-noble metal-based ORR catalyst in both acidic and basic media.226–230 Recently, a porous Fe3C embedded in N-doped CNFs has been developed through electrospinning, chemical vapor polymerization, and pyrolysis processes for ORR applications.226 During the electrospinning procedure and pyrolysis, PVDF was used not only as a carbon source but also for providing a porous structure because HF was generated from PVDF thermal decomposition. The resultant Fe3C@N-doped CNFs that were pyrolyzed at 900 °C showed an outstanding ORR performance in both alkaline and acidic electrolytes. In 0.1 M KOH aqueous solution, the catalyst showed onset and half-wave (E1/2) potentials of −0.035 and −0.121 V, which are more positive than those of the Pt/C catalyst, while in 0.1 M HClO4 solution, the onset and half-wave (E1/2) potentials of the catalyst were calculated to be 0.532 and 0.342 V, which were comparable with those of the Pt/C catalyst. These results demonstrated the excellent ORR activity. Additionally, the prepared Fe3C@N-doped CNFs exhibited higher stability and tolerance to the methanol crossover than the commercial Pt/C catalyst, which were attributed to the protection of graphitic carbon layers on the surface of active Fe3C nanoparticles.
3.4 Application of electrospun nanomaterials towards the CO2RR
It is well known that the increase in the CO2 concentration in the atmosphere results in a phenomenon of global warming. Therefore, much effort has been focused on CO2 capture, storage and utilization in the past few decades.231 Most importantly, it is a meaningful objective to convert CO2 to hydrocarbon fuels via sustainable technologies such as photochemical, electrochemical and biochemical reactions. Among these techniques, the electrocatalytic CO2RR is one of the sustainable techniques with high conversion efficiency to selectively produce hydrocarbon fuels. However, it is still a great challenge to efficiently activate the inert and stable CO2 molecules. Generally, current research is focused on the development of high-performance electrocatalysts for the CO2RR to improve their catalytic kinetics. A typical electrocatalytic CO2RR process usually includes three steps.232 First, CO2 molecules are adsorbed on the surface of the catalyst. Then, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds are dissociated through electron and proton transfer, which can generate C–O and C–H bonds. Finally, the product is desorbed from the surface of the catalyst. Because of the high activity of the intermediate of CO2˙− from an independent electron transfer process, a series of products such as CO, HCOOH, HCHO, CH4, C2H4, CH3OH, and C2H5OH can be obtained through a thermodynamically favorable proton-coupled electron transfer process. Therefore, it is a great challenge to improve the selectivity of the product by modulating the electronic structure of the catalyst.
O bonds are dissociated through electron and proton transfer, which can generate C–O and C–H bonds. Finally, the product is desorbed from the surface of the catalyst. Because of the high activity of the intermediate of CO2˙− from an independent electron transfer process, a series of products such as CO, HCOOH, HCHO, CH4, C2H4, CH3OH, and C2H5OH can be obtained through a thermodynamically favorable proton-coupled electron transfer process. Therefore, it is a great challenge to improve the selectivity of the product by modulating the electronic structure of the catalyst.
Pt-based nanomaterials are a type of important electrocatalyst for the CO2RR, showing advantages including high catalyst productivity, good selectivity and favorable stability.233,234 He and co-workers demonstrated a facile electrospinning process combined with a carbonization process to prepare Pt nanoparticles embedded in N-doped CNFs on flexible carbon cloth (Pt-NP@NCNFs@CC) for the CO2RR.233 During the electrospinning process, ZnCl2 was additionally added into the precursor solution to generate a porous structure of Pt-NP@NCNFs@CC, which exhibit a specific surface area of 233.5 m2 g−1 with a pore size of 4–5 nm. In addition, the size of Pt nanoparticles in the nanofibers was estimated to be 5 nm. The prepared Pt-NP@NCNFs@CC showed a higher reductive current under a CO2 atmosphere compared to that under a N2 atmosphere in 0.1 M KHCO3 solution, confirming an excellent electrochemical CO2RR (Fig. 8a). Furthermore, the onset potential was −0.46 VRHE at the Pt-NP@NCNFs@CC electrode, which was much lower than that at a pure CC electrode (−0.7 V) (Fig. 8a and b). As shown in Fig. 8c, a high faradaic efficiency of 91% to produce a formate was observed at −0.5 VRHE, which was much better than that using pure Pt nanoparticles and NCNFs@CC. In addition, the catalyst showed good recyclability with a remaining faradaic efficiency of 90% at least ten times (Fig. 8d). Therefore, it could be proposed that CO2 molecules might be adsorbed onto the carbon atom close to the pyridinic N, followed by the formation of *COOH with Pt–H from the Pt surface. On the other hand, the electrocatalytic carboxylation of CO2 to generate 2-phenylpropionic acid has also been attained in 1-phenylethyl bromide in acetonitrile solution, providing a faradaic efficiency of 99%. Importantly, the selectivity of the CO2RR to hydrocarbons and alcohols is very important for their applications in the energy field. Suitable electrocatalysts such as Cu-based materials have been proved to be good candidates for the CO2RR to produce alcohols. Interestingly, when the Pt-NP@NCNFs are prepared on Cu foil, they have been found to show a selective CO2RR to generate a formate at −0.6 VRHE with 93% faradaic efficiency and alcohols with 35% faradaic efficiency at a −1.0 VRHE cathode potential.234
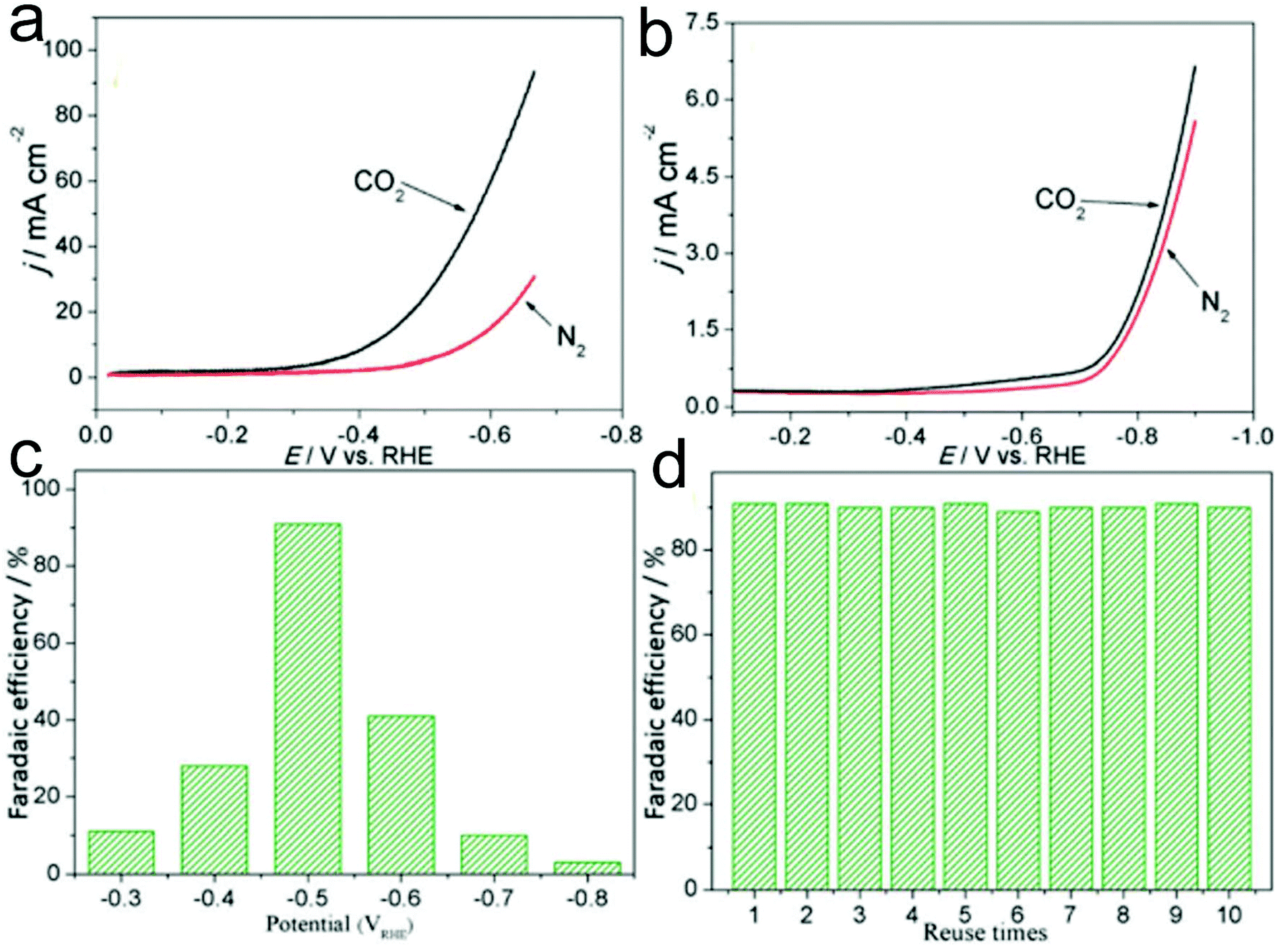 | ||
| Fig. 8 LSV recorded at a sweep rate of 0.1 V s−1 in 0.1 M KHCO3 medium at (a) Pt-NP@NCNFs@CC and (b) CC electrodes. (c) Faradaic efficiency of the Pt-NP@NCNFs@CC catalyst at potentials from −0.3 to −0.8 VRHE. (d) Recyclability measurement of the catalyst at −0.5 VRHE. Reprinted with permission from ref. 233. Copyright 2018, Royal Society of Chemistry. | ||
In addition to the formate and alcohols, CO is also a typical product during the electrocatalytic CO2RR. CuInS2 nanofibers have been reported to show excellent CO2RR activity due to their suitable band gap (1.3 eV) and large catalytically active sites.235 The CuInS2 nanofibers were prepared through electrospinning and subsequent annealing and sulfurization processes. The prepared CuInS2 nanofibers delivered an outstanding faradaic efficiency of 77 ± 4% for the reduction of CO2 to CO, which was four times higher than that of the solution processed CuInS2 sample. This result confirmed that the fibrous structure of CuInS2 is beneficial for the CO2RR to CO. Furthermore, the electrocatalyst showed outstanding stability compared with other catalysts. Recently, electrospun Co3O4 nanofibers have also been used as efficient electrocatalysts for the CO2RR to CO with a faradaic efficiency of 65%.236 The mechanism for the CO2RR was considered to be the adsorption of CO2 on the surface of Co3O4 nanofibers and subsequent formation of COO*−via one electron transfer from the electrode. In the following, CO could be produced from an oxygen–carbon coupling of COO*− with CO2. It was specially pointed out that a by-product of the formate is also obtained in the CO2RR process, which is related to the protonation of COO*− from the trace water in the electrolyte and subsequent electron transfer. However, the faradaic efficiency for the by-product formation of the formate was only 27%, which was much lower than that of the generation of CO.
Sn-based materials have now attracted increasing attention because of their high efficiency and excellent selectivity to generate C1 products from an electrocatalytic CO2RR process.237–239 The electrocatalytic activity of the Sn-based catalysts is highly sensitive to their composition, morphology and amount of catalytically active sites. 1D nanomaterials usually show large surface areas and thus present abundant active sites. Additionally, the 1D structure offers a unique charge transfer ability at the interfaces between the electrode and electrolyte, which improves the electrocatalytic activity. 1D nanomaterials also possess a superior structure stability over bulk catalysts during electrochemical reactions. Typically, 1D electrospun wire-in-tube (WIT) structured SnO2 nanofibers have been demonstrated to be efficient CO2RR electrocatalysts.237 During the electrospinning and subsequent calcination processes, a larger density of structural defects such as grain boundaries (GBs) has been achieved in the SnO2 nanofibers, which provide enhanced catalytically active sites. Furthermore, the unique 1D WIT structure of SnO2 nanofibers favors the electron transport. Therefore, the prepared 1D WIT SnO2 nanofibers delivered a maximum faradaic efficiency of 93% to produce C1 products (HCOOH and CO) and a suppressed faradaic efficiency of hydrogen generation at a higher current density of −0.99 V, which was much better than that of the SnO2 nanoparticle electrode. The superiority in the CO2RR has been ascribed to the large surface area of the 1D WIT structure, the stabilization of the active surface by GBs within the nanopores, and the field-induced reagent concentration for the stabilization of adsorbed COO*− intermediates.
To further increase the electrocatalytic activity and selectivity of the CO2RR, partially reduced Sn/SnO2 porous nanofibers have been prepared via a similar electrospinning and calcination procedure but with a reduction process.238 The reduction process to form Sn increases the oxygen vacancies of SnO2 nanofibers, which is beneficial for improving the electrocatalytic performance. Then, the synthesized Sn/SnO2 porous nanofibers delivered a faradaic efficiency of 82.1% to generate the formate with a current density of about 22.9 mA cm−2. Recently, single-atom catalysts have become a hot topic in heterogeneous catalysis due to their ultrahigh catalytic activity. It has been reported that atomically dispersed Sn species show a completely different selectivity towards the CO2RR.239 In detail, Sn modified N-doped porous CNFs have been prepared through an electrospinning and pyrolysis process. It is found that N-doped CNF supported Sn nanoparticles delivered a partial high current density of 11 mA cm−2 and a faradaic efficiency of 82.1% at a overpotential of 690 mV to produce the formate. However, atomically dispersed Sn species on CNFs changed to selectively produce CO during the CO2RR, exhibiting a faradaic efficiency of 91% at an overpotential of 490 mV. The Sn–N moieties in the Sn atom modified N-doped porous CNFs contribute to the high activity and selectivity for CO generation.
3.5 Application of electrospun nanomaterials towards the NRR
Electrocatalytic fixing of N2 to generate NH3via the NRR is a facile, economical and sustainable route for large-scale NH3 production compared with the traditional Haber–Bosch process.240 However, it is significantly necessary to develop an efficient NRR catalyst to increase the production rate and faradaic efficiency for NH3 synthesis. Until now, a variety of noble metals, transition metal oxides, nitrides, sulfides and carbides have been used as electrocatalysts for the NRR. Importantly, the morphology, crystallinity, heteroatom doping and even defect engineering of the catalysts strongly influence their NRR activities. Generally, there are two types of mechanisms to investigate the NRR process on different catalysts. In the dissociative pathway, the N2 triple bond is broken first and then the N atom is adsorbed on the surface of the catalyst to achieve hydrogenation to generate NH3.241 However, an extremely high energy input is required to overcome the dissociation energy of the N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond. For the associative mechanism, N2 molecules are adsorbed on the surface of the catalyst, and are able to accept protons from the electrolyte and electrons from the catalyst to form adsorbed species and other intermediates through alternating and distal pathways, and finally produce NH3. During the associative pathway, it is not necessary to break the first bond in N2, decreasing the energy input for the NRR. The electrocatalytic NRR mainly undergos an associative mechanism, enabling the realization of the NRR under ambient conditions.
N bond. For the associative mechanism, N2 molecules are adsorbed on the surface of the catalyst, and are able to accept protons from the electrolyte and electrons from the catalyst to form adsorbed species and other intermediates through alternating and distal pathways, and finally produce NH3. During the associative pathway, it is not necessary to break the first bond in N2, decreasing the energy input for the NRR. The electrocatalytic NRR mainly undergos an associative mechanism, enabling the realization of the NRR under ambient conditions.
1D electrospun nanomaterials show a large surface active area and excellent charge transfer, which are beneficial for the NRR efficiency. As mentioned, defect engineering is usually used to guide the manipulation of the construction of NRR electrocatalysts. It has been reported that the introduction of oxygen vacancies is able to weaken the NN triple bond because of the ability of the trapping electrons to be injected into an antibonding orbital of the N2 molecule. Bismuth vanadate (Bi4V2O11) possesses intrinsic oxygen vacancies, and is anticipated to be a good candidate as an efficient NRR catalyst. Recently, Bi4V2O11/CeO2 hybrid nanofibers have been prepared through an electrospinning and calcination approach.242 Interestingly, the amorphization of the hybrid was much related to the mole ratio of Ce/Bi. When the molar ratio of Ce/Bi was larger than 1:2, amorphous Bi4V2O11/CeO2 hybrid nanofibers were obtained. However, a crystalline Bi4V2O11 phase was observed after the molar ratio of Ce/Bi reduced to 1:4. The XPS results revealed the formation of high-valence bismuth defects for the amorphous sample during the calcination process, inducing enriched oxygen vacancies. Furthermore, based on the Mott–Schottky measurement and UV/vis diffusion reflection spectra, a type I band alignment of the Bi4V2O11/CeO2 hybrid nanofibers was formed (Fig. 9a–c). Therefore, the localized electron transfer from CeO2 to Bi4V2O11 in the amorphous sample was enhanced, which was beneficial for the activation of the NN triple bond and promoted NRR performance (Fig. 9d). As a result, the prepared Bi4V2O11/CeO2 hybrid nanofibers delivered a high NH3 yield and faradaic efficiency of 23.21 μg h−1 mg−1cat and 10.16%, respectively, at −0.2 VRHE (Fig. 9e). In addition, the catalyst also exhibited excellent stability during NH3 production (Fig. 9f). In contrast, the prepared amorphous Bi4V2O11/CeO2 hybrid nanofibers showed a much better NRR activity than pristine CeO2, Bi4V2O11, and even crystalline Bi4V2O11/CeO2 hybrid nanofibers, implying their efficient electron transfer and multiple defects (Fig. 9g and h).
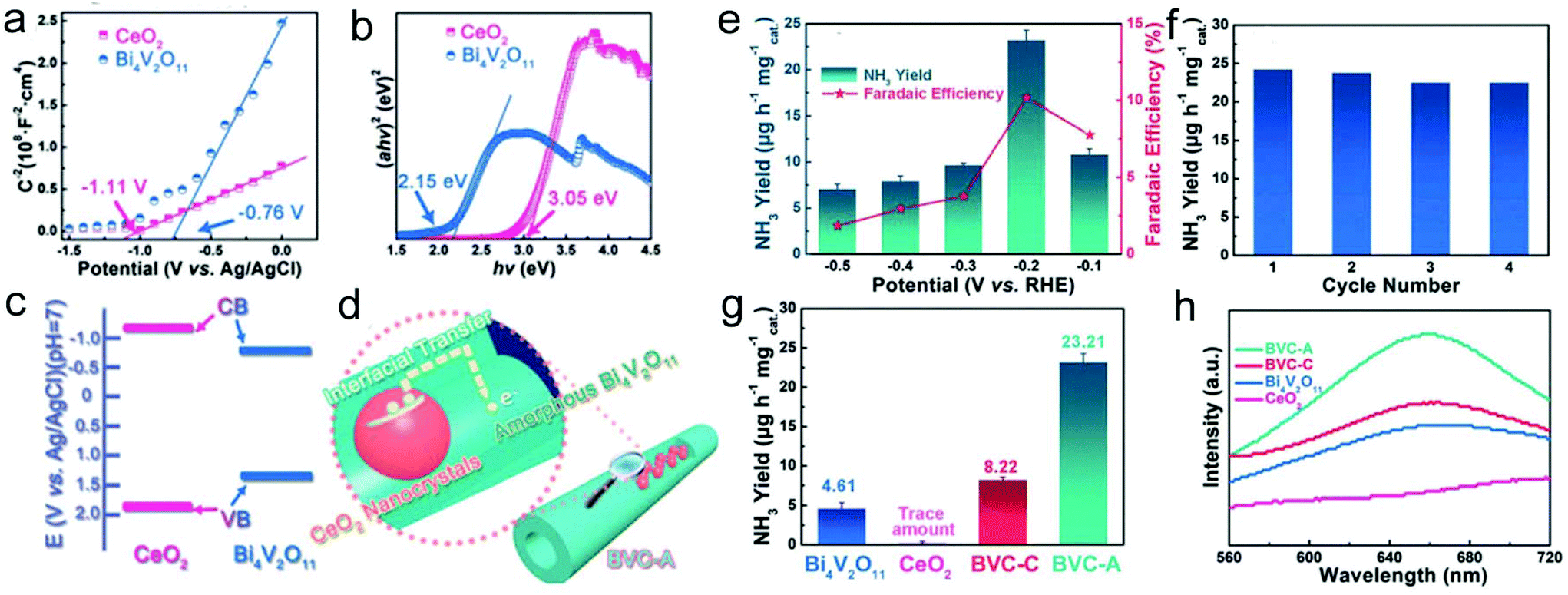 | ||
| Fig. 9 (a) Mott–Schottky plot and (b) Kubelka–Munk plot of Bi4V2O11 and CeO2. (c) Band alignment of Bi4V2O11 and CeO2. (d) Illustration of the charge transfer in BVC-A. (e) Yield of NH3 and faradaic efficiency at different potentials in the electrocatalytic NRR of BVC-A. (f) Cycling test of BVC-A. (g) Yield of NH3 with various catalysts at −0.2 VRHE. (h) UV/vis absorption spectra of the electrolyte stained with an indophenol indicator. BVC-A represents amorphous Bi4V2O11/CeO2 hybrid nanofibers and BVC-C stands for crystalline Bi4V2O11/CeO2 hybrid nanofibers. Reprinted with permission from ref. 242. Copyright 2018, John Wiley and Sons. | ||
Nb compounds have aroused great interest owing to their promising applications in solid acid catalysis. For example, the production yield of the chemical synthesis of NH3 using FeO as the catalyst has been efficiently accelerated with an Nb2O5 component. Recently, electrospun Nb2O5 nanofibers have also been found to show an outstanding NRR activity.243 The Nb2O5 nanofibers showed a porous structure with a 1D feature and a BET surface area of 122.1 m2 g−1. The prepared catalyst showed a much higher NH3 yield (43.6 μg h−1 mg−1cat) and faradaic efficiency (9.26%) in 0.1 M HCl solution than other NRR catalysts such as Au/TiO2, MoO3, γ-Fe2O3etc. Interestingly, in addition to the acidic electrolyte, the catalyst also delivered favorable NH3 yields (22.3 and 11.45 μg h−1 mg−1cat) and faradaic efficiencies (6.2% and 6.7%) in 0.1 M Na2SO4 and 0.1 M NaOH solution. The Nb2O5 nanofibers possessed remarkable selectivity and excellent long-term stability. Furthermore, the prepared Nb2O5 nanofibers exhibited a higher NRR activity than a commercial Nb2O5 sample, which was attributed to the good diffusion of N2 and the product as well as the exposure of more active sites due to the highly porous 1D structure. DFT calculations have been carried out to explore the mechanism for the NRR (Fig. 10). Since the first hydrogenation step to form NNH* showed no barrier with a Gibbs free energy of −0.73 eV for N2 molecules adsorbed on the surface of Nb2O5 nanofibers, it revealed that the formation of the NNH2* species was the potential-limiting step, showing a energy barrier of 0.56 eV. The theoretical calculation results were well consistent with the experimental observations.
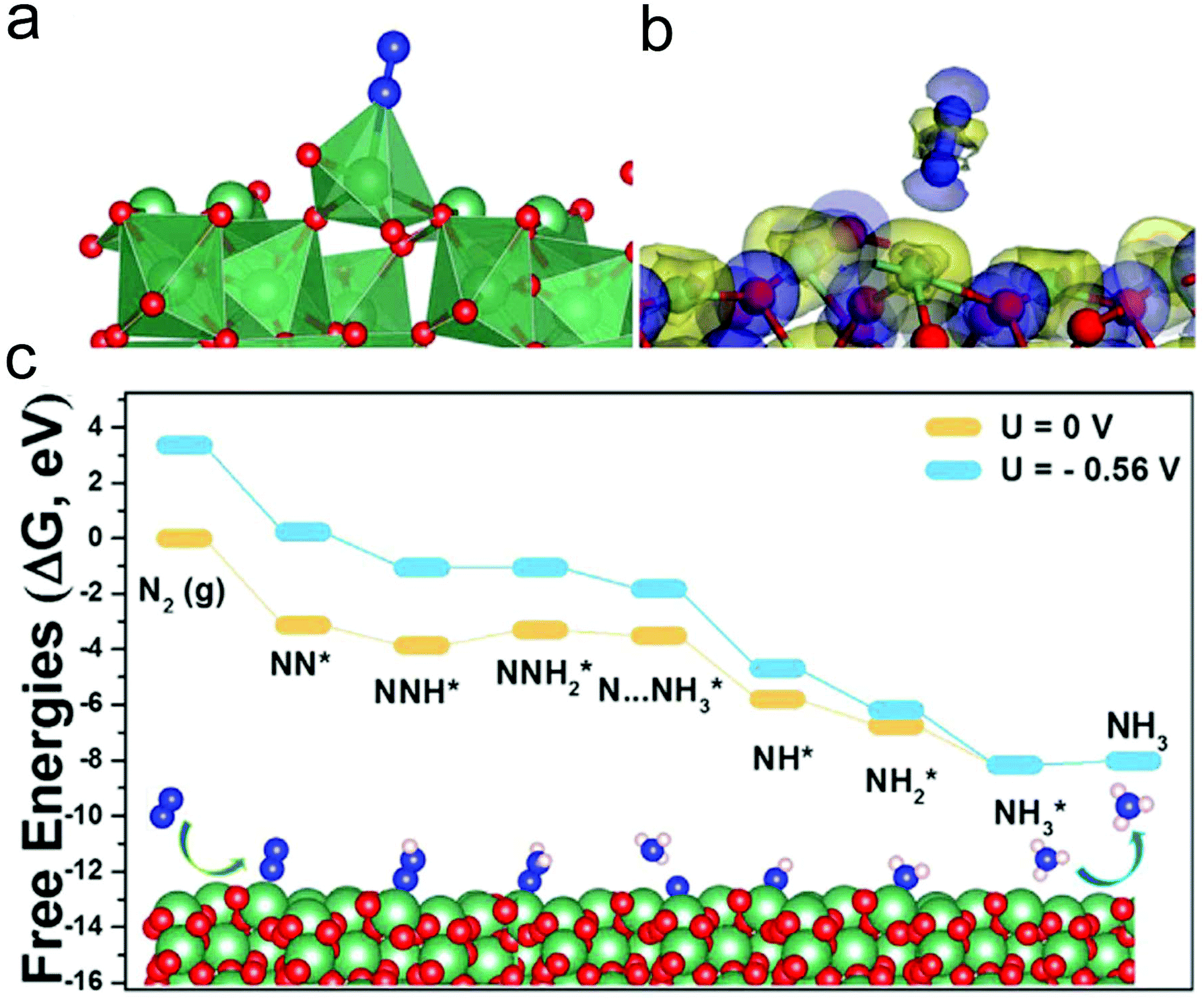 | ||
| Fig. 10 (a) Optimized structure and (b) the charge density difference of the configuration of N2 adsorbed on the Nb2O5 (181) surface. Blue and yellow colours stand for the charge depletion zones and charge accumulation. (c) Free energy diagram and optimized structures for the NRR on the Nb2O5 (181) surface at zero and applied potentials (limiting potential) through distal mechanisms. Reprinted with permission from ref. 243. Copyright 2018, Elsevier. | ||
3.6 Application of electrospun nanomaterials towards small molecule oxidation reactions
Fuel cells have been attracting significant research interest in the field of energy storage and conversion. The typical electrocatalytic processes on the cathode of a fuel cell usually include the methanol oxidation reaction (MOR), ethanol oxidation reaction (EOR), urea oxidation reaction (UOR) etc.244–258 In previous studies, the most widely studied electrocatalyst for the MOR is Pt-based nanomaterials. Through electrospinning and a subsequent two-step calcination process under N2 and air, polycrystalline Pt nanowires with a diameter of 40–90 nm have been fabricated toward the MOR.244 The prepared Pt nanowires showed a higher MOR peak current density and lower peak potential, confirming a larger specific activity than commercial Pt black. Furthermore, the Pt nanowires displayed a higher ratio of the forward peak current density to backward one (If/Ib) (0.73) than the Pt black catalyst (0.65), implying the better tolerance for the poisoning species of the Pt nanowires. However, the mass activity of Pt nanowires was smaller than that of Pt black, which should be improved in the future studies. Similarly, binary metallic nanowires that were synthesized via electrospinning and heat treatment have also been used for the MOR.245 The prepared PtRh and PtRu nanowires with a 1:1 atomic ratio showed a superior MOR mass activity and long-term stability over commercial PtRu nanoparticles on carbon matrices.
Generally, the interactions between the support and the active catalyst play a key role in the catalytic and electrocatalytic performance.246–248 Transition metal oxides are usually used as efficient supports to load noble metal nanoparticles to enhance the catalytic activity and selectivity as well as long-term stability during the catalytic process. Zhang, Yuan and co-workers studied the influence of MnO2 on the MOR activity of Pt nanoparticles on N-doped CNFs.246 Pt/MnO2-CNFs could be prepared through electrospinning, carbonization, adsorption and reduction processes (Fig. 11a). The resulting Pt/MnO2-CNFs displayed a MOR peak current density of 263.2 mA cm−2, which was around 3.0 and 4.0-fold compared with those of Pt/CNFs and Pt/XC72R, confirming the enhanced MOR activity via the addition of the MnO2 component (Fig. 11b). Additionally, the If/Ib value for Pt/MnO2-CNFs (1.12) was much higher than those of Pt/CNFs (0.86) and Pt/XC72R (0.55), demonstrating a better tolerance for poisoning species due to the synergistic effect between Pt and Mn elements (Fig. 11c). Importantly, the Pt/MnO2-CNFs also showed excellent durability (Fig. 11d). The ECSA could still remain at 77% after 1500 cycles at the Pt/MnO2-CNF electrode, which was better than that of Pt/CNFs (Fig. 11e).
 | ||
| Fig. 11 (a) Schematic illustration of the synthetic procedure for Pt/MnO2-CNFs. CV curves of the prepared Pt/MnO2-CNF catalyst and other control samples in (b) 0.5 M H2SO4 and (c) 0.5 M H2SO4 + 1 M CH3OH aqueous solution at a scan rate of 50 mV s−1. (d) CV curve of the Pt/MnO2-CNF catalyst in 0.5 M H2SO4 at a scan rate of 50 mV s−1 and (e) its normalized ECSA values at different cycles. Reprinted with permission from ref. 246. Copyright 2015, Elsevier. | ||
Although Pt-based nanomaterials display a superior MOR performance, their high cost extensively restricts their broad and practical applications. Therefore, the development of cheap non-precious electrocatalysts has attracted increasing attention.249–251 Co and Ni-based nanomaterials have been proved to show certain MOR activity. For example, Co-incorporated N-doped CNFs have been prepared through electrospinning and subsequent carbonization treatment for the MOR in an alkaline medium.249 The prepared Co/N-CNFs exhibited the advantages of high conductivity and excellent wettability due to the graphite-like hexagonal framework of CNFs, which contributed to the improved MOR performance. As a result, the Co/N-CNFs showed a maximum current density of 100.84 mA cm−2, which was higher than that of N-free Co/CNFs (63.56 mA cm−2). Furthermore, the catalyst showed better stability, with the steady current density remaining at 3.96 mA cm−2 after 1000 s, compared to N-free Co/CNFs (2.94 mA cm−2).
Compared with a direct methanol fuel cell (DMFC), a direct ethanol fuel cell (DEFC) possesses the advantages of low toxicity and high theoretical energy density, and is considered as a more promising fuel cell. Therefore, EOR catalysts have attracted increasing attention in recent years. Similar to the case of the MOR, the transition metal and metal oxides are good candidates as EOR catalysts.252–254 For example, Pt-SnO2/CNFs have been prepared through an electrospinning and carbonization process for the EOR.254 The introduction of PVDF in the electrospun precursor provided the porous structure of the Pt-SnO2/CNFs with a large BET surface area of 374.85 m2 g−1. Owing to the increased active sites, the prepared Pt-SnO2/CNF catalyst showed an ethanol oxidation peak current value of 140.14 mA cm−2, and a maximum power density of 18.1 mW cm−2 was achieved at a potential of 0.7 V (oxygen flow rate: 100 mL min−1, temperature: 80 °C). This result demonstrates that the electrospun Pt-SnO2/CNFs are efficient EOR catalysts.
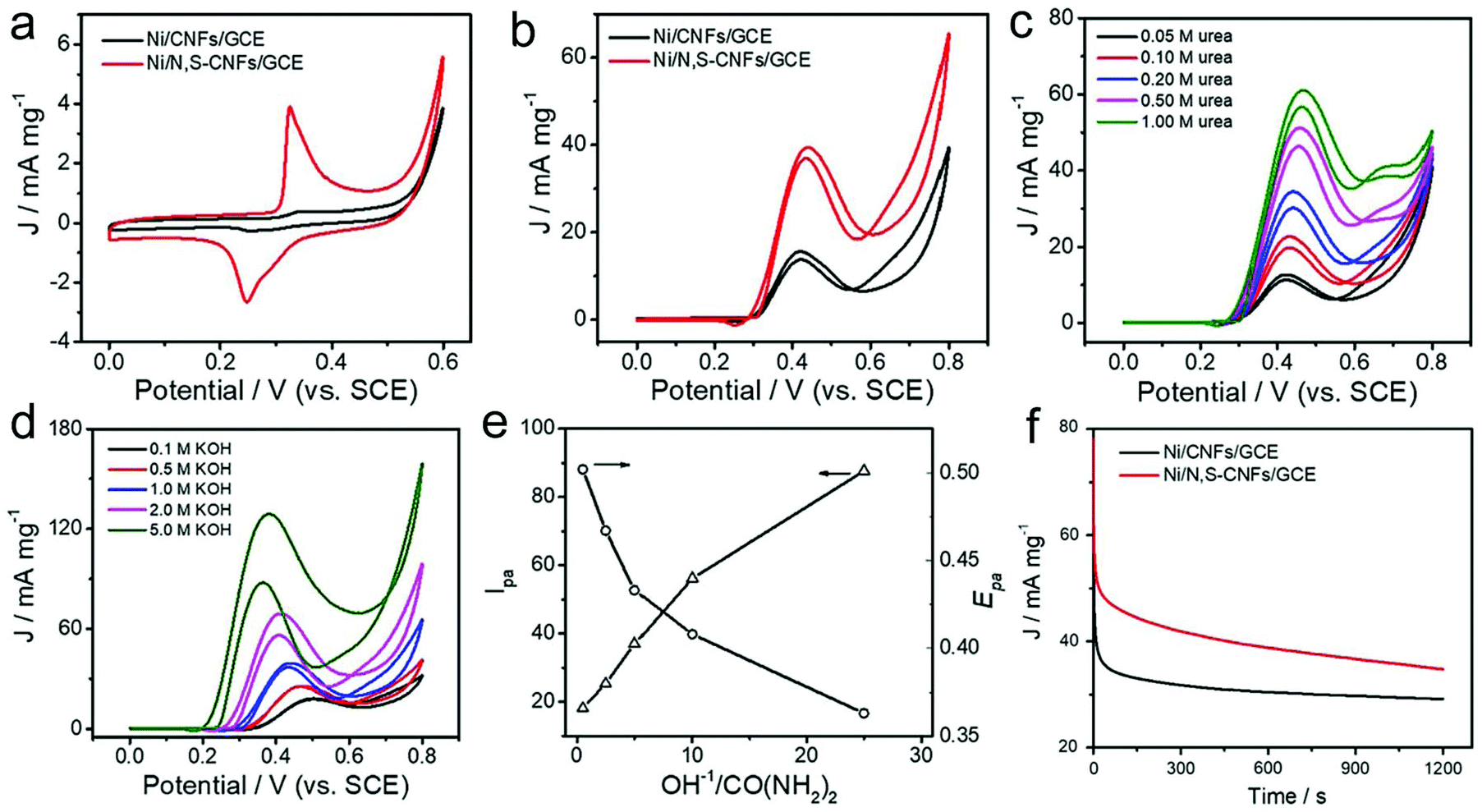
In addtion to the MOR and EOR, the UOR is another anodic reaction in fuel cells. Compared with liquid hydrogen, urea shows a larger value of energy density. Thus the investigation of the UOR is important in the energy conversion field. Pt and Rh are efficient electrocatalysts for the UOR, but their high price limits their practical applications. Recently, Ni-based materials have been found to show a favorable UOR activity, and they also possess the advantage of low cost.255–258 For example, N-doped and Ni-decorated CNFs that are synthesized via an electrospinning and carbonization process have been used as electrocatalysts for the UOR.255 For these catalysts, N-doping was beneficial for the formation of a NiOOH layer on the surface of Ni nanoparticles, which promoted the electrocatalytic activity for the UOR. In detail, the increment of the N content in the Ni-decorated CNFs reduced the onset potential. Similarly, Ni nanowires encapsulated in N and S-codoped CNFs (Ni/N,S-CNFs) have also been prepared via an electrospinning and carbonization process, which could be used as efficient electrocatalysts for the UOR.256 As shown in Fig. 12a, the CV curve of Ni/N,S-CNFs showed a pair of redox peaks at 0.326 V and 0.248 V with a peak separation (ΔEp) of 78 mV in 1 M KOH solution. The ΔEp value of the Ni/N,S-CNFs was much smaller than that of Ni/CNFs, implying faster charge-transfer kinetics. The prepared Ni/N,S-CNFs delivered a peak current density of 37.0 mA mg−1 at 0.42 V (vs. SCE), which was two times higher than that of Ni/CNFs (Fig. 12b). In Fig. 12c, it was found that the anodic peak current density (Ipa) increases with an increase in the urea concentration at a Ni/N,S-CNF modified GCE, demonstrating an indirect or catalyst regeneration mechanism. In addition, the Ipa also increased with an increase in the concentration of KOH (Fig. 12d). The Ipa value at Ni/N,S-CNFs was much higher than that at the Ni/CNF modified GCE, while the peak oxidation potential (Epa) value at Ni/N,S-CNFs showed a positive shift compared with that at the Ni/CNF modified GCE, confirming the superior electrocatalytic activity of the prepared Ni/N,S-CNFs (Fig. 12e). Furthermore, the stability of this catalyst was also superior to that of the Ni/CNFs (Fig. 12f). This result revealed that N,S co-doping strongly enhanced the electrocatalytic performance.
 | ||
| Fig. 12 CV curves of Ni/CNF and Ni/N,S-CNF modified GEs in 1 M KOH solution in the (a) absence and (b) presence of 0.2 M urea. (c) The response of urea with various concentrations at the Ni/N,S-CNF modified GE. Scan rate: 0.02 V s−1. (d) The response at the Ni/N,S-CNF modified GE for 0.2 M urea with various concentrations of KOH. (e) The electrochemical response at the Ni/N,S-CNF modified GE in N2-saturated 1 M KOH with different concentrations of urea. Scan rate: 0.02 V s−1. (f) I–t curves at the Ni/N,S-CNF modified GE in 1 M KOH in the presence of 0.2 M urea at 0.437 V. Scan rate: 0.02 V s−1. Reprinted with permission from ref. 256. Copyright 2018, Elsevier. | ||
3.7 Application of electrospun nanomaterials towards bi-/multifunctional electrocatalysis
As discussed, it is noticed that usually a special electrocatalyst is suitable for a special electrocatalytic reaction. However, some electrocatalysts have shown electrocatalytic activities in dual or multiple reactions.259–272 This suggests that rechargeable batteries can be constructed by using the same electrocatalysts as both cathodes and anodes. For instance, a water electrolyzer cell has been fabricated with a bifunctional electrocatalyst for both the HER and OER. We have demonstrated the preparation of Ni and Mo2C nanoparticles encapsulated in N-doped CNFs (Ni/Mo2C-NCNFs) via an electrospinning and carbonization process for overall water splitting (Fig. 13a).259 As shown in Fig. 13b and c, it is found that Ni and Mo2C nanoparticles were well distributed throughout the porous CNFs. The resulting Ni/Mo2C-NCNFs were proved to be efficient HER and OER catalysts. Furthermore, the electrocatalytic activities were much influenced by the proportion of Ni and Mo2C precursors, carbonization temperature, and weight ratios between metal precursors and PAN. Thus, the optimized Ni/Mo2C-NCNFs exhibited an excellent HER activity with a low overpotential of 143 mV to reach a current density of 10 mA cm−2 and a Tafel slope of 57.8 mV dec−1 in an alkaline solution (Fig. 13d and e). The superior HER activity was attributed to the synergistic effect of the components in the catalyst and the more exposed active sites, which was reflected by the large double-layer capacitance (Cdl) value (29.41 mF cm−2) of the catalyst (Fig. 13f). On the other hand, the prepared Ni/Mo2C-NCNFs also showed an outstanding OER activity with overpotentials of 288 and 432 mV to reach a current density of 10 and 100 mA cm−2, respectively (Fig. 13g). These values were also much better than those of the commercial RuO2 catalyst, demonstrating the superior OER activity of the Ni/Mo2C-NCNFs. A Tafel slope of 78.4 mV dec−1 was obtained, displaying fast OER kinetics (Fig. 13h). Fig. 13i also shows the favorable stability of the catalyst. The excellent OER performance of the Ni/Mo2C-NCNFs could be attributed to the electron transfer between Ni and Mo2C, resulting in an improved OH− affinity on the Ni surface. In addition, the N-doped CNFs enabled electron transport between Ni and Mo2C nanoparticles, also contributing to the enhanced OER properties. An alkaline electrolyzer has been constructed to evaluate the overall water splitting performance, representing an operating voltage of 1.64 V at a current density of 10 mA cm−2 (Fig. 13j). The device showed a comparable electrocatalytic activity and much better long-term stability than the benchmark composed of Pt/C∥RuO2 electrodes (Fig. 13k). The morphology of the Ni/Mo2C-NCNFs did not change much after the OER, which would contribute to the outstanding long-term stability of the alkaline electrolyzer (Fig. 13l). | ||
| Fig. 13 (a) Schematic illustration of the synthetic process of Ni/Mo2C-NCNFs. (b) SEM and (c) TEM images of the synthesized Ni/Mo2C-NCNFs. (d) HER polarization curves, (e) Tafel plots, and (f) capacitive currents at various scan rates for Ni/Mo2C-NCNFs and control samples. (g) OER polarization curves and (h) Tafel slopes for Ni/Mo2C-NCNFs and control samples. (i) The stability test for Ni/Mo2C-NCNFs on Ni foam. (j) LSC curves of an overall water splitting device by using Ni/Mo2C-NCNFs∥Ni/Mo2C-NCNFs and Pt/C∥RuO2 electrodes. (k) Chronoamperometry curves of the Ni/Mo2C-NCNFs∥Ni/Mo2C-NCNFs and Pt/C∥RuO2 electrodes; the inset shows the bubbles generated from the electrodes. (l) TEM image of the Ni/Mo2C-NCNFs after the OER process. Reprinted with permission from ref. 259. Copyright 2019, John Wiley and Sons. | ||
Perovskite oxides have also been developed as bifunctional electrocatalysts for both the HER and OER.260 For instance, the electrospun SrNb0.1Co0.7Fe0.2O3−δ perovskite nanorods showed outstanding HER and OER performance in an alkaline medium. The prepared SrNb0.1Co0.7Fe0.2O3−δ catalyst delivered an overpotential of 262 mV for the HER and 390 mV for the OER to reach a current density of 10 mA cm−2. By using the same catalyst as the cathode and anode, an alkaline electrolyzer has been constructed, representing a voltage of around 1.68 V to deliver a water-splitting current density of 10 mA cm−2. Under a higher applied voltage above 1.78 V, the constructed electrolyzer exhibited a superior electrocatalytic activity to the device composed of Pt/C∥RuO2 electrodes. A mechanism for the origin of the superior HER and OER properties has been demonstrated. It was considered that the B-site metal cations and oxygen anions in the SrNb0.1Co0.7Fe0.2O3−δ sample acted as active sites contributing to the OER performance, and the oxygen vacancies might also be responsible for the OER activities. In addition, the B-site metal cations and oxygen anions were the main active sites for the HER. From the Tafel slope value, the Volmer reaction was found to be the RDS during the HER on the SrNb0.1Co0.7Fe0.2O3−δ catalyst.
In addition to the bifunctional catalyst with both HER and OER activities, another bifunctional catalyst for the ORR and OER has also attracted significant attention for its promising applications in developing metal–air batteries.273–295 Jiao and co-workers reported the fabrication of mesoporous thin-walled CuCo2O4@C nanotubes via a coaxial electrospinning technique as a bifunctional oxygen electrocatalyst.273 The resulting CuCo2O4@C nanotubes showed an excellent electrocatalytic activity and stability, delivering a positive onset potential of 0.951 V, which was comparable with that of the commercial Pt/C catalyst. The n value was calculated to be 3.9, indicating an efficient four electron reaction. Furthermore, the catalyst also showed favorable OER properties, displaying a low overpotential (327 mV at 10 mA cm−2) with a Tafel slope of 74.0 mV dec−1. Owing to the superior ORR and OER properties, a primary Zn–air battery has been assembled by using CuCo2O4@C nanotubes as the cathode (Fig. 14a). From the discharge–charge polarization curves, the prepared CuCo2O4@C nanotubes showed a lower open circuit voltage (OCV) of 1.37 V than Pt/C-IrO2 (1.45 V), which could be ascribed to the remarkable oxygen/hydroxyl diffusion from the unique channel structure (Fig. 14b). Importantly, a better cycling stability of the CuCo2O4@C nanotubes up to 160 cycles was observed compared to that of the Pt/C-IrO2 electrode (Fig. 14c). In a word, the outstanding ORR and OER performance for Zn–air batteries could be attributed to the following reasons: (1) the hollow structure with mesopores provides efficient channels for oxygen/hydroxyl diffusion and abundant active sites, which promotes the catalytic efficiency. (2) The CuCo2O4 nanoparticles with a small size offer a large utilization rate. (3) The 3D conductive network contributes to an excellent electronic contact. (4) N-Doping also promotes the oxygen redox catalysis (Fig. 14d).
 | ||
| Fig. 14 (a) Schematic illustration of the fabrication of rechargeable Zn–air batteries by using CuCo2O4@C nanotubes as the cathode and Zn foil as the anode. (b) Charge and discharge polarization curves of different catalysts. (c) Galvanostatic discharge–charge cycling curves at 2 mA cm−2 using CuCo2O4@C nanotubes and Pt/C + IrO2 electrodes. The inset shows the LED illuminated by using CuCo2O4@C nanotubes as air electrodes. (d) Schematic drawing of the advantages of the CuCo2O4@C nanotube electrodes. In the figures, CCO stands for CuCo2O4. Reprinted with permission from ref. 273. Copyright 2017, American Chemical Society. | ||
In recent years, Ni-, Co- and Fe-based phosphides have also been used as multifunctional catalysts.296,297 For example, NiCoP nanoparticles embedded in CNFs (NiCoP/CNFs) that were prepared via the electrospinning technique showed excellent ORR, OER and HER activities in KOH electrolytes.296 For ORR applications, the catalyst exhibited a half-wave potential and limited diffusion current density of 0.82 V versus a reversible hydrogen electrode and 7.16 mA cm−2. The n value was calculated to be around 4.0, confirming an efficient four electron reaction. For OER applications, the catalyst delivered a low onset potential of 1.43 V and an overpotential of 268 mV to reach 10 mA cm−2 with a minimum Tafel slope of 83 mV dec−1. For HER applications, the catalyst delivered a low overpotential of 130 mV to reach 10 mA cm−2 with a Tafel slope of 83 mV dec−1. In addition to be used towards the ORR, OER and HER, the prepared NiCoP/CNFs have also been constructed to be a supercapacitor, displaying a superior specific capcitance (333 F g−1 at 2 A g−1) and rate capability (87%), large energy density at a power density of 4000 W kg−1 and remarkable cycling stability (25000 cycles). Due to the multifunctional electrocatalytic properties, this kind of catalyst showed a bright prospect for the energy storage and conversion applications.
4. Summary and outlook
1D nanomaterials with a unique architecture represent a new class of highly efficient and low-cost electrocatalysts in the field of electrochemical energy storage and conversion. In this review, we have summarized the recent developments in the synthesis and electrocatalytic applications of electrospun nanomaterials. Through the structural design, composition tailoring, and electronic modulation, a large number of electrospun nanomaterials including metals, metal oxides, metal sulfides, metal nitrides, metal phosphides, metal carbides, and their hybrids as efficient electrocatalysts in the HER, OER, ORR, CO2RR, NRR and small molecule oxidation reaction with outstanding activity and stability have been discussed. Despite having achieved much progress in regulating the dynamics and activities of the electrospun nanomaterials, several challenges and perspectives are required to be taken into consideration.Firstly, the electrospinning technique has been widely used to prepare a large number of 1D nanomaterials for electrocatalytic applications. Regardless of the method, more unique nanostructures with regulated compositions such as nanotubes, multichannel tubes, core–sheath structures, fiber-in-tubes, tube-in-tubes and Janus nanobelts should be explored for the HER, OER, ORR, CO2RR, NRR and small molecule oxidation reaction. These nanostructures not only provide more active sites, but also contribute to the improved electron and mass transport, thus enhancing the electrocatalytic activity. Despite some success, some challenges with respect to the synthetic procedure still remain. For example, for the preparation of nanotubes, multichannel tubes, core–sheath structures, fiber-in-tubes, and tube-in-tubes, coaxial electrospinning is usually employed. However, the precise construction of such structures with a unique morphology, and controllable inner and outer diameters in a large scale is still challenging. Furthermore, to achieve such nanostructures of the ceramic materials, a post-calcination process is usually required. During such a treatment process, the preservation of the abundant active sites is required to enable high-performance electrocatalytic activity.
Secondly, most of the previously reported nanocatalysts for electrocatalysis are in the form of powder. For practical applications to fabricate electrodes, a conductive support or insulating polymer binders are usually necessary, and always result in an enlarged interfacial resistance and the hindering of the active sites. The electrospinning technique shows a great advantage to produce self-supported membrane-like materials consisting of 1D nanomaterials. After post-treatment, flexible and self-supported functional carbon and ceramic as well as their hybrid membranes are expected to be used as efficient electrocatalysts. In addition, the electrospun 1D nanomaterials can be assembled into 3D structures.298 The spatial effect of the membrane-like and 3D structures composed of electrospun 1D nanomaterials enables better dispersion of the active catalysts to provide more active sites, higher conductivity due to the lower interfacial resistance, and excellent electrolyte transport compared with individual powdery electrospun nanomaterials. Therefore, a higher electrocatalytic performance of such structured materials is anticipated.
Thirdly, from the economical viewpoint, the solution electrospinning technique to produce most of the 1D nanomaterials requires a large amount of an organic solvent, which not only increases the production cost but also easily causes environmental pollution. Compared with solution electrospinning, the melt electrospinning technique affords a facile and green route to prepare electrospun nanomaterials without the usage of any solvent. However, the fiber refinement and productivity are the main problems because of the high system viscosity. Through optimization of the polymer solution with low-molecular-weight or basic materials with low-viscosity, electrospun polymeric and derived functional inorganic nanomaterials with diameters around 100 nm from the melt electrospinning approach are expected.
Further studies are desired to develop a large number of highly efficient and low-cost electrospun nanomaterial electrocatalysts and understand their kinetics and mechanisms during the catalytic reactions. It is anticipated that these novel electrocatalysts with unprecedented catalytic properties realize a great advance in the field of electrochemical energy storage and conversion.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (51773075 and 21875084) and the Project of the Department of Science and Technology of Jilin Province (20190101013JH). The project was also supported by the Open Research Fund of the State Key Laboratory of Polymer Physics and Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences.References
- N. Kittner, F. Lill and D. M. Kammen, Nat. Energy, 2017, 2, 17125 CrossRef.
- D. Gielen, F. Boshell and D. Saygin, Nat. Mater., 2016, 15, 117 CrossRef CAS PubMed.
- V. Thavasi, G. Singh and S. Ramakrishna, Energy Environ. Sci., 2008, 1, 205 RSC.
- Z. Dong, S. J. Kennedy and Y. Wu, J. Power Sources, 2011, 196, 4886 CrossRef CAS.
- X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148 RSC.
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2015, 44, 2060 RSC.
- D. Chen, C. Chen, Z. M. Baiyee, Z. Shao and F. Ciucci, Chem. Rev., 2015, 115, 9869 CrossRef CAS PubMed.
- M. K. Birhanu, M. C. Tsai, A. W. Kahsay, C. T. Chen, T. S. Zeleke, K. B. Ibrahim, C. J. Huang, W. N. Su and B. J. Hwang, Adv. Mater. Interfaces, 2018, 5, 1800919 CrossRef.
- S. Wang, F. Ichihara, H. Pang, H. Chen and J. Ye, Adv. Funct. Mater., 2018, 28, 1803309 CrossRef.
- C. Hu and L. Dai, Adv. Mater., 2019, 31, 1804672 CrossRef PubMed.
- M. Kuang, P. Han, L. Huang, N. Cao, L. Qian and G. Zheng, Adv. Funct. Mater., 2018, 28, 1804886 CrossRef.
- Q. Shao, P. Wang and X. Huang, Adv. Funct. Mater., 2019, 29, 1806419 CrossRef.
- Y. Peng, B. Lu and S. Chen, Adv. Mater., 2018, 30, 1801995 CrossRef PubMed.
- K. Liu, H. Zhong, S. Li, Y. Duan, M. Shi, X. Zhang, J. Yan and Q. Jiang, Prog. Mater. Sci., 2018, 92, 64 CrossRef CAS.
- J. Liu, Q. Ma, Z. Huang, G. Liu and H. Zhang, Adv. Mater., 2018, 30, 1800696 Search PubMed.
- S. Dou, X. Wang and S. Wang, Small Methods, 2019, 3, 1800211 CrossRef.
- R. Prasannachandran, T. V. Vineesh, A. Anil, B. M. Krishna and M. M. Shaijumon, ACS Nano, 2018, 12, 11511 CrossRef CAS PubMed.
- J. Du, Y. Pan, T. Zhang, X. Han, F. Cheng and J. Chen, J. Mater. Chem., 2012, 22, 15812 RSC.
- S. Chen, Z. Kang, X. Hu, X. Zhang, H. Wang, J. Xie, X. Zheng, W. Yan, B. Pan and Y. Xie, Adv. Mater., 2017, 29, 1701687 CrossRef PubMed.
- G. Fu, X. Yan, Y. Chen, L. Xu, D. Sun, J. M. Lee and Y. Tang, Adv. Mater., 2018, 30, 1704609 CrossRef PubMed.
- Y. Xia, P. Yang, Y. Sun, Y. Wu, B. Mayers, B. Gates, Y. Yin, F. Kim and H. Yan, Adv. Mater., 2003, 15, 353 CrossRef CAS.
- J. Yuan, Y. Xu and A. H. Müller, Chem. Soc. Rev., 2011, 40, 640 RSC.
- X. Lu, W. Zhang, C. Wang, T. C. Wen and Y. Wei, Prog. Polym. Sci., 2011, 36, 671 CrossRef CAS.
- Z. Huang, Y. Zhang, M. Kotaki and S. Ramakrishna, Compos. Sci. Technol., 2003, 63, 2223 CrossRef CAS.
- D. Li and Y. Xia, Adv. Mater., 2004, 16, 1151 CrossRef CAS.
- X. Lu, C. Wang and Y. Wei, Small, 2009, 5, 2349 CrossRef CAS PubMed.
- A. Greiner and J. H. Wendorff, Angew. Chem., Int. Ed., 2007, 46, 5670 CrossRef CAS PubMed.
- H. Wang, S. Yuan, D. Ma, X. Zhang and J. Yan, Energy Environ. Sci., 2015, 8, 1660 RSC.
- X. Lu, C. Wang, F. Favier and N. Pinna, Adv. Energy Mater., 2017, 7, 1601301 CrossRef.
- J. Xue, T. Wu, Y. Dai and Y. Xia, Chem. Rev., 2019, 119, 5298 CrossRef CAS PubMed.
- W. Song, B. Zhao, C. Wang, Y. Ozaki and X. Lu, J. Mater. Chem. B, 2019, 7, 850 RSC.
- W. Song, B. Zhao, C. Wang and X. Lu, Compos. Commun., 2019, 12, 1 CrossRef.
- X. Wang, B. Ding, G. Sun, M. Wang and J. Yu, Prog. Polym. Sci., 2013, 58, 1173 CAS.
- J. Zeleny, Phys. Rev., 1917, 10, 1 CrossRef.
- D. H. Reneker and I. Chun, Nanotechnology, 1996, 7, 216 CrossRef CAS.
- R. Kessick, J. Fenn and G. Tepper, Polymer, 2004, 45, 2981 CrossRef CAS.
- L. Song, Y. Zhu, Z. Yang, C. Wang and X. Lu, J. Mater. Chem. B, 2018, 6, 5931 RSC.
- J. Shui and J. Li, Nano Lett., 2009, 9, 1307 CrossRef CAS PubMed.
- H. Ji, R. Zhao, N. Zhang, C. Jin, X. Lu and C. Wang, NPG Asia Mater., 2018, 10, 749 CrossRef CAS.
- G. Taylor, Proc. R. Soc. London, Ser. A, 1964, 280, 383 CrossRef.
- P. Heikkila and A. Harlin, Eur. Polym. J., 2008, 44, 3067 CrossRef CAS.
- S. A. Theron, A. L. Yarin, E. Zussman and E. Kroll, Polymer, 2005, 46, 2889 CrossRef CAS.
- M. Yu, R. Dong, Y. Yan, G. Yu, M. You, X. Ning and Y. Long, Macromol. Mater. Eng., 2017, 302, 1700002 CrossRef.
- O. Dosunmu, G. G. Chase, W. Kataphinan and D. Reneker, Nanotechnology, 2006, 17, 1123 CrossRef CAS PubMed.
- D. Tian, X. Lu, Y. Zhu, M. Li and C. Wang, J. Power Sources, 2019, 413, 50 CrossRef CAS.
- M. Chi, S. Chen, M. Zhong, C. Wang and X. Lu, Chem. Commun., 2018, 54, 5827 RSC.
- N. Song, F. Ma, Y. Zhu, S. Chen, C. Wang and X. Lu, ACS Sustainable Chem. Eng., 2018, 6, 16766 CrossRef CAS.
- X. Zhang, W. Fan, H. Li, S. Zhao, J. Wang, B. Wang and C. Li, J. Mater. Chem. A, 2018, 6, 21458 RSC.
- X. Zhang, W. Fan, S. Zhao, R. Cao and C. Li, Catal. Sci. Technol., 2019, 9, 1998 RSC.
- S. Zhao, X. Wu, L. Wang and Y. Huang, J. Appl. Polym. Sci., 2004, 91, 242 CrossRef CAS.
- C. M. Wu, H. G. Chiou, S. L. Lin and J. M. Lin, J. Appl. Polym. Sci., 2012, 126, E89 CrossRef CAS.
- T. Mazoochi, M. Hamadanian, M. Ahmadi and V. Jabbari, Int. J. Ind. Chem., 2012, 3, 2 CrossRef.
- S. Zarham, S. Bazgir, A. Tavakoli, A. S. Rashidi and R. Damerchely, J. Eng. Fibers Fabr., 2012, 7, 42 Search PubMed.
- Z. Sun, E. Zussman, A. L. Yarin, J. H. Wendorff and A. Greiner, Adv. Mater., 2003, 15, 1929 CrossRef CAS.
- Y. Z. Zhang, Z. M. Huang, X. J. Xu, C. T. Lim and S. Ramakrishna, Chem. Mater., 2004, 16, 3406 CrossRef CAS.
- D. Li, A. Babel, S. A. Jenekhe and Y. Xia, Adv. Mater., 2004, 16, 2062 CrossRef CAS.
- D. Li and Y. Xia, Nano Lett., 2004, 4, 933 CrossRef CAS.
- Y. Zhao, X. Cao and L. Jiang, J. Am. Chem. Soc., 2007, 129, 764 CrossRef CAS PubMed.
- P. Gupta and G. L. Wilkes, Polymer, 2003, 44, 6353 CrossRef CAS.
- T. Lin, H. Wang and X. Wang, Adv. Mater., 2005, 17, 2699 CrossRef CAS.
- Z. Y. Liu, D. D. Sun, P. Guo and J. O. Leckie, Nano Lett., 2007, 7, 1081 CrossRef CAS PubMed.
- Q. Ma, J. Wang, X. Dong, W. Yu and G. Liu, Adv. Funct. Mater., 2015, 25, 2436 CrossRef CAS.
- M. Bognitzki, W. Czado, T. Frese, A. Schaper, M. Hellwig, M. Steinhart, A. Greiner and J. H. Wendorff, Adv. Mater., 2001, 13, 70 CrossRef CAS.
- B. H. Kim, K. S. Yang and H. G. Woo, Mater. Lett., 2013, 93, 190 CrossRef CAS.
- C. Ma, Y. J. Li, J. L. Shi, Y. Song and L. Liu, Chem. Eng. J., 2014, 249, 216 CrossRef CAS.
- H. Y. Chen, J. C. Di, N. Wang, H. Dong, J. Wu, Y. Zhao, J. H. Yu and L. Jiang, Small, 2011, 7, 1779 CrossRef CAS PubMed.
- Q. Gao, W. Zhang, Z. Shi, L. Yang and Y. Tang, Adv. Mater., 2019, 31, 1802880 CrossRef PubMed.
- A. B. Laursen, S. Legnes, S. Dahl and I. Chorkendorff, Energy Environ. Sci., 2012, 5, 5577 RSC.
- C. G. Morales-Guio, L. A. Stern and X. L. Hu, Chem. Soc. Rev., 2014, 43, 6555 RSC.
- T. Yang, M. Du, H. Zhu, M. Zhang and M. Zou, Electrochim. Acta, 2015, 167, 48 CrossRef CAS.
- Z. Chen, X. Duan, W. Wei, S. Wang and B. Ni, J. Mater. Chem. A, 2019, 7, 14971 RSC.
- G. Zhao, K. Rui, S. Dou and W. Sun, Adv. Funct. Mater., 2018, 28, 1803291 CrossRef.
- M. Miao, J. Pan, T. He, Y. Yan, B. Xia and X. Wang, Chem. – Eur. J., 2017, 23, 10947 CrossRef CAS PubMed.
- H. Du, R. Kong, X. Guo, F. Qu and J. Li, Nanoscale, 2018, 46, 21617 RSC.
- Y. Guo, T. Park, J. Yi, J. Henzie, J. Kim, Z. Wang, B. Jiang, Y. Bando, Y. Sugahara, J. Tang and Y. Yamauchi, Adv. Mater., 2019, 31, 1807134 CrossRef PubMed.
- N. Han, P. Liu, J. Jiang, L. Ai, Z. Shao and S. Liu, J. Mater. Chem. A, 2018, 6, 19912 RSC.
- Q. Ding, M. Liu, Y. Miao, Y. Huang and T. Liu, Electrochim. Acta, 2015, 159, 1 CrossRef CAS.
- T. Li, G. Luo, K. Liu, X. Li, D. Sun, L. Xu, Y. Li and Y. Tang, Adv. Funct. Mater., 2018, 28, 1805828 CrossRef.
- J. H. Lee, M. J. Jang, Y. S. Park, S. M. Choi, Y. D. Kim and K. H. Lee, J. Korean Inst. Surf. Eng., 2017, 50, 322 Search PubMed.
- L. Zhang, S. Zhu, S. Dong, N. J. Woo, Z. Xu, J. Huang, J. K. Kim and M. Shao, J. Electrochem. Soc., 2018, 165, J3271 CrossRef CAS.
- J. Wang, H. Zhu, J. Chen, B. Zhang, M. Zhang, L. Wang and M. Du, Int. J. Hydrogen Energy, 2016, 41, 18044 CrossRef CAS.
- M. Li, Y. Zhu, N. Song, C. Wang and X. Lu, J. Colloid Interface Sci., 2018, 514, 199 CrossRef CAS PubMed.
- J. Wang, J. Chen, J. Chen, H. Zhu, M. Zhang and M. Du, Adv. Mater. Interfaces, 2017, 4, 1700005 CrossRef.
- J. Wang, H. Zhu, D. Yu, J. Chen, J. Chen, M. Zhang, L. Wang and M. Du, ACS Appl. Mater. Interfaces, 2017, 9, 19756 CrossRef CAS PubMed.
- B. Zhang, H. Zhu, M. Zou, X. Liu, H. Yang, M. Zhang, W. Wu, J. Yao and M. Du, J. Mater. Sci., 2017, 52, 8207 CrossRef CAS.
- G. Yanalak, A. Aljabour, E. Aslan, F. Ozel, I. H. Patir, M. Kus and M. Ersoz, Electrochim. Acta, 2018, 291, 311 CrossRef CAS.
- J. Chen, D. Yu, W. Liao, M. Zheng, L. Xiao, H. Zhu, M. Zhang, M. Du and J. Yao, ACS Appl. Mater. Interfaces, 2016, 8, 18132 CrossRef CAS PubMed.
- Y. Cho, A. Yu, C. Lee, M. H. Kim and Y. Lee, ACS Appl. Mater. Interfaces, 2018, 10, 541 CrossRef CAS PubMed.
- C. Wu, C. Li, B. Yang, S. Zhou, D. Shi, Y. Wang, G. Yang, J. He and Y. Shan, Mater. Res. Express, 2016, 3, 095018 CrossRef.
- M. Wu, L. Fan, R. Ma, J. Zhu, S. Gu, T. Wang, D. Gong, Z. Xu and B. Lu, Mater. Lett., 2016, 182, 15 CrossRef CAS.
- C. Wu, J. Li, D. Zhang, B. Yang, L. Li, T. Zhou, C. Zhang, G. Yang and Y. Shan, Int. J. Hydrogen Energy, 2016, 41, 13915 CrossRef CAS.
- S. F. Anis, B. S. Lalia, A. O. Mostafa and R. Hashaikeh, J. Mater. Sci., 2017, 52, 7269 CrossRef CAS.
- K. Ketpang, M. Kim, S. Kim and S. Shanmugam, Int. J. Hydrogen Energy, 2013, 38, 9732 CrossRef CAS.
- A. Chinnappan, D. Ji, W. A. D. M. Jayathilaka, C. Baskar, X. Qin and S. Ramakrishna, Int. J. Hydrogen Energy, 2018, 43, 15217 CrossRef CAS.
- Z. Zhang, Y. Wang, X. Leng, V. H. Crespi, F. Kang and R. Lv, ACS Appl. Energy Mater., 2018, 1, 1268 CrossRef CAS.
- M. Wan, J. Li, T. Li, H. Zhu, W. Wu and M. Du, Nanotechnology, 2018, 29, 385602 CrossRef PubMed.
- J. H. Lee, Y. S. Park, M. J. Jang and S. M. Park, Korean J. Met. Mater., 2018, 56, 885 CrossRef CAS.
- X. Zhu, L. Mo, Y. Wu, F. Lai, X. Han, X. Ling, T. Liu and Y. Miao, Compos. Commun., 2018, 9, 86 CrossRef.
- S. Kang, J. Jang, S. Ahn and C. S. Lee, Dalton Trans., 2019, 48, 2170 RSC.
- H. Zhu, M. Du, M. Zhang, M. Zou, T. Yang, Y. Fu and J. Yao, J. Mater. Chem. A, 2014, 2, 7680 RSC.
- M. Wan, D. Yu, H. Zhu, M. Zhang and M. Du, Chin. J. Inorg. Chem., 2017, 33, 595 CAS.
- J. Li, W. Wu, M. Wan, L. Gu, J. Wang, T. Li, H. Zhu, M. Zhang and M. Du, Int. J. Electrochem. Sci., 2017, 12, 4563 CrossRef CAS.
- Y. Wen, H. Zhu, L. Zhang, S. Zhang, M. Zhang and M. Du, Mater. Res. Bull., 2019, 112, 46 CrossRef CAS.
- Y. Zhu, L. Song, N. Song, M. Li, C. Wang and X. Lu, ACS Sustainable Chem. Eng., 2019, 7, 2899 CrossRef CAS.
- Y. Huang, Y. Miao, L. Zhang, W. Tjiu, J. Pan and T. Liu, Nanoscale, 2014, 6, 10673 RSC.
- X. Zhang, L. Li, Y. Guo, D. Liu and T. You, J. Colloid Interface Sci., 2016, 472, 69 CrossRef CAS PubMed.
- Y. Rheem, Y. Han, K. H. Lee, S. M. Choi and N. V. Myung, Nanotechnology, 2017, 28, 105605 CrossRef.
- H. Gu, Y. Huang, L. Zuo, W. Fan and T. Liu, Electrochim. Acta, 2016, 219, 604 CrossRef CAS.
- S. Wan, Y. Liu, G. Li, X. Li, D. Wang and X. Zou, Catal. Sci. Technol., 2016, 6, 4545 RSC.
- J. Yan, Y. Zhang, Y. Huang, Y. Miao and T. Liu, Adv. Mater. Interfaces, 2017, 4, 1600825 CrossRef.
- H. Gu, Y. Huang, L. Zuo, W. Fan and T. Liu, Inorg. Chem. Front., 2016, 3, 1280 RSC.
- M. Zou, Y. Jiang, M. Wan, M. Zhang, H. Zhu, T. Yang and M. Du, Electrochim. Acta, 2015, 176, 255 CrossRef CAS.
- H. Gu, W. Fan and T. Liu, Nanoscale Horiz., 2017, 2, 277 RSC.
- X. Chia, Z. Sofer, J. Luxa and M. Pumera, Chem. – Eur. J., 2017, 23, 11719 CrossRef CAS PubMed.
- Y. Huang, Y. Miao, J. Fu, S. Mo, C. Wei and T. Liu, J. Mater. Chem. A, 2015, 3, 16263 RSC.
- F. Ozel, A. Yar, E. Aslan, E. Arkan, A. Aljabour, M. Can, I. H. Patir, M. Kus and M. Ersoz, ChemNanoMat, 2015, 1, 477 CrossRef CAS.
- P. Liu, J. Li, Y. Lu and B. Xiang, Int. J. Hydrogen Energy, 2018, 43, 72 CrossRef CAS.
- S. Yu, J. Kim, K. R. Yoon, J. W. Jung, J. Oh and I. D. Kim, ACS Appl. Mater. Interfaces, 2015, 7, 28116 CrossRef CAS PubMed.
- M. Zou, J. Chen, L. Xiao, H. Zhu, T. Yang, M. Zhang and M. Du, J. Mater. Chem. A, 2015, 3, 18090 RSC.
- J. Tian, Q. Liu, A. M. Asiri and X. Sun, J. Am. Chem. Soc., 2014, 136, 7587 CrossRef CAS PubMed.
- T. Liu, D. Liu, F. Qu, D. Wang, L. Zhang, R. Ge, S. Hao, Y. Ma, G. Du, A. M. Asiri, L. Chen and X. Sun, Adv. Energy Mater., 2017, 7, 1700020 CrossRef.
- Y. Miao, F. Li, Y. Zhou, F. Lai, H. Lu and T. Liu, Nanoscale, 2017, 9, 16313 RSC.
- M. Wang, Y. Cui, H. Liu, M. Xu and S. Bao, Angew. Chem., Int. Ed., 2018, 57, 1963 CrossRef CAS PubMed.
- Y. Li, H. Li, K. Cao, T. Jin, X. Wang, H. Sun, J. Ning, Y. Wang and L. Jiao, Energy Storage Mater., 2018, 12, 44 CrossRef.
- M. Streckova, R. Orinakova, E. Mudra, Z. Dankova, M. Sabalova, V. Girman, A. Kovalcikova, J. Hovancova, M. Heckova, F. Kalavsky and J. Dusza, Energy Technol., 2018, 6, 1310 CrossRef CAS.
- Q. Mo, W. Zhang, L. He, X. Yu and Q. Gao, Appl. Catal., B, 2019, 244, 620 CrossRef CAS.
- M. Streckova, E. Mudra, R. Orniakova, L. Markusova-Buckova, M. Sebek, A. Kovalcikova, T. Sopcak, V. Girman, Z. Dankova, M. Micusik and J. Dusza, Chem. Eng. J., 2016, 303, 167 CrossRef CAS.
- J. Wang, W. Cui, Q. Liu, Z. Xing, A. Asiri and X. Sun, Adv. Mater., 2016, 28, 215 CrossRef CAS PubMed.
- H. Lin, W. Zhang, Z. Shi, M. Che, X. Yu, Y. Tang and Q. Gao, ChemSusChem, 2017, 10, 2597 CrossRef CAS PubMed.
- X. Liu, L. Zhang, X. Lan and X. Hu, Electrochim. Acta, 2018, 274, 23 CrossRef CAS.
- F. Lyu, Q. Wang, S. M. Choi and Y. Yin, Small, 2019, 15, 1804201 CrossRef PubMed.
- T. Ma, J. Bai, Q. Wang and C. Li, Dalton Trans., 2018, 47, 10240 RSC.
- X. An, D. Shin, J. Jeong and J. Lee, ChemElectroChem, 2016, 3, 1720–1724 CrossRef CAS.
- C. Li, Z. Zhang, M. Wu and R. Liu, Mater. Lett., 2019, 238, 138 CrossRef CAS.
- D. Shin, M. Choun, H. C. Ham, J. K. Lee and J. Lee, Phys. Chem. Chem. Phys., 2017, 19, 21987 RSC.
- X. An, D. Shin, J. D. Ocon, J. K. Lee, Y. Son and J. Lee, Chin. J. Catal., 2014, 35, 891 CrossRef CAS.
- B. Jeong, D. Shin, J. K. Lee, D. H. Kim, Y. D. Kim and J. Lee, Phys. Chem. Chem. Phys., 2014, 16, 13807 RSC.
- Z. Zhang, J. Zhang, T. Wang, Z. Li, G. Yang, H. Bian, J. Li and D. Gao, RSC Adv., 2018, 8, 5338 RSC.
- L. Wu, L. Shi, S. Zhou, J. Zhao, X. Miao and J. Guo, Energy Technol., 2018, 6, 2350 CrossRef CAS.
- T. Li, Y. Lv, J. Su, Y. Wang, Q. Yang, Y. Zhang, J. Zhou, L. Xu, D. Sun and Y. Tang, Adv. Sci., 2017, 4, 1700226 CrossRef PubMed.
- S. Hyun, V. Ahilan, H. Kim and S. Shanmuam, Electrochem. Commun., 2016, 63, 44 CrossRef CAS.
- H. Zhu, D. Yu, S. Zhang, J. Chen, W. Wu, M. Wan, L. Wang, M. Zhang and M. Du, Small, 2017, 13, 1700468 CrossRef PubMed.
- M. Li, Y. Xiong, X. Liu, X. Bo, Y. Zhang, C. Han and L. Guo, Nanoscale, 2015, 7, 8920 RSC.
- H. Zhu, L. Gu, D. Yu, Y. Sun, M. Wan, M. Zhang, L. Wang, L. Wang, W. Wu, J. Yao, M. Du and S. Guo, Energy Environ. Sci., 2017, 10, 321 RSC.
- M. S. Won, M. J. Jang, K. H. Lee, Y. D. Kim and S. M. Choi, J. Korean Inst. Surf. Eng., 2016, 49, 539 CrossRef.
- A. Yu, C. Lee, M. H. Kim and Y. Lee, ACS Appl. Mater. Interfaces, 2017, 9, 35057 CrossRef CAS PubMed.
- S. Moon, Y. B. Cho, A. Yu, M. H. Kim, C. Lee and Y. Lee, ACS Appl. Mater. Interfaces, 2019, 11, 1979 CrossRef CAS PubMed.
- G. Liu, J. Xu, Y. Wang and X. Wang, J. Mater. Chem. A, 2015, 3, 20791 RSC.
- C. Wu, C. Li, B. Yang, S. Zhou, D. Shi, Y. Wang, G. Yang, J. He and Y. Shan, Mater. Res. Express, 2016, 3, 095018 CrossRef.
- H. Chen, X. Huang, L. Zhou, G. Li, M. Fan and X. Zou, ChemCatChem, 2016, 8, 992 CrossRef CAS.
- M. Wan, H. Zhu, S. Zhang, H. Jin, Y. Wen, L. Wang, M. Zhang and M. Du, Electrochim. Acta, 2018, 279, 301 CrossRef CAS.
- Z. Wang, S. Tan, Y. Xiong and J. Wei, Prog. Nat. Sci., 2018, 28, 399 CrossRef CAS.
- Z. Wang, M. Li, C. Liang, L. Fan, J. Han and Y. Xiong, RSC Adv., 2016, 6, 69251 RSC.
- D. Zhen, B. Zhao, H. C. Shin, Y. Bu, Y. Ding, G. He and M. Liu, Adv. Mater. Interfaces, 2017, 4, 1700146 CrossRef.
- R. Mo, S. Wang, H. Li, J. Li, S. Yang and J. Zhong, Electrochim. Acta, 2018, 290, 649 CrossRef CAS.
- Y. Li, S. Chen, D. Xi, Y. Bo, R. Long, C. Wang, L. Song and Y. Xiong, Small, 2018, 14, 1702109 CrossRef PubMed.
- Y. Zhao, J. Zhang, X. Guo, H. Fan, W. Wu, H. Liu and G. Wang, J. Mater. Chem. A, 2017, 5, 19672 RSC.
- J. S. Lee, G. Nam, J. Sun, S. Higashi, H. W. Lee, S. Lee, W. Chen, Y. Cui and J. Cho, Adv. Energy Mater., 2016, 6, 1601052 CrossRef.
- L. Li, J. He, Y. Wang, X. Lv, X. Gu, P. Dai, D. Liu and X. Zhao, J. Mater. Chem. A, 2019, 7, 1964 RSC.
- J. I. Shui, C. Chen and J. C. M. Li, Adv. Funct. Mater., 2011, 21, 3357 CrossRef CAS.
- X. Deng, S. Yin, X. Wu, M. Sun, Z. Xie and Q. Huang, Electrochim. Acta, 2018, 283, 987 CrossRef CAS.
- K. K. Karuppanan, A. V. Raghu, M. K. Panthalingal, S. Ramanathan, T. Kumaresan and B. Pullithadathil, J. Mater. Chem. A, 2018, 6, 12768 RSC.
- M. Kim, C. Kwon, K. Eom, J. Kim and E. Cho, Sci. Rep., 2017, 7, 44411 CrossRef CAS PubMed.
- K. Senevirathne, R. Hui, S. Campbell, S. Ye and J. Zhang, Electrochim. Acta, 2012, 59, 538 CrossRef CAS.
- G. Cognard, G. Ozouf, C. Beauger, I. Jiménez-Morales, S. Cavaliere, D. Jones, J. Roziére, M. Chatenet and F. Maillard, Electrocatalysis, 2017, 8, 51 CrossRef CAS.
- C. Lai, P. Kolla, Y. Zhao, H. Fong and A. L. Smirnova, Electrochim. Acta, 2014, 130, 431 CrossRef CAS.
- T. H. Vu, D. Shin, J. Jeong and J. Lee, J. Phys. Chem. C, 2016, 120, 22342 CrossRef CAS.
- A. Jindal, D. K. Gautam and S. Basu, J. Electroanal. Chem., 2016, 775, 198 CrossRef CAS.
- Y. Qiu, J. Yu, T. Shi, X. Zhou, X. Bai and J. Y. Huang, J. Power Sources, 2011, 196, 9862 CrossRef CAS.
- Y. Chen, Q. Liu and J. Wang, J. Mater. Chem. A, 2016, 4, 5553 RSC.
- D. Shin, B. Jeong, B. S. Mun, H. Jeon, H. J. Shin, J. Baik and J. Lee, J. Phys. Chem. C, 2013, 117, 11619 CrossRef CAS.
- J. Guo, J. Liu, H. Dai, R. Zhou, T. Wang, C. Zhang, S. Ding and H. Wang, J. Colloid Interface Sci., 2017, 507, 154 CrossRef CAS PubMed.
- J. Yin, Y. Qiu, J. Yu, X. Zhou and W. Wu, RSC Adv., 2013, 3, 15655 RSC.
- D. Liu, X. Zhang, Z. Sun and T. You, Nanoscale, 2013, 5, 9528 RSC.
- Y. Wang, J. Jin, S. Yang, G. Li and J. Jiang, Int. J. Hydrogen Energy, 2016, 41, 11174 CrossRef CAS.
- M. Mooste, E. K. Pőldsepp, L. Matisen, M. Merisalu, M. Kook, V. Kiand, V. Vassiljeva, A. Krumme, V. Sammelselg and K. Tammeveski, Catal. Lett., 2018, 148, 1815 CrossRef CAS.
- G. S. Park, J. S. Lee, S. T. Kim, S. Park and J. Cho, J. Power Sources, 2013, 243, 267 CrossRef CAS.
- J. Yin, Y. Qiu and J. Yu, J. Electroanal. Chem., 2013, 702, 56 CrossRef CAS.
- Y. Qiu, J. Yin, H. Hou, J. Yu and X. Zuo, Electrochim. Acta, 2013, 96, 225 CrossRef CAS.
- Q. Guo, D. Zhao, S. Liu, S. Chen, M. Hanif and H. Hou, Electrochim. Acta, 2013, 138, 318 CrossRef.
- S. Wang, C. Dai, J. Li, L. Zhao, Z. Ren, Y. Ren, Y. Qiu and J. Yu, Int. J. Hydrogen Energy, 2015, 40, 4673 CrossRef CAS.
- D. S. Yang, S. Chaudhari, K. P. Rajesh and J. S. Yu, ChemCatChem, 2014, 6, 1236 CAS.
- M. A. Abdelkareem, D. Takino, T. Ishikawa, T. Tsujiguchi and N. Nakagawa, Key Eng. Mater., 2012, 497, 73 CAS.
- S. Wang, Z. Cui, J. Qin and M. Cao, Nano Res., 2016, 9, 2270 CrossRef CAS.
- W. Yang, L. Chen, X. Liu, X. Yue, C. Liu and J. Jia, J. Mater. Chem. A, 2016, 4, 5834 RSC.
- Q. Shi, Y. Lei, Y. Wang, H. Wang, L. Jiang, H. Yuan, D. Fang, B. Wang, N. Wu and Y. Gou, Curr. Appl. Phys., 2015, 15, 1606 CrossRef.
- Y. Zhao, Q. Lai, Y. Wang, J. Zhu and Y. Liang, ACS Appl. Mater. Interfaces, 2017, 9, 16178 CrossRef CAS PubMed.
- K. R. Yoon, J. Choi, S. H. Cho, J. W. Jung, C. Kim, J. Y. Cheong and I. D. Kim, J. Power Sources, 2018, 380, 174 CrossRef CAS.
- I. T. Kim, M. J. Song, S. Shin and M. W. Shin, Appl. Surf. Sci., 2018, 435, 1159–1167 CrossRef CAS.
- S. Wang, Z. Cui and M. Cao, Chem. – Eur. J., 2015, 21, 2165 CrossRef CAS PubMed.
- W. Yang, X. Liu, L. Chen, L. Liang and J. Jia, Chem. Commun., 2017, 53, 4034 RSC.
- K. Liu, S. Kattel, V. Mao and G. Wang, J. Phys. Chem. C, 2016, 120, 1586 CrossRef CAS.
- C. Shang, M. Li, Z. Wang, S. Wu and Z. Lu, ChemElectroChem, 2016, 3, 1437 CrossRef CAS.
- Z. Wang, P. Zuo, L. Fan, J. Han, Y. Xiong and G. Yin, J. Power Sources, 2016, 311, 68 CrossRef CAS.
- G. H. An, E. H. Lee and H. J. Ahn, J. Alloys Compd., 2016, 682, 746 CrossRef CAS.
- J. P. McClure, R. Jiang, D. Chu and P. S. Fedkiw, Carbon, 2014, 79, 457 CrossRef CAS.
- W. Yang, Y. Zhang, C. Liu and J. Jia, J. Power Sources, 2015, 274, 595 CrossRef CAS.
- Q. Liu, S. Cao, Y. Qiu and L. Zhao, Mater. Sci. Eng., B, 2017, 223, 159 CrossRef CAS.
- X. Yan, K. Liu, T. Wang, Y. You, J. Liu, P. Wang, X. Pan, G. Wang, J. Luo and J. Zhu, J. Mater. Chem. A, 2017, 5, 3336 RSC.
- Q. Liu, S. Cao and Y. Qiu, Int. J. Hydrogen Energy, 2017, 42, 29274 CrossRef CAS.
- Q. Liu, S. Cao, Y. Fu, Y. Guo and Y. Qiu, J. Electroanal. Chem., 2018, 813, 52 CrossRef CAS.
- B. Jeong, D. Shin, M. Choun, S. Maurya, J. Baik, B. S. Mun, S. H. Moon, D. Su and J. Lee, J. Phys. Chem. C, 2016, 120, 7705 CrossRef CAS.
- C. Liu, J. Wang, J. Li, R. Luo, X. Sun, J. Shen, W. Han and L. Wang, Carbon, 2017, 114, 706 CrossRef CAS.
- Y. Qiu, J. Yu, W. Wu, J. Yin and X. Bai, J. Solid State Electrochem., 2013, 17, 565 CrossRef CAS.
- X. Yan, Y. Yao and Y. Chen, Nanoscale Res. Lett., 2018, 13, 218 CrossRef PubMed.
- J. Yan, H. Lu, Y. Huang, J. Fu, S. Mo, C. Wei, Y. Miao and T. Liu, J. Mater. Chem. A, 2015, 3, 23299 RSC.
- L. Dong, W. Wang, J. Zang, Y. Zhang, Z. Wang, J. Su and Y. Wang, Int. J. Hydrogen Energy, 2018, 43, 14273 CrossRef CAS.
- Y. W. Ju, S. Yoo, C. Kim, S. Kim, I. Y. Jeon, J. Shin, J. B. Boek and G. Kim, Adv. Sci., 2016, 3, 1500205 CrossRef PubMed.
- P. Zamani, D. Higins, F. Hassan, G. Jiang, J. Wu, S. Abureden and Z. Chen, Electrochim. Acta, 2014, 139, 111 CrossRef CAS.
- J. Guo, Q. Niu, Y. Yuan, I. Maitlo, J. Nie and G. Ma, Appl. Surf. Sci., 2017, 416, 118 CrossRef CAS.
- Q. Shi, Y. Lei, Y. Wang and Z. Wang, J. Inorg. Mater., 2016, 31, 351 CrossRef CAS.
- C. Liu, J. Wang, J. Li, R. Luo, X. Sun, J. Shen, W. Han and L. Wang, Carbon, 2017, 114, 706 CrossRef CAS.
- Y. Zhao, Q. Lai, J. Zhu, J. Zhong, Z. Tang, Y. Luo and Y. Liang, Small, 2018, 14, 1704207 CrossRef PubMed.
- C. Liu, J. Wang, J. Li, J. Liu, C. Wang, X. Sun, J. Shen, W. Han and L. Wang, J. Mater. Chem. A, 2017, 5, 1211 RSC.
- Q. Niu, J. Guo, B. Chen, J. Nie, X. Guo and G. Ma, Carbon, 2017, 114, 250 CrossRef CAS.
- Q. Niu, B. Chen, J. Guo, J. Nie, X. Guo and G. Ma, Nano-Micro Lett., 2019, 11, 8 CrossRef CAS.
- Q. Bai, F. Shen, S. Li, J. Liu, L. Dong, Z. Wang and Y. Lan, Small Methods, 2018, 2, 1800049 CrossRef.
- D. Yin, C. Han, X. Bo, J. Liu and L. Guo, J. Colloid Interface Sci., 2019, 533, 578 CrossRef CAS.
- Z. Zeng, T. Zhang, Y. Liu, W. Zhang, Z. Yin, Z. Ji and J. Wei, ChemSusChem, 2018, 11, 580 CrossRef CAS.
- H. Tang, W. Chen, J. Wang, T. Dugger, L. Cruz and D. Kisailus, Small, 2018, 14, 1703459 CrossRef PubMed.
- L. Delmondo, J. A. Munoz-Tabares, A. Sacco, N. Garino, M. Castellino, G. P. Salvador, C. F. Pirri, M. Qunglio and A. Chiodoni, Phys. Chem. Chem. Phys., 2017, 19, 28781 RSC.
- D. Hassen, M. A. Shenashen, S. A. El-Safty, M. M. Selim, H. Isago, A. Elmarakbi, A. El-Safty and H. Yamaguchi, J. Power Sources, 2016, 330, 2923 CrossRef.
- S. Pei, Z. Zhou, X. Chen, X. Huang, T. Liu, B. Cao and F. Wang, Int. J. Electrochem. Sci., 2016, 11, 8994 CrossRef CAS.
- C. Alegre, C. Busacca, O. D. Blasi, V. Antonucci, A. S. Aricò, A. D. Blasi and V. Baglio, J. Power Sources, 2017, 364, 101 CrossRef CAS.
- J. Guo, B. Chen, Q. Hao, J. Nie and G. Ma, Appl. Surf. Sci., 2018, 456, 959 CrossRef CAS.
- G. Ren, X. Lu, Y. Li, Y. Zhu, L. Dai and L. Jiang, ACS Appl. Mater. Interfaces, 2016, 8, 4118 CrossRef CAS.
- R. Zhong, Y. Wu, Z. Liang, W. Guo, C. Zhi, C. Qu, S. Gao, B. Zhu, H. Zhang and R. Zou, Carbon, 2019, 142, 115 CrossRef CAS.
- G. H. An, Y. G. Lee and H. J. Ahn, J. Alloys Compd., 2018, 746, 177 CrossRef CAS.
- Q. Li, J. Zhao, M. Wu, C. Li, L. Han and R. Liu, ChemistrySelect, 2019, 4, 722–728 CrossRef CAS.
- Y. G. Lee, G. H. An and H. J. Ahn, Korean J. Mater. Res., 2018, 28, 640 CrossRef.
- H. Mistry, A. S. Varela, S. Kühl, P. Strasser and B. R. Cuenya, Nat. Rev. Mater., 2016, 1, 16009 CrossRef CAS.
- Y. Wang, P. Han, X. Lv, L. Zhang and G. Zheng, Joule, 2018, 2, 2551 CrossRef CAS.
- H. Yang, Q. Lin, H. Zhang, G. Li, L. Fan, X. Chai, Q. Zhang, J. Liu and C. He, Chem. Commun., 2018, 54, 4108 RSC.
- H. Yang, Q. Lin, H. Zhang, Y. Wu, L. Fan, X. Chai, Q. Zhang, J. Liu and C. He, Electrochem. Commun., 2018, 93, 138 CrossRef CAS.
- A. ALjabour, D. H. Apaydin, H. Coskun, F. Ozel, M. Ersoz, P. Stadler, N. S. Sariciftci and M. Kus, ACS Appl. Mater. Interfaces, 2016, 8, 31695 CrossRef CAS PubMed.
- A. Aljabour, H. Coskun, D. H. Apaydin, F. Ozel, A. W. Hassel, P. Stadler, N. S. Sariciftci and M. Kus, Appl. Catal., B, 2018, 229, 163 CrossRef CAS.
- L. Fan, Z. Xia, M. Xu, Y. Lu and Z. Li, Adv. Funct. Mater., 2018, 28, 1706289 CrossRef.
- H. Hu, L. Gui, W. Zhou, J. Sun, J. Xu, Q. Wang, B. He and L. Zhao, Electrochim. Acta, 2018, 285, 70 CrossRef CAS.
- Y. Zhao, J. Liang, C. Wang, J. Ma and G. C. Wallace, Adv. Energy Mater., 2018, 8, 1702524 CrossRef.
- L. Zhang, X. Ji, X. Ren, Y. Ma, X. Shi, Z. Tian, A. M. Asiri, L. Chen, B. Tang and X. Sun, Adv. Mater., 2018, 30, 1800191 CrossRef.
- J. Zhao and Z. Chen, J. Am. Chem. Soc., 2017, 139, 12480 CrossRef CAS PubMed.
- C. Lv, C. Yan, G. Chen, Y. Ding, J. Sun, Y. Zhou and G. Yu, Angew. Chem., Int. Ed., 2018, 130, 6073 CrossRef.
- J. Han, Z. Liu, Y. Ma, G. Gui, F. Xie, F. Wang, Y. Wu, S. Gao, Y. Xu and X. Sun, Nano Energy, 2018, 52, 264 CrossRef CAS.
- J. M. Kim, H. I. Joh, S. M. Jo, D. J. Ahn, H. Y. Ha, S. A. Hong and S. K. Kim, Electrochim. Acta, 2010, 55, 4827 CrossRef CAS.
- Y. S. Kim, S. H. Nam, H. S. Shim, H. J. Ahn, M. Anand and W. B. Kim, Electrochem. Commun., 2008, 10, 1016 CrossRef CAS.
- C. Wang, H. Gao, X. Chen, W. Yuan and Y. Zhang, Electrochim. Acta, 2015, 152, 383 CrossRef CAS.
- B. Liu, H. Wang, Y. Chen, J. Wang, L. Peng and L. Li, J. Alloys Compd., 2016, 682, 584 CrossRef CAS.
- J. Chen, Q. Niu, G. Chen, J. Nie and G. Ma, J. Phys. Chem. C, 2017, 121, 1463 CrossRef CAS.
- B. M. Thamer, M. H. El-Newehy, S. S. Al-Deyab, M. A. Abdelkareem, H. Y. Kim and N. A. M. Barakat, Appl. Catal., A, 2015, 498, 230 CrossRef CAS.
- S. Uhm, B. Jeong and J. Lee, Electrochim. Acta, 2011, 56, 9186 CrossRef CAS.
- A. M. Al-Enizi, A. A. Elzatahry, A. M. Abdullah, A. Vinu, H. Iwai and S. S. Al-Deyab, Appl. Surf. Sci., 2017, 401, 306 CrossRef CAS.
- I. M. A. Mohamed, A. S. Yasin, N. A. M. Barakat, S. A. Song, H. E. Lee and S. S. Kim, Appl. Surf. Sci., 2018, 435, 122 CrossRef CAS.
- N. A. M. Barakat, H. M. Moustafa, M. M. Nassar, M. A. Abdelkareem, M. S. Mahmoud, A. A. Almajid and K. A. Khalil, Electrochim. Acta, 2015, 182, 143 CrossRef CAS.
- X. Wang, X. Hu, J. Huang, W. Zhang, W. Ji, Y. Hui and X. Yao, Solid State Sci., 2019, 94, 64 CrossRef.
- N. A. M. Barakat, M. A. Yassin, A. S. Yasin and S. Al-Meer, Int. J. Hydrogen Energy, 2017, 42, 21741 CrossRef CAS.
- D. Liu, W. Li, L. Li, H. Ling and T. You, J. Colloid Interface Sci., 2018, 529, 337 CrossRef CAS PubMed.
- K. J. Babu, N. Senthilkumar, A. R. Kim and G. G. Kumar, Sens. Actuators, B, 2017, 241, 541 CrossRef CAS.
- Y. Nabil, S. Cavaliere, I. A. Harkness, J. D. B. Sharman, D. J. Jones and J. Rozière, J. Power Sources, 2017, 363, 20 CrossRef CAS.
- M. Li, Y. Zhu, H. Wang, C. Wang, N. Pinna and X. Lu, Adv. Energy Mater., 2019, 9, 1803185 CrossRef.
- Y. Zhu, W. Zhou, Y. Zhong, Y. Bu, X. Chen, Q. Zhong, M. Liu and Z. Shao, Adv. Energy Mater., 2017, 7, 1602122 CrossRef.
- G. George, L. Elias, A. C. Hegde and S. Anandhan, RSC Adv., 2015, 5, 40940 RSC.
- D. Ji, S. Peng, L. Fan, L. Li, X. Qin and S. Ramakrishna, J. Mater. Chem. A, 2017, 5, 23898 RSC.
- Y. Zhao, J. Zhang, K. Li, Z. Ao, C. Wang, H. Liu, K. Sun and G. Wang, J. Mater. Chem. A, 2016, 4, 12818 RSC.
- L. Gu, H. Zhu, D. Yu, S. Zhang, J. Chen, J. Wang, M. Wan, M. Zhang and M. Du, Part. Part. Syst. Charact., 2017, 34, 1700189 CrossRef.
- D. Ji, S. Peng, J. Lu, L. Li, S. Yang, G. Yang, X. Qin, M. Sriniasan and S. Ramakrishna, J. Mater. Chem. A, 2017, 5, 7507 RSC.
- H. Wang, C. Sun, Y. Cao, J. Zhu, Y. Chen, J. Guo, J. Zhao, Y. Sun and G. Zou, Carbon, 2017, 114, 628 CrossRef CAS.
- B. Patil, B. Satilmis and T. Uyar, ChemSusChem, 2019, 12, 1469 CrossRef CAS PubMed.
- J. Chen, J. Chen, D. Yu, M. Zhang, J. Zhu and M. Du, Electrochim. Acta, 2017, 246, 17 CrossRef CAS.
- X. Liu, M. Zhang, T. Yang, L. Wang, H. Zhu, S. Wang and M. Du, Mater. Des., 2016, 109, 162 CrossRef CAS.
- J. Chen, J. Wang, J. Chen and L. Wang, J. Mater. Sci., 2017, 52, 1306 Search PubMed.
- B. Hua, M. Li, Y. Zhang, Y. Sun and J. Luo, Adv. Energy Mater., 2017, 7, 1700666 CrossRef.
- S. Surendran, S. Shanmugapriya, P. Zhu, C. Yan, R. H. Vignesh, Y. S. Lee, X. Zhang and R. K. Selvan, Electrochim. Acta, 2019, 296, 1083 CrossRef CAS.
- X. Wang, Y. Li, T. Jin, J. Meng, L. Jiao, M. Zhu and J. Chen, Nano Lett., 2017, 17, 7989 CrossRef CAS PubMed.
- D. Ji, L. Fan, L. Li, N. Mao, X. Qin, S. Peng and S. Ramakrishna, Carbon, 2019, 142, 379 CrossRef CAS.
- R. Singhal and V. Kalra, ChemPhysChem, 2017, 18, 223 CrossRef CAS PubMed.
- H. Wu, W. Sun, J. Shen, Z. Mao, H. Wang, H. Cai, Z. Wang and K. Sun, ACS Sustainable Chem. Eng., 2018, 6, 15180 CrossRef CAS.
- S. Peng, X. Han, L. Li, S. Chou, D. Ji, H. Huang, Y. Du, J. Liu and S. Ramakrishna, Adv. Energy Mater., 2018, 8, 1800612 CrossRef.
- C. Li, M. Wu and R. Liu, Appl. Catal., B, 2019, 244, 150 CrossRef CAS.
- D. Ji, S. Peng, D. Safanama, H. Yu, L. Li, G. Yang, X. Qin, M. Srinivasan, S. Adams and S. Ramakrishna, Chem. Mater., 2017, 29, 1665 CrossRef CAS.
- Z. Wang, M. Li, L. Fan, J. Han and Y. Xiong, Appl. Surf. Sci., 2017, 401, 89 CrossRef CAS.
- J. Shim, K. J. Lopez, H. J. Sun, G. Park, J. C. An, S. Eom, S. Shimpalee and J. W. Weidner, J. Appl. Electrochem., 2015, 45, 1005 CrossRef CAS.
- H. Gong, T. Wang, H. Guo, X. Fan, X. Liu, L. Song, W. Xia, B. Gao, X. Huang and J. He, J. Mater. Chem. A, 2018, 6, 16943 RSC.
- X. Zhang, Y. Gong, S. Li and C. Sun, ACS Catal., 2017, 7, 7737 CrossRef CAS.
- H. Wu, W. Sun, J. Shen, C. Lu, Y. Wang, Z. Wang and K. Sun, Nanoscale, 2018, 10, 13149 RSC.
- K. N. Jung, S. M. Hwang, M. S. Park, K. J. Kim, J. G. Kim, S. X. Dou, J. H. Kim and J. W. Lee, Sci. Rep., 2015, 5, 7665 CrossRef CAS PubMed.
- Y. Wang, J. Fu, Y. Zhang, M. Li, F. M. Hassan, G. Li and Z. Chen, Nanoscale, 2017, 9, 15865 RSC.
- M. Wu, Y. Wang, Z. Wei, L. Wang, M. Zhuo, J. Zhang, X. Han and J. Ma, J. Mater. Chem. A, 2018, 6, 10918 RSC.
- Y. Fu, H. Yu, C. Jiang, T. Zhang, R. Zhan, X. Li, J. Li, J. Tian and R. Yang, Adv. Funct. Mater., 2018, 28, 1705094 CrossRef.
- C. Alegre, E. Modica, A. Di Blasi, O. Di Blasi, C. Busacca, M. Ferraro, A. S. Aricò, V. Antonucci and V. Baglio, Renewable Energy, 2018, 125, 250 CrossRef CAS.
- B. Li, S. W. Chien, X. Ge, J. Chai, X. Goh, K. T. Nai, T. S. A. Hor, Z. Liu and Y. Zong, Mater. Chem. Front., 2017, 1, 677 RSC.
- B. M. Thamer, M. H. El-Newehy, N. A. M. Barakat, M. A. Abdelkareem, S. S. Al-Deyab and H. Y. Kim, Int. J. Hydrogen Energy, 2015, 40, 14845 CrossRef CAS.
- J. Guo, M. Gao, J. Nie, F. Yin and G. Ma, J. Colloid Interface Sci., 2019, 544, 112–120 CrossRef CAS PubMed.
- Y. Bu, O. Gwon, G. Nam, H. Jang, S. Kim, Q. Zhong, J. Cho and G. Kim, ACS Nano, 2017, 11, 11594 CrossRef CAS PubMed.
- Y. Gong, X. Zhang, Z. Li, Z. Wang, C. Sun and L. Chen, ChemNanoMat, 2017, 3, 485 CrossRef CAS.
- H. W. Park, D. U. Lee, P. Zamani, M. H. Seo, L. F. Nazar and Z. Chen, Nano Energy, 2014, 10, 192 CrossRef CAS.
- S. Surendran, S. Shanmugapriya, A. Sivanantham, S. Shanmugam and A. K. Selvan, Adv. Energy Mater., 2018, 8, 1800555 CrossRef.
- M. Wang, C. Zhang, T. Meng, Z. Pu, H. Jin, D. He, J. Zhang and S. Mu, J. Power Sources, 2019, 413, 367 CrossRef CAS.
- Y. Si, J. Yu, X. Tang, J. Ge and B. Ding, Nat. Commun., 2014, 5, 5802 CrossRef PubMed.
| This journal is © the Partner Organisations 2019 |