A mid-IR flow-through sensor for direct monitoring of enzyme catalysed reactions. Case study: measurement of carbohydrates in beer
M.
Haberkorn
,
P.
Hinsmann
and
B.
Lendl
*
Institute of Analytical Chemistry, Vienna University of Technology, Getreidemarkt 9/151, A-1060 Vienna, Austria. E-mail: blendl@mail.zserv.tuwien.ac.at; Fax: +43-1-58801-15199
First published on 12th December 2001
Abstract
A novel mid-IR flow-through sensor for in situ monitoring of the enzymatic reaction of amyloglucosidase with carbohydrates was developed. Amyloglucosidase was immobilised on agarose beads with N-ethyl-N′-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC) and directly placed in a conventional IR flow-through cell. The carbohydrate content of various beer samples was then determined by following the enzymatic hydrolytic cleavage of carbohydrates to glucose with Fourier-transform infrared (FTIR) spectroscopy. The whole procedure was done in an automated way operating in the stopped flow mode by incorporating the flow-through sensor in a sequential injection (SI) manifold. As the immobilised enzyme was directly probed by the IR beam, an in situ study of the enzymatic reaction was possible enabling determination of the Michaelis–Menten constant of the immobilised enzyme. A linear calibration curve was recorded using maltose standards in the range between 0.86 and 7.13 g L−1. The proposed method was successfully applied to the determination of the carbohydrate content of four different beer samples by the standard addition method. Moreover experiments showed the possibility of monitoring in situ the immobilisation of the enzyme as well as a small organic acid (malic acid) on the agarose beads using EDC.
1. Introduction
Flow-through sensor systems have gained great interest in the past few years especially in the field of biochemical systems1 and combinatorial chemistry2–4 due to their versatility and robustness compared to stand alone sensors. A characteristic of the latter is the permanent contact to the matrix of the species to be analysed which can lead to ageing effects or baseline instabilities when used for long-time applications. Flow-through sensors can be operated with a defined time of interaction to the analyte also allowing regeneration between measurements. Most of these sensors developed so far rely on the principle of retaining the analyte on a solid support and measuring the obtained signal by UV/VIS or fluorescence detection. Several flow-through sensor systems have been presented for the determination of traces of metals such as aluminium,5–7 cadmium8 and iron.9 C18 and ion exchanger based sensors have also been applied to the determination of phosphate,10,11 amines,12 pesticides13 and sulfonamides.14 Further work has been done on biochemical sensors by immobilising specific enzymes on the solid support and tracing the catalysed reaction15–17 mostly using an additional indicator dye reaction. These systems represent very sensitive methods but lack the great advantage of structural information. Fourier-transform infrared (FTIR) spectroscopy overcomes this drawback by providing molecule-specific information. However, the construction of a FTIR flow-through sensor requires a complicated construction of the flow-through cell, as the pathlength of the device must be kept equal or below 50 μm when measuring in aqueous solutions due to the high absorption of water in the mid-IR range. Nevertheless sensor systems based on FTIR detection have recently been developed based on the principle of retaining the analyte of interest by specific interaction with the functionalised polymer beads to enhance the sensitivity and selectivity of the method. With the structural information obtained it was possible to simultaneously determine acetic and malic acid by multivariate data evaluation.18 Furthermore this approach was applied to the determination of water hardness with an indirect method by measuring the difference in absorption on protonated and metal loaded ion exchanger beads.19 In the presented paper we describe the immobilisation of 1,4-α-D-glucan glucohydrolase (amyloglucosidase) on agarose polymer beads which were then placed in the flow-through cell for the determination of the carbohydrate content in beer samples20 by following the enzymatic hydrolytic cleavage reaction. Amyloglucosidase hydrolyses 1,4-linked α-D-glucose chains by cleaving off β-D-glucose from the non-reducing ends of the chain.21 In previous work, both the sucrose content of different real samples22 as well as the enzyme activity of sugar reacting enzymes23,24 have been determined by FTIR spectroscopy by monitoring the formation of products during reaction. With the proposed method we were able to investigate the reaction in situ by placing the layer of enzyme-immobilised beads directly into the IR beam thereby ensuring that the measurements be carried out at the place of reaction.2. Experimental
2.1. Reagents
All reagents were of analytical grade. Amyloglucosidase from Aspergillus niger (E.C. number 3.2.1.3) and TRIS (2-amino-2-(hydroxymethyl)-1,3-propanediol) were supplied by Boehringer Mannheim, N-ethyl-N′-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC) by Sigma and maltose monohydrate by Merck. The polymer beads (4% agarose beads, particle size 45–165 μm, spacer arm 10-carbon with 7–11 μmol amino groups per mL drained gel) were purchased from Pharmacia Biotech.2.2. Enzyme batch mode immobilisation procedure
Approximately 2 mL of the gel suspension were washed with 150 mL 0.5 M sodium chloride solution and with the same amount of water. Afterwards the beads were suspended in 25 mL 0.1 M EDC solution (adjusted to pH 8 with HCl) containing 50 mg amyloglucosidase and mixed with an eccentric rotator for 18 h. Subsequently the beads were washed alternately with 0.1 M sodium acetate (pH 4) and 0.1 M TRIS (pH 8.3), both containing 0.5 M sodium chloride and stored in the refrigerator in 0.1 M disodium hydrogen phosphate solution at pH 7.2.3. In situ monitoring of enzyme immobilisation
The SI-FTIR system was used to carry out immobilisation of amyloglucosidase on the beads and to follow this reaction step in situ by FTIR spectroscopy. Therefore the flow-through sensor described below, was filled with untreated beads and placed in the presented SI manifold.The same procedure as for manual immobilisation was used except for the fact that no eccentric rotator was used for the actual immobilisation step. Instead the coupling solution was pumped through the cell at 125 μL min−1 leading the used solution back to the flask with the stock solution. That way the coupling solution was circulated through the IR cell for 18 h.
2.4. Flow-through sensor cell
The enzyme-immobilised beads were put between the CaF2 windows (thickness 2 mm) of a conventional Perkin-Elmer flow-through cell, equipped with a 50 μm Teflon spacer. Care had to be taken to apply a small amount of beads suspended in the storage solution in a way that ensured that a homogenous layer of beads covered the whole width of the spacer channel (approximately 5 mm) thereby preventing the analyte liquid stream from passing through partly without contact with the enzyme. It is important that the beads are not gathering together in clusters, but rather homogenously fill the IR probed flow channel section.2.5. SI system and FTIR instrument
The SI manifold (Fig. 1) was set up with a Cavro (Sunnyvale, CA, USA) XP 3000 syringe pump (syringe size 2500 μL) and a Valco (Houston, TX, USA) 14 port selection valve. Poly(tetrafluoroethylene) (PTFE) tubing (id: 0.75 mm, length: 300 mm from valve to flow-through sensor) and fittings were obtained from Global FIA (Gig Harbour, WA, USA). A 5 mL syringe with a magnetic stirrer served as a mixing chamber to prepare appropriate dilutions. | ||
| Fig. 1 Scheme of the SIA manifold. | ||
For all the experiments carried out a Bruker (Karlsruhe, Germany) Equinox 55 FTIR spectrometer with a narrow band mercury cadmium telluride (MCT) detector was used. Spectra were recorded by coadding 128 scans at a resolution of 8 cm−1 and a scanner velocity of 100 kHz HeNe frequency.
The whole set-up was controlled automatically by coupling the AnalySIA control software (Center for Biotechnology, Åbo Akademi University, Turku, Finland) of the SI manifold with the OPUS 3.1 software (Bruker, Karlsruhe, Germany) from the spectrometer.
2.6. Measurement sequence
For the measurements of the calibration set (0.86–7.13 g L−1) a maltose stock solution of 9.5 g L−1 in 0.1 M sodium acetate buffer adjusted to pH 4.75 was prepared. The appropriate dilutions were prepared automatically in the SI system, injected into the sensing cell and upon reaching the sensor the flow was stopped. Spectra were recorded for ten minutes to trace the formation of the product. For the standard addition method of real sample measurements the beer was degassed in an ultrasonic bath for one hour. Then with the help of the automated SI manifold, solutions of 0–3.8 g L−1 of maltose were added to the samples and the resulting mixture was diluted with buffer to gain a final dilution of 1:10 of the beer samples. The analysis was carried out in the same way as described above.3. Results and discussion
3.1. Batch immobilisation
In Fig. 2 the absorbance spectra of Sepharose beads with and without batch mode immobilised amyloglucosidase are shown. Comparing these two spectra, it can be seen that the spectrum of the enzyme coupled beads shows an additional band at 1540 cm−1, the “Amide II” band. As the peptide bond presents the backbone of enzymes, the appearance of the “Amide II” band suggests that the batch mode immobilisation procedure was successful. | ||
| Fig. 2 Absorbance spectra of batch mode immobilised amyloglucosidase. In addition the spectra of the Sepharose beads and a 10 g L−1 solution of amyloglucosidase (referring to the secondary axis) are given. | ||
The spectrum of the 10 g L−1 solution of amyloglucosidase also proves the above made conclusion, as the “Amide II” band of amyloglucosidase indeed occurs at 1540 cm−1 wavenumbers.
Comparing the intensity of the “Amide II” band of the enzyme solution with the intensity of the peak in the spectrum of the immobilised enzyme, it can be estimated that the concentration of the enzyme on the beads corresponds to a solution of approximately 28 g L−1 amyloglucosidase.
3.2. In situ immobilisation approach
To carry out in situ immobilisation of the enzyme on the beads, the agarose resins were placed in the flow cell and the coupling reaction was performed by supplying the reagents with the SI manifold. In general it was expected to see the “Amide II” band of amyloglucosidase at 1540 cm−1 rise significantly with proceeding time as it should get bound to the beads, but only a small peak at 1540 cm−1 corresponding to a local enzyme concentration of approximately 0.6 g L−1 could be obtained, which compared to the spectrum of the batch mode coupled beads represents an ineffective immobilisation procedure.The reason that the automated immobilisation of the enzyme occurred only with small yield could result from the slow diffusion of the enzyme. Changing the enzyme solution’s residence times by alteration of flow rates during immobilisation however did not provide any significant improvements.
The enzymes are coupled via their carboxylic groups to the amino moieties of the beads. In order to further investigate the effect of diffusion on the in situ immobilisation reaction a faster diffusing small organic acid (malic acid) was selected and subjected to the immobilisation procedure instead of the enzyme.
Fig. 3 (a) shows the progress of immobilisation, with the background spectrum taken after the coupling solution had already entered the cell. Starting from the first difference spectrum at 0 immobilisation time, the following 12 spectra were measured at equal time intervals of 3 min and finally the last three spectra were recorded after 60, 90 and 120 min of reaction time. As can be seen, with proceeding immobilisation time, the peak at 1393 cm−1 (C–O symmetric stretching vibration) rises, indicating enrichment of malic acid in the flow through cell. Fig. 3 (b) shows the spectra of untreated Sepharose beads as well as malic acid coupled beads after rinsing with water and their calculated difference spectra. Comparing the calculated difference spectrum with the spectrum of 1.5 g L−1 malic acid solution (referring to the secondary absorbance axis) clearly demonstrates that the compound causing the peak at 1393 cm−1 wavenumbers is malic acid and that it is present at an amount correlating to an aqueous solution of approximately 2 g L−1. This result supports the assumption that the small diffusion coefficient of the enzyme somehow hampers the immobilisation process inside the flow-through cell.
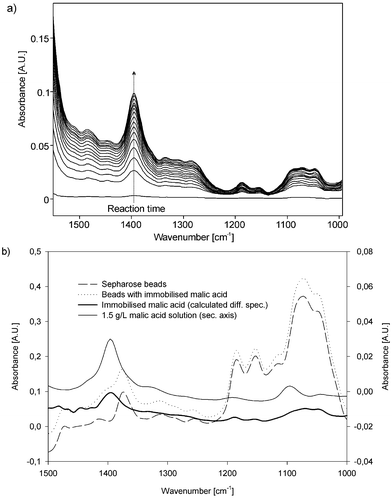 | ||
| Fig. 3 (a) Difference spectra of online immobilisation of malic acid on the Sepharose beads. (b) Malic acid coupled to Sepharose beads using a SIA manifold. | ||
3.3. Data evaluation
In order to quantify the amount of carbohydrates in the sample, difference spectra were recorded by taking the background immediately after stopping the flow and starting the reaction. The evaluation was done by calculating the difference in absorption between the decreasing band at 1149 cm−1 and the increasing band at 1084 cm−1 wavenumbers (Fig. 4). | ||
| Fig. 4 Spectra of a synthetic maltose sample before and after the reaction and the appropriate difference spectrum. | ||
3.4. Determination of the Michaelis–Menten constant of the immobilised enzymes
For the determination of the Michaelis–Menten constant conditions of zero order kinetics with respect to the substrate concentration are needed.25 As this can reasonably be assumed at the beginning of the reaction, the difference spectra after 30 s reaction time when analysing the calibration set were taken. By evaluating the peak height difference in the difference spectra between 1080 and 1149 cm−1 for each concentration of the calibration set the concentration dependent reaction rate was determined. Using a Lineweaver–Burk plot25 the Michaelis–Menten constant of the immobilised enzyme for maltose was determined to be 6.69 mmol L−1 (2.41 g L−1).3.5. Determination of carbohydrates in beer
Due to the limited sensitivity of MIR detection the analyte concentration could not be diluted much below the Michaelis–Menten constant of the immobilised enzyme in order to allow relating the observed initial reaction rate to the amount of present carbohydrates with sufficient precision. For quantitative analysis experimental conditions had to be adapted which assured complete hydrolysis of the analytes. The experiments were thus carried out in the stopped flow mode as described in section 2.6. and the finally obtained difference spectra were taken for data evaluation. Even for the more highly concentrated solutions no significant increase in the concentration of glucose was observed after ten minutes reaction time. Hence data evaluation was based on the difference spectra obtained after 10 min reaction time. The results of the linear calibration are summarised in Table 1. The reason for choosing maltose as a calibration standard for the carbohydrate determination of beer samples is the fact that maltose represents the highest fraction of carbohydrates in beer. Furthermore, all remaining carbohydrates with a higher degree of polymerisation are subject to the same reaction principle (hydrolysis of terminal 1,4-linked α-D-glucose units) as maltose.| Analyte | Equation parameters | r | r 2 | Determined concentration/g L−1 |
|---|---|---|---|---|
| Calibration set (0.86–7.13 g L−1) | b = 0.00033 | 0.9979 | 99.58 | |
| Beer sample 1 | a = 0.00902 b = 0.00029 | 0.9981 | 99.61 | 31.3 ± 0.9 |
| Beer sample 2 | a = 0.00826 b = 0.00031 | 0.9997 | 99.95 | 26.4 ± 0.8 |
| Beer sample 3 | a = 0.00788 b = 0.00031 | 0.9976 | 99.53 | 25.6 ± 0.8 |
| Beer sample 4 | a = 0.00900 b = 0.00033 | 0.9974 | 99.48 | 27.1 ± 0.8 |
As the carbohydrate content was expected in the range of 20–40 g L−1 (expressed in g L−1 maltose) the degassed beer samples were diluted 1:10 and analysed in the same way as the standard solutions. Independent of the beer matrix the same spectral features were obtained (as shown in Fig. 5 (a)) as the enzymatic reaction is specific for terminal 1,4-linked α-D-glucose carbohydrate chains which are hydrolysed releasing β-D-glucose. Small differences between the difference spectra of maltose standards and beer samples (Fig. 5 (b)) are due to the conversion of 1,4-linked α-D-glucose carbohydrates with a higher degree of polymerisation present in the analysed natural sample. Bellon-Maurell et al.26 and the work of Schindler et al.27 describe the effect of different degrees of polymerisation of α-D-glucose chains on the spectral features in the mid-IR.
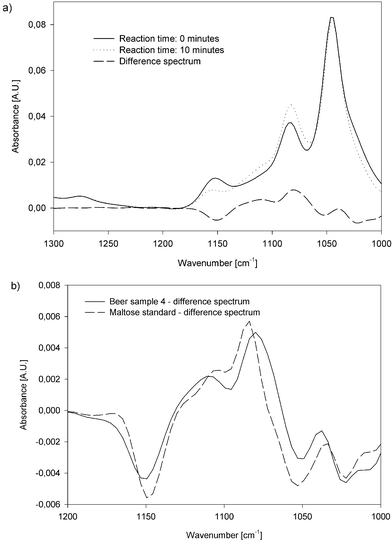 | ||
| Fig. 5 (a) Spectra of a beer sample before and after the reaction and the appropriate difference spectrum. (b) Difference spectra before and after the reaction of a synthetic maltose solution and a beer sample. | ||
In order to take account of the peak shifts due to the degree of polymerisation of the substrate, the evaluation of the difference spectra was carried out so that the peak height difference between the absorbance value at 1049 cm−1 wavenumbers and the maximum absorbance value in the range from 1090 to 1070 cm−1 was used for data evaluation. The implementation of the variable peak maximum in the evaluation procedure was used in order to compensate the small peak shift. Four beer samples from different Austrian breweries were analysed and the results of the standard addition experiments are listed in Table 1. The g L−1 maltose unit in this case is an index representative for the carbohydrate content of the samples.
4. Conclusion
The presented work demonstrates the possibility of on-bead reaction monitoring in aqueous phase using FTIR spectroscopy using the example of derivatisation of agarose beads with malic acid and to a smaller extent with amyloglucosidase. Within the case study of carbohydrate determination in beers the Michaelis–Menten constant of the immobilized amyloglucosidase could be determined. Furthermore the analyte could be successfully quantified in different natural samples. The principles of the proposed mid-IR flow-through sensor can be extended to different enzyme systems but also be used as a general tool for monitoring of chemical reactions taking place between dissolved reagents and proteins immobilized on agarose beads in aqueous phase.Acknowledgement
The authors thankfully acknowledge the financial support received from the Austrian Science Fund within project P13686 ÖCH.References
- M. Valcárcel and M. D. Luque de Castro, Flow through (Bio)chemical sensors, Elsevier, Amsterdam, 1994 Search PubMed.
- D. E. Pivonka, K. Russel and T. Gero, Appl. Spectrosc., 1996, 50(12), 1471 Search PubMed.
- D. E. Pivonka and D. L. Palmer, J. Comb. Chem., 1999, 1, 294 CrossRef CAS.
- D. E. Pivonka, J. Comb. Chem., 2000, 2, 33 CrossRef CAS.
- P. Cañizares, M. D. Luque de Castro and M. Valcárcel, Anal. Lett., 1994, 27(2), 247 CAS.
- M. R. Pereiro García, M. E. Díaz García and A. Sanz-Medel, Analyst, 1990, 115, 575 RSC.
- P. Cañizares and M. D. Luque de Castro, Anal. Chim. Acta, 1994, 295, 59 CrossRef CAS.
- P. Richter, M. D. Luque de Castro and M. Valcárcel, Anal. Lett., 1993, 26(4), 733 CAS.
- F. Lazaro, M. D. Luque de Castro and M. Valcárcel, Anal. Chim. Acta, 1989, 219, 231 CrossRef CAS.
- N. Lacy, G. D. Christian and J. Ruzicka, Anal. Chem., 1990, 62, 1482 CrossRef CAS.
- K. Yoshimura, S. Nawata and G. Kura, Analyst, 1990, 115, 843 RSC.
- B. Fernández-Band, F. Lázaro, M. D. Luque de Castro and M. Valcárcel, Anal. Chim. Acta, 1990, 229, 177 CrossRef CAS.
- B. Fernández-Band, P. Linares, M. D. Luque de Castro and M. Valcárcel, Anal. Chem., 1991, 63, 1672 CrossRef CAS.
- M. T. Tena, M. D. Luque de Castro and M. Valcárcel, Analyst, 1994, 119, 1625 RSC.
- P. Linares, M. D. Luque de Castro and M. Valcárcel, Anal. Chim. Acta, 1990, 230, 199 CrossRef CAS.
- T. D. Yerian, G. D. Christian and J. Ruzicka, Anal. Chem., 1988, 60, 1250 CrossRef CAS.
- J. M. Fernández-Romero and M. D. Luque de Castro, Anal. Chem., 1993, 65, 3048 CrossRef CAS.
- B. Lendl and R. Schindler, Vibr. Spectrosc., 1999, 19, 1 CrossRef CAS.
- A. Pérez-Ponce and B. Lendl, Appl. Spectrosc., 2000, 54(5), 676 Search PubMed.
- M. Gallignani, S. Garrigues and M. de la Guardia, Anal. Chim. Acta, 1994, 269, 155 CrossRef CAS.
- Enzyme Handbook, ed. D. Schomburg and M. Salzmann, Springer, Berlin, 1991, pp. 3.2.1.3 Search PubMed.
- B. Lendl and R. Kellner, Mikrochim. Acta, 1995, 119, 73 CAS.
- R. Schindler, B. Lendl and R. Kellner, Anal. Chim. Acta, 1998, 366, 35 CrossRef CAS.
- R. Schindler, H. Le Thanh, B. Lendl and R. Kellner, Vibr. Spectrosc., 1998, 16, 127 CrossRef CAS.
- Methods in Enzymatic Analysis, ed. H. U. Bergmeyer, Verlag Chemie, Weinheim, 3rd edn., 1983, vol. 1, pp. 68–103 Search PubMed.
- V. Bellon-Maurell, C. Vallant and D. Goffinet, Appl. Spectrosc., 1995, 49, 556 Search PubMed.
- R. Schindler, B. Lendl and R. Kellner, Analyst, 1997, 122, 531 RSC.
| This journal is © The Royal Society of Chemistry 2002 |