Synthesis of novel polyurethane polyesters using the enzyme Candida antarctica lipase B
Richard W.
McCabe
* and
Alan
Taylor
Centre for Materials Science, University of Central Lancashire, Preston, UK PR1 2HE. E-mail: rwmccabe@uclan.ac.uk; ataylor3@uclan.ac.uk; Fax: +44 1772 892903; Tel: +44 1772 893533
First published on 8th April 2002
Abstract
A novel enzymatic route has been used to synthesise standard and unusual polyester polyurethanes without employing the usual highly toxic isocyanate intermediates
The majority of reported enzymatic syntheses using lipases, are concerned either with the synthesis of optically active esters or alcohols, where the enantioselectivity of the enzyme steers the reaction product to a particular isomer,1 or with the synthesis of polyesters.2,3,4 Apart from the synthesis of sugar esters,5 the hydrolysis6 and transesterification7 of sensitive esters (e.g. prostaglandin esters) and the work of Harffey et al. with epoxide esters,8 the mild, low temperature aspects of enzymatic synthesis have not been exploited to full advantage as yet.
Polyester based polyurethanes have numerous applications in surface and textile coatings, adhesives and elastomers. These materials are manufactured from hydroxy terminated polyester resins made by high temperature Lewis acid catalysed condensation of a diacid and diol.9 Subsequent reaction with highly toxic diisocyanates produce a polyurethane polymer.10 The diisocyanates are derived from the even more toxic phosgene and the corresponding diamine.11 Thus, the production of a narrow range of less toxic, low volatility (mainly aromatic) diisocyanates is limited to only a few companies in the world who are capable of operating the process safely.
There have been many attempts at non-phosgenation routes to diisocyanates and to the synthesis of polyurethanes without isocyanates. None of these has been successful commercially. In the conventional process the addition of the isocyanate must occur after esterification because the carbamate group begins decomposing at 160–180 °C,12,13 well below the esterification temperature (typically 220 °C). However, the use of enzymatic methods allows us to reverse the conventional process by creating the urethane first and then using a low temperature enzymatic polyester synthesis to build the polymer. Thus, we were able to synthesise a novel series of biscarbamate esters and polyesters.
It was known from the work of Delaby et al.14 in the 1950’s that the carbamate group could be synthesised by the ring opening addition of a cyclic carbonate, such as ethylene carbonate 1a, with a primary diamine. The product of this reaction being the bis(hydroxyethyl) carbamate (e.g.Scheme 1).
 | ||
| Scheme 1 Synthesis of diurethane diols. | ||
These diols have been used to form polymers by reacting the biscarbamates with methylol melamine to give cross-linked urethane containing polyether polymers.15 These polymers had some of the properties of a polyurethane, but the need for high temperature stoving meant that some degradation took place.
Enzyme catalysed esterification of bis(hydroxyethyl) carbamates derived from readily available diamines, affords a low temperature route to polyester polyurethanes avoiding the use of diisocyanates. This idea suggested two possible applications, firstly less hazardous syntheses of polymers (avoiding diisocyanates) that are analogues of existing polymers, secondly synthesis of novel polyester polyurethanes where the requisite diamine was available, but the diisocyanate was not, e.g. ethylenediamine is readily available, but ethylenediisocyanate is not as it is extremely toxic due to its volatility. Reactions with aliphatic diamines were performed as aliphatic diisocyanates are much more expensive and hazardous than aromatic ones.
Delaby et al. synthesised, bis(hydroxyethyl) hexamethylene carbamate 3a, using ethylene carbonate 1a and hexamethylenediamine 2a.16 Modifying their procedure slightly (omission of cooling), we produced compound 3a (mp 94 °C) in 60% yield and high purity; by gel permeation chromatography (GPC) and 1H NMR spectroscopy (Table 1).
| Compound | R | X |
|---|---|---|
| 1a | H | — |
| 1b | CH3 | — |
| 2a | — | –(CH2)6– |
| 2b | — | –(CH2)2– |
| 2c | — |

|
| 2d | — |
 n ∼ 3
n ∼ 3 |
| 2e | — |
 n
∼1 or 2
n
∼1 or 2 |
| 3a, 4a | H | –(CH2)6– |
| 3b, 4b | H | –(CH2)2– |
| 3c, 4c | CH3 |
|
| 3d, 4d | H |
n
∼ 3 |
| 3e, 4e | H |
n
∼1 or 2 |
As the desired physical properties of the finished polyester polyurethane meant that the biscarbamate 3a was unlikely to be used as the only diol component of the polyester, a co-polyester 4a with butane-1,4-diol and adipic acid was synthesised using the biscarbamate as about 10% of the total diol (Scheme 2). The biscarbamate 3a was dissolved in butane-1,4-diol 5 at 90 °C under nitrogen, cooled to 60 °C and reacted with adipic acid 6 using Novozyme 435, a commercial preparation of supported Candida antarctica lipase B, as catalyst. The adipic acid 6 was added in portions so as not to inhibit the enzyme and the water formed was removed at about 100 mmHg over 2 days. GPC analysis using a 1000 Å column gave the molecular weight of the resulting polyester polyurethane 4a as 9350 Daltons, compared to a polystyrene standard, with a dispersity of 1.75.
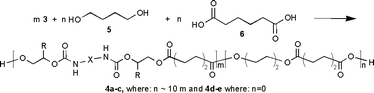 | ||
| Scheme 2 Synthesis of a polyester polyurethane co-polymer. | ||
The polyester polyurethane 4a is the analogue of a polybutane adipate polyester that has been partially chain extended with hexamethylene diisocyanate and which could be chain extended once more by the addition of further diisocyanate.
Next, the method was extended to the synthesis of urethane polyesters for which no equivalent isocyanate is available. Ethylenediamine 2b was reacted with ethylene carbonate 1a to give bis(hydroxyethyl) ethane carbamate 3b, identical to a carbamate based on ethylene diisocyanate. The mp of the white crystalline compound was 93 °C. The yield was 60% after recrystallization; losses being due to the slight solubility of the product during a cold ethanol wash. Chromatography showed a single product free from starting materials. Once again, a butane-1,4-diol, adipic acid co-polyester 4b was synthesised using the biscarbamate 3b as 10% of the diol component. The molecular weight was found to be 4500 Daltons by GPC, with a dispersity of 2.4. This novel polyester was found to be extremely water soluble, due to the preponderance of ethane groups in the polymer. Such a water soluble polymer may well have applications in water soluble polyurethane coatings or adhesives.
One major problem associated with the commercialisation of a new process using novel intermediates is the need for costly toxicological testing of the compounds. The EINECS regulations are relaxed if the novel compound does not leave the reactor and if the final product is a high molecular weight polymer. Thus, as butane-1,4-diol 5 does not react with either of the reactants, the toluene solvent can be replaced with butane-1,4-diol 5. IR spectroscopy and GPC analysis confirmed completion of the reaction with diamine 2b; the product 3b being a clear solution at 60 °C and a white waxy solid on cooling. Novozyme 435 and the requisite amount of adipic acid 6 were added to the reaction product 3b to give a polyester 4b with a molecular weight of about 1500 Daltons. GPC gave the molecular weight as 2200 Mn, 4640 Mw with a dispersity of 2.1. The acid number was 0.7 mg KOH g−1 and the hydroxyl number 78 mg KOH g−1; this end group analysis gave a molecular weight of 1488 Daltons. There is no reason why this principle of using a diol from the second stage esterification as the diluent in the formation of the biscarbamate cannot be extended to the synthesis of any biscarbamate.
As it provides a useful polymer substituent, isophorone diamine 2c was reacted with propylene carbonate 1b to give the biscarbamate 3c. GPC analysis showed that the reaction had gone to completion with no reactants remaining. This biscarbamate 3c was converted to a co-polyester polyurethane 4c in the usual manner. GPC gave the molecular weight, Mw, as 6000 and the dispersity as 2.14. The acid number was 2.0 mg KOH g−1.
α,ω-Polytetramethylene ether diols are used extensively in the manufacture of high performance polyurethane elastomers and coatings. There are no equivalent diisocyanates available, but the related diamine 2d is available by the reaction of an α,ω-polytetramethylene ether diol with acrylonitrile followed by hydrogenation to give the bis(1-aminoprop-2-yl)polytetramethylene ether 2d of molecular weight 350. The biscarbamate 3d of diamine 2d was synthesised by reaction with ethylene carbonate 1a.
The product 3d was a reddish viscous liquid. NMR analysis showed that all the ethylene carbonate 1a had reacted, however there was a trace of unreacted amine remaining. Because of the substantial polyether backbone of the diamine 2d it was not thought necessary to add any butane-1,4-diol 5 to the biscarbamate 3d in order to form a useful polyester polyurethane. Therefore, adipic acid 6 and Novozyme 435 were added and after heating at 60 °C under reduced pressure for 48 hours the final polymer 4d had a molecular weight of 6500 by GPC and an acid number of 5.0 mg KOH g−1. The combination of the ester groups and the ether backbone gave a polymer that was not soluble in any of the common solvents. It was thought that this material would make an excellent intermediate in the manufacture of solvent resistant coatings.
The above reaction was extended to the related polyoxypropyleneamine 2e, Jeffamine D230. The amine was added to the ethylene carbonate 1a as before, however, the exotherm was substantially less than with any of the previous amines and thus the reaction was maintained at 80°C overnight. TLC and 1H NMR spectroscopy indicated that the reaction had gone to completion with only a trace of residual amine remaining. This biscarbamate 3e was also converted to polyester in the same manner as the others and as for polymer 4d, the finished polyester 4e was a brown viscous liquid the molecular weight was 6500 Daltons by GPC and the acid number was 2 mg KOH g−1.
We would like to thank Baxenden Chemicals Ltd., Accrington, UK, who have filed a patent on these reactions and the resulting compounds, for their support.
Notes and references
- e.g. C. Orrenius, D. Rotticci, A. Mattson, K. Hult and T. Norin, Tetrahedron: Asymmetry, 1995, 6, 1217–1220 Search PubMed.
- E. M. Brazwell, D. Y. Filos and C. J. Morrow, J. Polym. Sci., Part A: Polym. Chem., 1995, 33, 89–95 Search PubMed.
- S. Kobayashi, H. Uyama and S. Namekawa, Polym. Degrad. Stab., 1998, 59, 195–201 CrossRef CAS.
- F. Binns, P. Harffey, S. M. Roberts and A. Taylor, J. Polym. Sci., Part. A: Polym. Chem., 1998, 36, 2069–2080 Search PubMed.
- P. Degn and W. Zimmerman, Biotechnol. Bioeng., 2001, 74, 483–491 CrossRef CAS.
- S. M. Roberts and N. M. Williamson, Curr. Org. Chem., 1997, 1, 1–20 Search PubMed and references therein.
- O. Parve, I. Jaerving, I. Martin, A. Metsala, I. Vallikivi, M. Aidnik, T. Pehk and N. Samel, Bioorg. Med. Chem. Lett., 1999, 9, 1853–1858 CrossRef CAS.
- F. Binns, P. Harffey, S. M. Roberts and A. Taylor, J. Chem. Soc., Perkin Trans. 1, 1999, 2671–2676 RSC.
- W. L. Chang and T. Karalis, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 493–504 Search PubMed.
- H. Ulrich, The Chemistry and Technology of Isocyanates, John Wiley, 1996, 347–352 Search PubMed.
- H. Ulrich, The Chemistry and Technology of Isocyanates, John Wiley, 1996, 342–347 Search PubMed.
- S. Petersen, Annalen, 1949, 562, 205–229 Search PubMed.
- Z. W. Wicks, Prog. Org. Coat., 1975, 3, 73–99 CrossRef CAS.
- R. Delaby, P. Chabrier and H. Najer, Memoires Presentes a la Societe Chimique, 1956, 1616 Search PubMed.
- K. Satterley and A. Taylor, Baxenden Chemicals Search PubMed , unpublished results.
- R. Delaby, A. Sekera, P. Chabrier and P. Pignaniol, Bull. Soc. Chem., 1953, 20, 278 Search PubMed.
| This journal is © The Royal Society of Chemistry 2002 |