Professor C. Robin Ganellin FRS![[hair space]](https://www.rsc.org/images/entities/h2_char_200a.gif) †
†
Professor C. Robin
Ganellin
*
First published on 18th June 2001
Abstract
Recipient of the RSC 1999 Adrian Albert Medal and Lectureship
Career
Robin Ganellin was born in east London, attended Harrow County Grammar School, and then studied at Queen Mary College, London where he received a first class BSc in chemistry and a PhD in 1958 in organic chemistry under Michael Dewar for research on tropylium chemistry. During this time he also collaborated with Dr Rowland Pettit and discovered the oxidative rearrangement of cyclooctatetraene to the tropylium cation. He spent a period in 1960 with A. C. Cope as a research associate at the Massachusetts Institute of Technology (MIT), where he devised the first direct optical resolution of chiral olefins using platinum complexes. He joined Smith Kline & French Laboratories (SK&F![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) ) in the UK as a medicinal chemist, and from 1966 collaborated with Sir James Black and led the chemical research for the discovery of the H2-receptor histamine antagonists. He is coinventor of the drug cimetidine (Tagamet®) which revolutionised the treatment of peptic ulcer
disease. He subsequently became Vice-President for Research at the company’s Welwyn facility. In 1986 he was awarded a DSc from London University for his published work on the medicinal chemistry of histamine and drugs acting at histamine receptors. In 1986 he was also made a Fellow of the Royal Society and appointed to the SK&F chair of medicinal chemistry at University College London, a position he still holds. He is author or co-author of more than 200 scientific publications and named coinventor on over 160 US patents.
) in the UK as a medicinal chemist, and from 1966 collaborated with Sir James Black and led the chemical research for the discovery of the H2-receptor histamine antagonists. He is coinventor of the drug cimetidine (Tagamet®) which revolutionised the treatment of peptic ulcer
disease. He subsequently became Vice-President for Research at the company’s Welwyn facility. In 1986 he was awarded a DSc from London University for his published work on the medicinal chemistry of histamine and drugs acting at histamine receptors. In 1986 he was also made a Fellow of the Royal Society and appointed to the SK&F chair of medicinal chemistry at University College London, a position he still holds. He is author or co-author of more than 200 scientific publications and named coinventor on over 160 US patents.
Professor Ganellin has received international recognition as aehemistry at the University of Kent at Canterbury (1979–89), Advisory Tutor in Chemistry at the Polytechnic of North London (1979–83) and Director of the Upjohn Discovery Unit at UCL (1987–94). Together with Dr A. M. Roe he initiated the biennial RSC Summer School in Medicinal Chemistry in 1981 in the format that exists to date and, indeed, he has lectured at every one of them.
He is a past Chairman of the Society for Drug Research and is currently President of the Medicinal Chemistry Section of IUPAC.
Research
The Adrien Albert Medal and Lectureship is awarded in recognition of the application of heterocyclic chemistry to problems of biological interest.Adrien Albert was an outstanding heterocyclic chemist, publishing over 120 papers on nitrogen heterocycles. He was also an outstanding medicinal chemist who was a pioneer in using physicochemical properties for relating chemical structure to biological activity.1 His book on this subject grew out of a course of lectures he gave at University College London in 1948, which is a nice coincidence.
Our research at UCL is in medicinal chemistry and is concerned with the design and synthesis of organic compounds as prototype drugs. In this work we have also relied heavily on the use of heterocyclic chemistry and physicochemical properties.
Having been trained as an organic chemist, knowledge of medicinal chemistry had to be acquired “on the job” when I joined SK&F. This is a long learning process but I was very fortunate in that it was accelerated when Dr J. W. Black (now Sir James Black, OM, FRS, Nobel Laureate) arrived to lead the pharmacology. The many discussions we had together introduced me to chemical questions of interest to pharmacologists and gave me a new insight into becoming a medicinal chemist.
The use of the word ‘prototype’ for a drug implies that a compound can be a useful chemical tool for pharmacologists to help them unravel the mechanistic intricacies of particular physiological processes, often related to disease states, whilst acknowledging that few, if any, such compounds actually become medicines to be used therapeutically.
At UCL we collaborate with biochemists and pharmacologists at the frontier of their subject to generate the chemical tools that will be used to definitively characterise a functional cell protein.
Our work has encompassed a wide range of biological applications, from G-protein coupled receptors (for histamine and serotonin), cholecystokinin-inactivating peptidase and HIV-aspartyl peptidase, potassium ion channels, through to phosphatidyl inositol transfer protein (PITP), Transport P and persistent sunscreens.
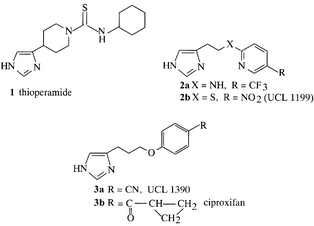
Histamine acts on four subtypes of histamine receptor, and the third subtype H3, are inhibitory presynaptic receptors which modulate the synthesis and release of histamine at histaminergic neurones in the central nervous system (CNS) and of certain non-histaminergic neurones both in the brain and periphery. Possible therapeutic applications of compounds which block H3 receptors include various CNS disorders. The prototype H3 antagonist is thioperamide (1) described2 in 1987, which is potent but was too toxic for clinical study. So far no other antagonist compound has entered beyond phase II clinical trial. We have been collaborating with J.-C. Schwartz and his laboratory at INSERM, Paris, where H3 receptors were first defined, and with W. Schunack in Berlin. Our first approach was to replace the potentially toxic thiourea moiety by aminoheterocycles and open the
piperidine ring3 (2). This led to potent phenoxypropylimidazoles4 which provided several candidate drugs (3). We have also obtained isomers (4) which are potent agonists and, remarkably do not have basic side chains. We also sought a non-imidazole H3-receptor antagonist which would have a greater propensity for brain penetration and have recently described5p-substituted phenoxyalkylamines (5); some newer analogues are very potent.
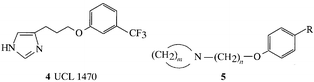
Serotonin acts on at least 14 different subtypes of receptors and we have been interested in ligands acting at the receptor designated 5-HT1. We have especially investigated naphthalene derivatives (6) as 5-HT1A partial agonists; these were also found to act at 5-HTIDα receptors.6 The latter also provided for an interesting structure–activity analysis whereby blocking potency at 5-HTIDα and 5-HTIDβ receptors correlated with molecular refractivity and the Verloop B1 size parameter.
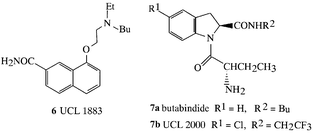
We have designed the first known inhibitor of the enzyme which inactivates the neurotransmitter peptide cholecystokinin-8 (CCK-8). The enzyme had not been fully purified but its activity was isolated from rat brain in the laboratory of J. C. Schwartz in Paris who characterised it as a serine peptidase and assayed the compounds synthesised at UCL. Our approach was to seek a reversible inhibitor since this would be more likely to be selective and non-toxic.7 This led to the indoline,8 butabindide (7a), a prototype drug which has Ki = 7 nM and is a selective competitive inhibitor which was shown to be active in potentiating the action of CCK-8 and to reduce food intake (satiating effect of CCK-8) in starved mice. Analogues of butabindide have yielded potent inhibitors having Ki as low as 0.4 nM, e.g. structure (7b).9
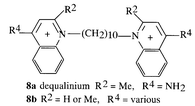
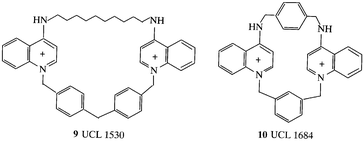
Ion channels selective for K+ form a large family and we have been synthesizing ligands for calcium-activated potassium ion channels in collaboration with D. H. Jenkinson and P. M. Dunn of UCL Pharmacology for the testing. The small conductance Ca2+-activated K+ channel (SKCa) is found in many cell types and selective blockers may have beneficial effects in, for example, myotonic muscular dystrophy, disorders of memory, narcolepsy, and in dismotilities of the gastrointestinal tract. Taking dequalinium (8) as a μM lead the pharmacophore was investigated10 and activity was inversely correlated with the energy of the LUMO.11 Dequalinium analogues were cyclised to give tetraazacyclophanes and 9 (UCL 1530)12 provided the first evidence for pharmacological differentiation between the
SKCa channels in liver and neuronal cells; 10 (UCL 1684) was the first non-peptidic nanomolar inhibitor13 (IC50 = 3 nM) and further developments have yielded an interesting series of bisalkane quinolinium cyclophanes, typified by 11 (UCL 1848) (IC50 = 2 nM).14
Interest in a persistent sunscreen led us to combine known protective chromophores with a group that would react with the keratin cysteine residues in skin. Thus (12) is a Michael base which would accept a thiol group and form a covalent bond, and hence it would bind and it also possesses a UV and visible light absorbing chromophore based on 4-methoxybenzophenone.15
References
- A. Albert, S. D. Rubbo, R. J. Goldacre, M. E. Davey and J. D. Stone, Br. J. Exp. Pathol., 1945, 26, 16 Search PubMed . See also A. Albert, Selective Toxicity, The Physico-chemical Basis of Therapy, 7th edn., Chapman and Hall, London, 1985 Search PubMed.
- J.-M. Arrang, M. Garbarg, J.-C. Lancelot, J.-M. Lecomte, H. Pollard, M. Robba, W. Schunack and J.-C. Schwartz, Nature (London), 1987, 327, 117 CrossRef CAS.
- C. R. Ganellin, S. K. Hosseini, Y. S. Khalaf, W. Tertiuk, J. M. Arrang, M. Garbarg, X. Ligneau and J.-C. Schwartz, J. Med. Chem., 1995, 38, 3342 CrossRef CAS.
- C. R. Ganellin, A. Fkyerat, B. Bang-Andersen, S. Athmani, W. Tertiuk, M. Garbarg, X. Ligneau and J.-C. Schwartz, J. Med. Chem., 1996, 39, 3806 CrossRef CAS.
- C. R. Ganellin, F. Leurquin, A. Piripitsi, J.-M. Arrang, M. Garbarg, X. Ligneau, W. Schunack and J.-C. Schwartz, Arch. Pharm. Pharm. Med. Chem., 1998, 331, 395 Search PubMed.
- K. Y. Wong, PhD Thesis, University of London, 1997. Design and Synthesis of 5-HT1A Receptor Ligands Search PubMed.
- C. R. Ganellin, P. B. Bishop, R. B. Bambal, S. M. T. Chan, J. K. Law, B. Marabout, P. Mehta Luthra, A. N. J. Moore, O. Peschard, P. Bourgeat, C. Rose, F. Vargas and J.-C. Schwartz, J. Med. Chem., 2000, 43, 664 CrossRef CAS.
- C. Rose, F. Vargas, P. Facchinetti, P. Bourgeat, R. B. Bambal, P. B. Bishop, S. M. T. Chan, A. N. J. Moore, C. R. Ganellin and J.-C. Schwartz, Nature (London), 1996, 380, 403 CrossRef CAS.
- C. Rose, J.-C. Schwartz, F. Vargas, Y. Chen, C. R. Ganellin, S. Samad and L. Zhao, Tripeptidylpeptidase Inhibitors. Patent PCT WO 99/33801 (1999) .
- D. Galanakis, J. A. D. Calder, C. R. Ganellin, C. S. Owen and P. M. Dunn, J. Med. Chem., 1995, 38, 3536 CrossRef CAS.
- D. Galanakis, C. A. Davis, C. R. Ganellin and P. M. Dunn, J. Med. Chem., 1996, 39, 359 CrossRef CAS.
- (a) J. Campos Rosa, D. Galanakis, C. R. Ganellin and P. M. Dunn, J. Med. Chem., 1996, 39, 4247 CrossRef CAS; (b) P. M. Dunn, D. C. H Benton, J. Campos Rosa, C. R. Ganellin and D. H. Jenkinson, Br. J. Pharmacol., 1996, 117, 35 CAS.
- J. Campos Rosa, D. Galanakis, C. R. Ganellin, P. M. Dunn and D. H. Jenkinson, J. Med. Chem., 1998, 41, 2 CrossRef.
- J.-Q. Chen, D. Galanakis, C. R. Ganellin, M. Dunn and D. H. Jenkinson, J. Med. Chem., 2000, 43, 3478 CrossRef CAS.
- B. E. Davison, B. C. N. M. Jones, C. R. Ganellin, P. B. Bishop and D. Jack, Compounds containing a Michael acceptor linked to a chromophore and their use in long lasting sunscreen compositions. Patent PCT WO 95/30646 (1995) .
Footnote |
| † Department of Chemistry, University College London, Christopher Ingold Laboratories, 20 Gordon Street, London, UK WC1H 0AJ. E-mail: c.r.ganellin@ucl.ac.uk |
| This journal is © The Royal Society of Chemistry 2001 |