
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Observation of super-Nernstian proton-coupled electron transfer and elucidation of nature of charge carriers in a multiredox conjugated polymer†
Duyen K.
Tran
 a,
Sarah M.
West
b,
Elizabeth M. K.
Speck
b and
Samson A.
Jenekhe
*ab
a,
Sarah M.
West
b,
Elizabeth M. K.
Speck
b and
Samson A.
Jenekhe
*ab
aDepartment of Chemical Engineering, University of Washington, Seattle, Washington 98195-1750, USA. E-mail: jenekhe@uw.edu
bDepartment of Chemistry, University of Washington, Seattle, Washington 98195-1750, USA
First published on 22nd April 2024
Abstract
Nernstian proton-coupled electron transfer (PCET) is a fundamental process central to many physical and biological systems, such as electrocatalysis, enzyme operation, DNA biosynthesis, pH-/bio-sensors, and electrochemical energy storage devices. We report herein the discovery of super-Nernstian PCET behavior with two protons per electron transferred in the electrochemical doping of a redox conjugated polymer, phenazine-substituted ladder poly(benzimidazobenzophenanthroline) (BBL-P), in aqueous electrolyte. We show that the super-Nernstian response originates from existence of multiredox centers that have a gradient of pKa on the conjugated polymer. Our use of various pH-dependent in operando techniques to probe the nature of charge carriers in n-doped BBL-P found that polarons are the charge carriers at low to intermediate levels of doping (0.1–1.0 electron per repeat unit (eru)) whereas at higher doing levels (1.3 eru), polarons, polaron pairs, and bipolarons co-exist, which evolve into strongly coupled polaron pairs at the highest doping levels (>1.5 eru). We show that PCET-assisted n-doping of BBL-P results in very high redox capacity (>1200 F cm−3) in acidic electrolyte. Our results provide important new insights into PCET in organic materials and the nature of charge carriers in n-doped conjugated polymers while having implications for various electrochemical devices.
Introduction
Proton-coupled electron transfer (PCET) is a fundamental process that lies at the heart of many physical and biological systems1–6 such as photosynthesis and respiration,3–5 DNA biosynthesis,3 enzyme operation,5,6 water splitting for fuel cells,4 nitrogen fixation,3 chemical sensors,5 electrochemical devices,5etc. Compared to the sole electron transfer, PCET has been shown to be a more thermodynamically favorable pathway as it effectively lowers the activation energy barriers between the reactants and the products.7 The majority of reported PCET processes feature stoichiometric proton/electron ratios as evidenced by a Nernstian shift (59.2 mV pH−1) in the Pourbaix diagram.4,8 Very few exceptions from the stoichiometric proton/electron ratios, or deviation from Nernstian behavior, have been observed.4,9–14 In particular, hydrated inorganic oxides with redox-active transition metal centers (e.g. nickel-borate, iridium, ruthenium) can afford a super-Nernstian shift (70–120 mV pH−1),9–12 whereby the additional proton uptake can be attributed to the presence of water molecules since thermal annealing of such oxides to rigorously remove excess moisture resulted in a conventional Nernstian shift.10 In addition to inorganic oxides, biological molecules such as iron-coordinating histidine based metalloprotein have also produced super-Nernstian shift4,13,14 due to its unique molecular structure which features separate electron sinks (i.e. ferrous/ferric redox centers) and proton sinks (i.e. histidine groups). These observed cases of multi-proton coupled electron transfers all require a transition metal center, which means that super-Nernstian PCET has yet to be discovered in an all-organic material. Fundamental questions, such as is it possible to achieve super-Nernstian PCET without metal centers? What are the structural features that could facilitate super-Nernstian PCET in organic materials? remain to be explored with potential implications for ultrasensitive pH sensing, enhanced electrochemical proton storage, proton batteries, and single-phase dual proton–electron conduction catalyst, among others.Redox π-conjugated polymers are an ideal platform to address the above research questions. Unlike nonconjugated redox polymers which typically contain pendant redox groups (e.g. quinone, imides, ketones, radicals, etc.),15 the class of redox π-conjugated polymers contain redox-active sites within the polymer backbone; thus, combining the robust charging/discharging nature of redox sites with the enhanced electronic charge delocalization and transport along the conjugated polymer backbone.16–22 Although traditional redox-active π-conjugated polymers are usually conducting polymers such as polyaniline (PANi), polypyrrole (PPy), or poly(3,4-ethylenedioxythiophene) (PEDOT) (Chart 1a)23 or metallopolymers24 (Chart 1a), current state-of-the-art redox π-conjugated polymers incorporate molecular redox sites into the backbone such as carbonyl group (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O–) containing polymers including naphthalene diimide (NDI) based copolymers16–19 (Chart 1a) and diketopyrrolopyrrole (DPP) based copolymers (Chart 1a).21,22 Although a detailed measurement and analysis of the Pourbaix diagram was absent in these previously reported redox π-conjugated polymers, they were assumed to follow the Nernstian response.16–22
O–) containing polymers including naphthalene diimide (NDI) based copolymers16–19 (Chart 1a) and diketopyrrolopyrrole (DPP) based copolymers (Chart 1a).21,22 Although a detailed measurement and analysis of the Pourbaix diagram was absent in these previously reported redox π-conjugated polymers, they were assumed to follow the Nernstian response.16–22
 | ||
| Chart 1 (a) Molecular structures of some previously reported redox π-conjugated polymers;16,18,22,23,26,27 (b) molecular structure of a multiredox π-conjugated polymer, BBL-P. | ||
The ability to store charges with excellent stability and robust reversibility in electrochemically n-doped redox π-conjugated polymers16,25 could also enable fundamental studies to explore the nature of charge carriers (e.g. electron-polarons, electron-bipolarons, etc.) in the materials. In fact, current understanding of the structure–electrochemical property-device performance relationships remain modest for n-type π-conjugated polymers, due in part to the polymer backbone unable to stabilize large amounts of injected electrons upon doping.16 As a result, there still exists a large knowledge gap regarding the electrochemical reactivity of the polymers and how tuning of such a material property influences the performance of electrochemical devices.
Here, we report use of a novel multiredox π-conjugated polymer, BBL-P (Chart 1b) to simultaneously investigate the PCET process and the nature of charge carriers in electrochemically n-doped BBL-P in aqueous electrolyte. The highly planar rigid-rod backbone of BBL-P facilitates large delocalization length that could stabilize the charged species upon formation, and the six total redox-active sites, consisting of two carbonyl groups and four imine sites (Chart 1b) can also facilitate multiple reversible and rapid electron injections. We hypothesize that the multiple redox sites of different pKa values could enable a super-Nernstian PCET response when BBL-P is electrochemically reduced in aqueous electrolyte. Furthermore, a high doping level could be achieved, which will aid in distinguishing whether polaron pairs or bipolarons are the multiply charged species in heavily doped BBL-P. Furthermore, the more disordered nature of BBL-P28 will help to stabilize the formation of multiply charged species,29,30 which otherwise would be harder in the more crystalline parent polymer, BBL (Chart 1a).31,32
We found that BBL-P undergoes three acid-base coupled redox reactions corresponding to the different molecular redox sites with decreasing pKa from naphthalene imine to phenazine imine to carbonyl oxygen. Each redox reaction is discovered to exhibit a super-Nernstian shift, involving two protons per one electron transferred, which is unusual and deviates from the commonly observed stoichiometric proton/electron ratio seen in most PCET processes.7,8,33 We similarly found that the electrochemical reduction of the parent polymer BBL (Chart 1a) thin films also show a super-Nernstian behavior albeit only 1.5 protons are coupled to every electron transferred. The observed super-Nernstian behavior is shown to originate from the multiredox centers with varying pKa in the conjugated polymer backbone. By using various pH-dependent in operando techniques to probe the nature of charge carriers in electrochemically n-doped BBL-P, we show the evolution of polarons at low to intermediate levels of doping (0.1–1.0 eru) to an equilibrium mixture of polarons, polaron pairs, and bipolarons at a higher level of doping (1.3 eru) and finally to polaron pairs at the highest level of doping (≥1.5 eru). Finally, we show that PCET-assisted n-doping of BBL-P in acidic electrolyte leads to a very high redox capacity (>1200 F cm−3).
Experimental methods
Chemicals
The phenazine-substituted poly(benzimidazobenzophenanthroline) ladder polymer, BBL-P, was synthesized according to our previously reported procedure.28 Methanesulfonic acid (MSA) was purchased from Sigma-Aldrich and used as received.Preparation of BBL-P films on electrodes
The BBL-P sample of 1.1 dL g−1 intrinsic viscosity was dissolved in methanesulfonic acid (MSA; 15 mg mL−1) and stirred at elevated temperature (>100 °C) for several days. The polymer solution was filtered through 1 μm pore size Grade GF/B Glass Microfiber Filters (Whatman GF/B w/GMF) immediately before use. The filtered polymer solution was spin-coated at 1500 rpm for 30 seconds onto ITO substrates for cyclic voltammetry, onto FTO substrates for spectroelectrochemistry, and onto gold coated glass substrates for in operando Raman spectroscopy. The acidic solvent was immediately removed by immersing the polymer films into a mixture of isopropanol (IPA) and ethylene glycol (EG) (IPA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EG = 1:1, v:v) several times over 12 hours. The polymer films were then dried in a vacuum oven at 90 °C overnight and then annealed at 170 °C on a hot plate for 10 minutes in ambient conditions.
:EG = 1:1, v:v) several times over 12 hours. The polymer films were then dried in a vacuum oven at 90 °C overnight and then annealed at 170 °C on a hot plate for 10 minutes in ambient conditions.
Cyclic voltammetry
Cyclic voltammetry was performed in conventional three-electrode cells with Ag/AgCl pellet as the reference electrode (World Precision Instrument) and Pt mesh as the counter electrode. Cyclic voltammetry measurements were performed at room temperature using EG&G Princeton Applied Research, model 273A potentiostat. The BBL-P film prepared as described above served as the working electrode. The supporting electrolyte consisted of 0.1 M KCl in deionized (DI) water, and its pH values were tuned by adding either KOH or HCl such that the final solution always contained at least 0.1 M KCl. The 0.1 M KCl(aq) electrolyte was purged with a nitrogen stream for at least 15 minutes before any measurements. A stream of N2 was also constantly flown on top of the electrolyte surface during measurements to minimize oxygen diffusion.Optical absorption and spectroelectrochemistry
Optical absorption spectra of the BBL-P films were taken on a PerkinElmer Lambda 900 spectrometer. For spectroelectrochemistry, BBL-P thin film was coated on FTO substrates which were inserted into a cuvette filled with 0.1 M KCl(aq) as the electrolyte. The polymer film area was 2.75 ± 0.18 cm2, and the polymer film thickness was 44.4 ± 8.8 nm. Three-electrode configuration containing Ag/AgCl pellet as the reference electrode, Pt mesh as the counter electrode, and FTO/BBL-P as the working electrode was used. A Metrohm Autolab PGSTAT302N potentiostat was used to control the potential via the Metrohm NOVA software (Version 2.1.6). The BBL-P working electrodes were biased at different potentials for 60 s for doping to be equilibrated before collecting the optical absorption spectra. The films were thoroughly de-doped by applying +0.5 V (vs. Ag/Ag+) between each doping cycle. Both the doping and the de-doping currents were simultaneously collected during the optical measurements for coulometry analysis. The electrolyte was degassed by purging with N2 stream for at least 20 min prior to measurements.In operando Raman spectroscopy
Raman spectra of BBL-P thin films were collected using a Thermo Scientific DXR2 Raman microscope. A 532 nm laser with a power of 2 mW was focused onto BBL-P film surface through a 10× objective lens. In operando Raman spectra were taken at different bias potentials and different pH values. A three-electrode cell consisting of BBL-P on gold coated glass substrate as the working electrode, Pt mesh as the counter electrode, and Ag/AgCl pellet as the reference electrode, and a Metrohm Autolab PGSTAT302N potentiostat was used to control the potential via the Metrohm NOVA software (Version 2.1.6).Results and discussion
pH-dependent cyclic voltammetry of BBL-P in aqueous electrolyte
Cyclic voltammetry (CV) was performed to investigate the electrochemical properties of BBL-P in an aqueous environment. A three-electrode electrochemical cell, consisting of a Ag/AgCl pellet reference electrode, Pt mesh as the counter electrode, and BBL-P thin film coated on a conducting ITO substrate as the working electrode, was used. The aqueous electrolyte always contained at least 0.1 M KCl(aq), and the pH of the electrolyte solution was varied by adding either hydrochloric acid (HCl) or potassium hydroxide (KOH). Additionally, acetonitrile was also titrated to the electrolyte at 10 vol% to poison the Pt electrode and suppress any hydrogen evolution reaction. The applied potentials were also kept within the stability window of water as a function of pH to ensure that no undesirable side reactions took place. The final electrolyte solution was de-oxygenated by purging with nitrogen (N2) for 20 minutes before any measurements, and a constant N2 stream was also maintained atop of the electrolyte surface to ensure an oxygen-free environment during the measurements. Cyclic voltammograms of BBL-P thin films with varying electrolyte's pH from pH = 4.11 to pH = 0.06 are shown in Fig. 1. Based on the number of observable features in the voltammograms, the pH-dependent electrochemical properties of BBL-P can be categorized into three separate regions: | ||
| Fig. 1 pH-dependent cyclic voltammograms of BBL-P thin films collected in 0.1 M KCl(aq) supporting electrolyte with a scan rate of 25 mV s−1 by using a 3-electrode configuration with ITO/BBL-P as the working electrode, Pt mesh as the counter electrode, and Ag/AgCl as the reference electrode: (a) pH = 4.11; (b) pH = 3.61; (c) pH = 3.13; (d) pH = 2.51; (e) pH = 2.14; (f) pH = 1.56; (g) pH = 1.07; (h) pH = 0.70; (i) pH = 0.29; and (j) pH = 0.06. | ||
Proton-coupled electron transfer (PCET) reactions in BBL-P
The above pH-dependent CV results suggest that BBL-P can undergo two acid-base equilibria in aqueous KCl solution with an average pKa of 2.5 and 1, which correspond respectively to the protonation at the naphthalene imine site (HBBL-P+) and the phenazine imine site (HBBL-P+’). Indeed, the parent BBL polymer has been shown to exhibit a pKa around 2.2 for the naphthalene imine site in aqueous tetrabutylammonium sulfate,26 whereas phenazine small molecules are known to undergo protonation with a pKa around 1.2.35,36Quantitative analysis of the pH-dependent formal potentials of each redox event via Pourbaix diagram (Fig. 2) revealed average slopes of −99.3 ± 0.8, −116.3 ± 1.1, and −109.6 ± 6.8 mV pH−1 for the first, second, and third redox reaction, respectively (Table 1). These results (|slope| > 59.2 mV pH−1) imply that BBL-P exhibits a super-Nernstian PCET behavior, where each redox reaction of BBL-P involves on average two protons per one electron (2H+/1e−). The numbers of protons per electrons (nH+/ne−) involved in each reduction event of BBL-P were also found to be independent of scan rates (Table 1). The observed super-Nernstian (|slope| > 59.2 mV pH−1) behavior of BBL-P, which deviates from the commonly observed one-proton/one-electron redox process, is very intriguing and worth further investigation. We conducted a similar experiment on the parent polymer BBL thin films, and the resulting cyclic voltammograms in aqueous electrolyte at various pH values are shown in Fig. S3.† Quantitative analysis of the Pourbaix diagram of BBL thin films (Fig. S4†) revealed average slopes of −89.2 ± 5.2 for the first redox process and −99.8 ± 3.9 mV pH−1 for the second redox process (Table S1†). These results thus suggest that BBL also exhibits a super-Nernstian behavior, featuring on average 1.5–1.7 protons per electron transferred in each reduction reaction. We note that in a previous attempt to understand the electrochemicaly doping reactions of BBL in aqueous electrolyte, a slightly smaller absolute value of slope of 78 mV pH−1 was observed, which corresponds to a nH+/ne− value of about 1.3.26 However, in that prior report, the process was thought to follow Nernstian response to the first approximation;26 thus, overlooking any possibility of multi-proton coupled electron transfer. The observed higher nH+/ne− of BBL thin film in our current study, which can be rationalized by subtle differences in film thickness, electrolytes, and polymer molecular weight, challenges the previously proposed PCET model of BBL.
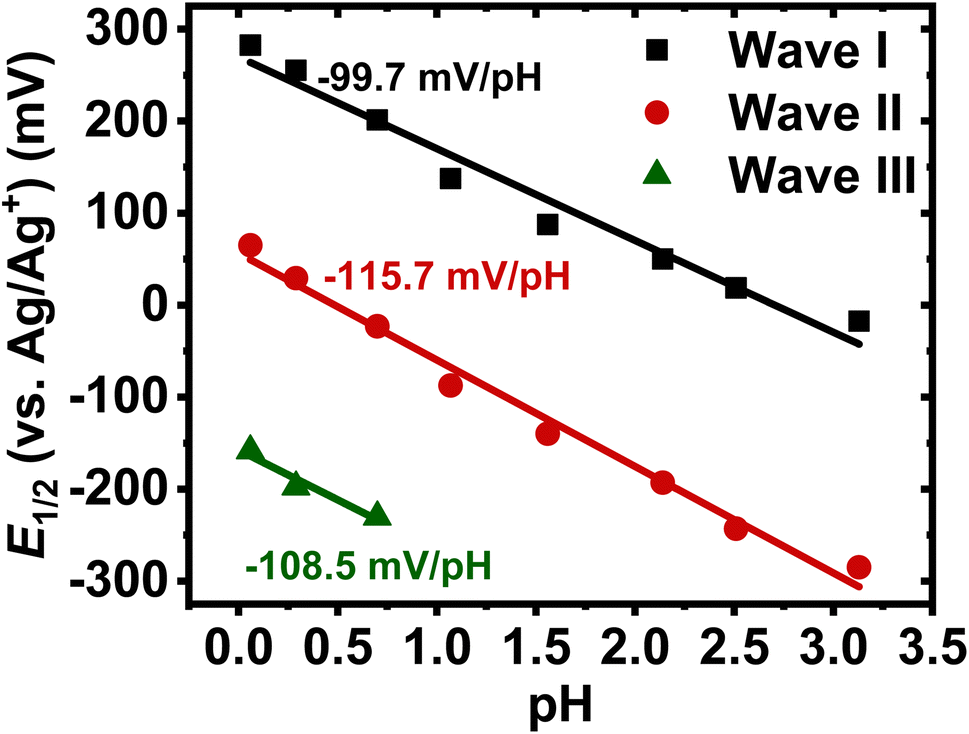 | ||
| Fig. 2 Representative Pourbaix diagram of BBL-P thin films in 0.1 M(aq) KCl at a scan rate of 25 mV s−1. | ||
| Scan rate (mV s−1) | Wave | Slopea (mV pH−1) | n H+/ne−b |
|---|---|---|---|
| a Slope, average slope calculated from Pourbaix diagram (+/− one standard deviation). b n H+/ne−, number of protons per electrons transferred in each reduction event of BBL-P (+/− one standard deviation). Average values and standard deviations were calculated from 5 different samples. | |||
| 25 | I | −99.3 ± 0.8 | 1.7 ± 0.01 |
| 25 | II | −116.3 ± 1.1 | 2.0 ± 0.02 |
| 25 | III | −109.6 ± 6.8 | 2.0 ± 0.10 |
| 50 | I | −97.8 ± 1.1 | 1.7 ± 0.02 |
| 50 | II | −115.1 ± 1.5 | 1.9 ± 0.02 |
| 50 | III | −116.9 ± 4.1 | 2.0 ± 0.10 |
| 100 | I | −97.3 ± 5.1 | 1.6 ± 0.10 |
| 100 | II | −113.3 ± 0.7 | 1.9 ± 0.01 |
| 100 | III | −119.3 ± 8.1 | 2.0 ± 0.14 |
| 150 | I | −100.1 ± 13.9 | 1.7 ± 0.21 |
| 150 | II | −116.1 ± 8.0 | 2.0 ± 0.02 |
| 150 | III | −119.5 ± 3.2 | 2.0 ± 0.1 |
Our results show that BBL-P as well as its parent ladder polymer BBL exhibit super-Nernstian PCET processes upon electrochemical doping, which is very interesting and has yet to be reported for redox polymers.15,25,37 We hypothesize that the super-Nernstian PCET response seen in BBL-P originates from its unique molecular structure, which features multiple protonatable redox sites of different pKa values in the polymer backbone. In particular, upon reduction of BBL-P at the carbonyl position, the electron density around both the naphthalene imine and the phenazine imine sites would increase owing to strong delocalization of π-electrons. As a result, the pKa values of both imine redox sites would be higher in the reduced states than those in the neutral state, which means that they are more susceptible to protonation with consequent observed super-Nernstian PCET behavior. It is important to note that small shifts of the pKa values of multiple protonatable sites in a molecular system is possible and has been previously reported in biologically relevant molecules, including Rieske iron-sulfur “charged ladder” metalloproteins.38
To confirm our hypothesis and arrive at a comprehensive mechanism underlying the observed super-Nernstian PCET-assisted electrochemical doping in BBL-P, we examined the thermodynamics and kinetics of each reduction process by density functional theory (DFT) calculations and variable scan rate cyclic voltammetry. Results from these analyses will provide macroscopic insights on the feasibility of the proposed mechanism. Furthermore, a combination of characterization techniques such as spectroelectrochemistry, coulometry, and in operando Raman spectroscopy were used to probe the evolution of optoelectronic properties, doping level, and molecular structure, all of which are discussed in detailed below to provide molecular insights into the electrochemical reduction processes. We propose that the extra proton involved in each electrochemical event of BBL-P is associated with the imine sites either on the naphthalene ring or on the phenazine unit, and the proposed multiple electrochemical reduction reactions of BBL-P in aqueous acidic electrolyte is shown in Scheme 1.
 | ||
| Scheme 1 Proposed electrochemical reduction reactions of BBL-P in aqueous KCl electrolyte. | ||
In the case of 3.1 < pH ≤ 8.1, the single redox wave (wave I) can be ascribed to the PCET reaction of BBL-P at the carbonyl oxygen as demonstrated in eqn 1A or 1B. As previously discussed, two pairs of well-resolved redox waves (wave I and wave II) were observed when 1 < pH < 3.1 (Fig. 1c–g). This result means that the reduced products of both reactions coexist in equilibrium whereby wave I corresponds to eqn 1A or 1B (Scheme 1), and wave II corresponds to the PCET reaction of BBL-P at the naphthalene imine site as shown in eqn 2 (Scheme 1). Similarly, when the pH of the electrolyte is less than 1, three pairs of pronounced and quasi-reversible redox waves, namely wave I, wave II, and wave III, were observed (Fig. 1h–g). In this case, three different reduced states of BBL-P coexist in equilibrium whereby wave I corresponds to eqn 1A or 1B (Scheme 1), wave II corresponds to eqn 2 (Scheme 1), and wave III corresponds to the reduction of BBL-P at the phenazine imine sites as demonstrated in eqn 3 (Scheme 1).
We emphasize that each reduction reaction is independent of one another and that the three redox waves are unlikely to be the sequential reduction reactions of BBL-P mainly due to the large energetic barrier required to inject additional electrons into the polymers upon the initial reduction. This phenomenon is in fact commonly observed for various organic redox molecules (M) where the potential of the second reduction reaction (Ṁ− → M2−) are more negative than that of the first reduction reaction (M → Ṁ−) (Table S2†), and the potential difference (ΔE) are relatively large of around 0.4–0.7 V (Table S2†). In our case, the ΔE values were approximately 0.2–0.3 V, which is much smaller than the commonly observed ΔE of redox small molecules and polymers. Therefore, we assigned each reduction wave to an independent reduction reaction of BBL-P at different redox sites as outlined in Scheme 1.
Thermodynamics of electrochemical doping reactions of BBL-P
We first examined the feasibility of the proposed electrochemical redox reactions of BBL-P from a thermodynamics perspective. Density functional theory (ωB97XD/6-31G (d,p)) of the dimers of the proposed structures (1–6) were performed to gain insights into the polymer chain conformations as well as their free energy. The optimized geometries of 1, 2, 4, and 6 structures all exhibited completely planar chain conformation (Fig. S5†), suggesting that the formation of polaron would not introduce any detrimental torsional defects into the rigid-rod polymer backbone.A quantitative comparison of the driving force for each of the reduction reaction is presented in Fig. S6.† The Gibbs free energy change (ΔG) of all three reactions was found to be negative indicating that the reduced products (2, 4, and 6) are thermodynamically favorable. Furthermore, the ΔG value was largest for the 1 → 2 reaction (ΔG12 = −84 eV), which means that the conversion from neutral BBL-P to its reduced unprotonated form (eqn 1) is readily accessible; thus, requiring a low doping potential. The ΔG value of the 1 → 4 reaction was intermediate (ΔG14 = −67 eV), implying that the observed second reduction wave corresponds to the reduction of protonated BBL-P at the naphthalene imine sites (eqn 2). Moreover, the smallest ΔG value (ΔG = −66 eV) that corresponds to the 1 → 6 conversion suggests that protonated BBL-P at the phenazine imine sites (eqn 3) would be slightly harder to reduce and thus require substantially more negative potentials than reduction of the protonated naphthalene imine. These findings are in excellent agreement with the experimental CV data and have confirmed that the proposed electrochemical reduction reaction scheme of BBL-P is valid from a thermodynamic standpoint.
Furthermore, we examined the DFT computed atomic charge distribution of BBL-P dimers in different protonated and reduced states (Tables S3 and S4†) to test our hypothesis of pKa shifts upon each PCET event. The electron density around both the naphthalene imine and phenazine imine was found to increase upon PCET; thus, making them susceptible to additional protonation. In particular, when the carbonyl oxygen underwent a 1H+/1e− transfer, the electron density at the naphthalene imine sites (N45, N17) as well as the phenazine imine sites (N82, N70) increased (Table S3†). The larger electron density at these sites is indicative of enhanced basicity and higher pKa values. As a result, these imine sites can be readily protonated, leading to the observed non-stoichiometric PCET. Similar phenomenon is also observed when the naphthalene imine nitrogen or phenazine imine nitrogen undergoes PCET-assisted electrochemical doping (Table S4†). For example, when the naphthalene imine undergoes a 1H+/1e− transfer, the electron density at the phenazine imine sites (N81, N82) increased as evidenced by the more negative atomic charge distribution (Table S4†). This result suggests that the pKa of these phenazine imine sites are higher compared to those in the neutral undoped BBL-P; thus, making them more susceptible to spontaneous protonation when 1 < pH < 3.
Kinetics of electrochemical reduction reactions of BBL-P
The kinetics of each electrochemical event of BBL-P was also investigated to quantitatively distinguish between the slow diffusion-limited and the fast surface-controlled processes. Representative cyclic voltammograms of BBL-P with varying scan rate (ν) collected in electrolyte solution of pH ranging from 4.1 to 0.06 are shown in Fig. 3a–j. Notably, the voltametric responses of BBL-P showed marginal changes with increasing scan rate, showing well-resolved cathodic and anodic peaks up to 150 mV s−1 at all pH values. A power law is applied to describe the electrochemical reduction kinetics:| i = avb |
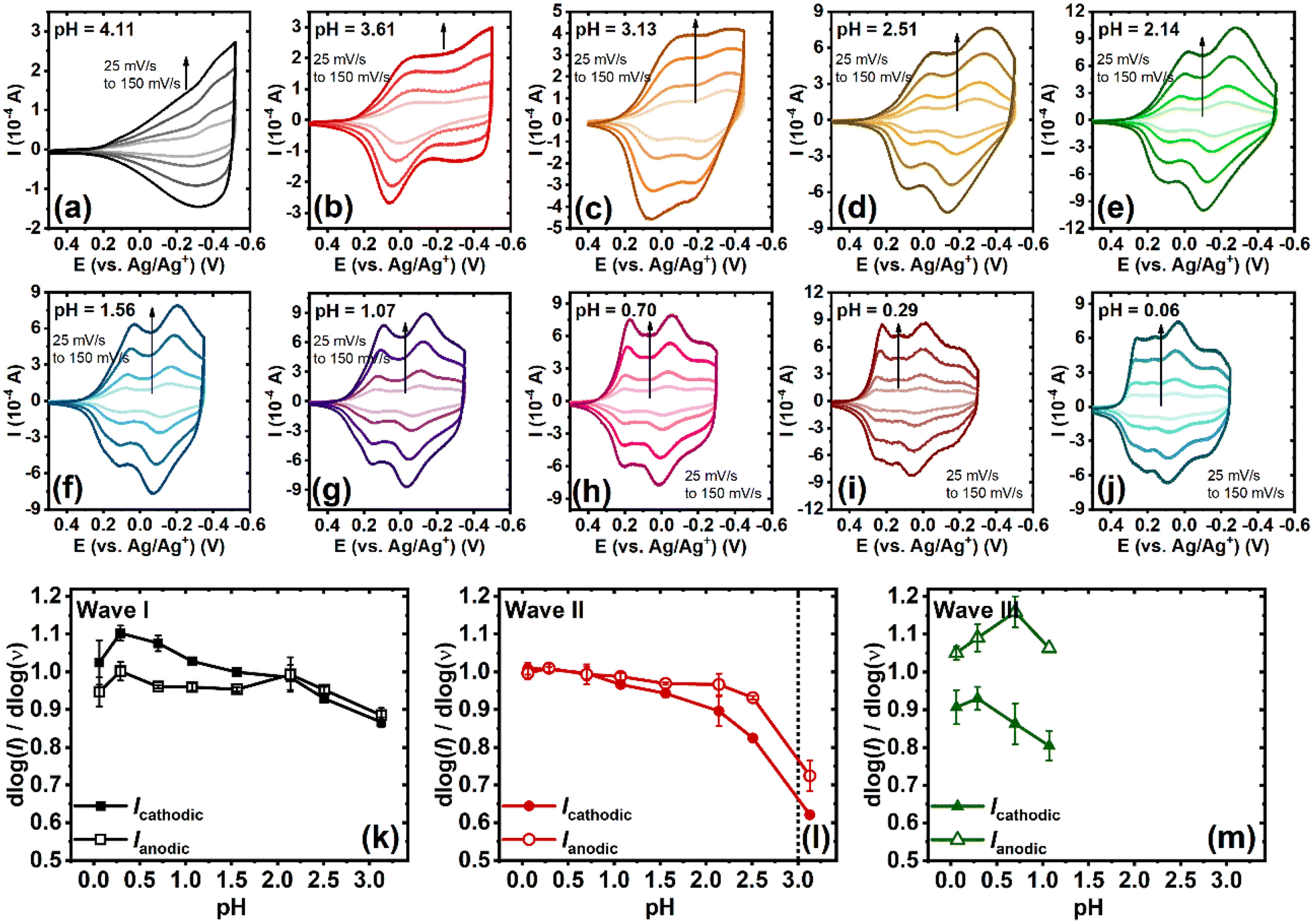 | ||
Fig. 3 (a–j) pH-dependent cyclic voltammograms of BBL-P thin films collected in 0.1 M KCl(aq) supporting electrolyte at various scan rates from 25 mV s−1 to 150 mV s−1. (k–m) Dependence of b-value 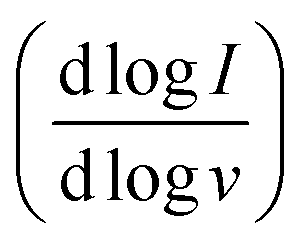 on the electrolyte pH for the first redox event (k), the second redox event (l), and the third redox event (m). on the electrolyte pH for the first redox event (k), the second redox event (l), and the third redox event (m). | ||
The relationship between log(i) and log(ν) with varying electrolyte solution pH are shown in Fig. S7,† whereby the slope of the linear fit represents the b-value. The pH-dependence of b-value for each electrochemical reduction event is presented in Fig. 3k–m. In general, a b-value of 1 indicates a rapid surface-controlled pseudocapacitive electrochemical process whereas a b-value of 0.5 represents a battery-like slow diffusion-limited process.39,40 In the case of the first reduction event, average b-values of both the cathodic peak and the anodic peak were found to gradually increase from b = 0.87–0.89 to near unity (b = 1.00) as the pH values were lowered from 3.1 to 0.06 (Fig. 3k). This result suggests that the reduction of unprotonated BBL-P showed a dominant pseudocapacitive behavior, especially in highly acidic electrolytes. In the case of the second reduction process, the b-value exhibited a steep increase from around 0.6–0.7 to 0.9–1.0 as the solution pH decreased from ∼3 to ∼2.5 (Fig. 3l), which indicates a transition from a slow diffusion-limited process to a fast surface-limited redox process and coincides with the transition from CV region 1 to region 2 discussed earlier. The charge storage mechanism remained pseudocapacitive with decreasing pH down to 0.06 as evidenced by the relatively constant b-value around 1.0 (Fig. 3l). The kinetics of the third reduction process also showed a dominant pseudocapacitive behavior with average b-value around 0.9–1.1 regardless of the electrolyte solution pH (Fig. 3m). These results collectively suggest that the kinetics of electrochemical reduction of BBL-P can be controlled by the electrolyte's pH where protonation of the polymer offers an effective means to overcome the kinetics barrier, leading to rapid reduction processes. Furthermore, the charge storage mechanism of BBL-P follows a pseudocapacitive behavior even at high scan rates, which indicates the remarkable rate capabilities of BBL-P.41 Therefore, ladder polymer BBL-P could be a promising electrode material for future applications in all-polymer proton supercapacitors42–44 or proton batteries.45–48
We have thus far demonstrated that the proposed electrochemical reduction reactions of BBL-P are valid from macroscopic thermodynamic and kinetic perspectives. To further our understanding of the multiple redox states of BBL-P from a molecular level, we employed a complement of in operando characterization techniques as discussed in the following sections.
Spectroelectrochemistry of BBL-P films in aqueous solution
We used spectroelectrochemistry to examine the evolution of optoelectronic properties upon electrochemical reduction as well as to probe the formation of polarons and bipolarons. The optical absorption spectra of undoped BBL-P with varying electrolyte's pH are shown in Fig. S8a.† Even when BBL-P is protonated (pH < 3 and pH < 1), the spectral shapes remain relatively constant with a main absorption feature centered at around 490–495 nm and a poorly resolved shoulder at around 630 nm. This result suggests that neither the electronic structures nor the polymer chain conformation were affected upon protonation, which is a unique characteristic of conjugated rigid-rod ladder polymers.49Spectroelectrochemistry of BBL-P was performed in a three-electrode configuration with BBL-P coated on conducting transparent substrate as the working electrode, Pt mesh as the counter electrode, and Ag/AgCl pellet as the reference electrode. The optical properties of BBL-P thin films at various doping potentials collected in aqueous KCl electrolyte solution of pH 0.16, 1.04, and 2.46 are shown in Fig. 4, S9, and S10,† respectively. The points on the voltammograms (Fig. 4a, S9a, and S10a†) indicate the potentials at which the films were doped before collecting the spectra. The current was monitored to ensure that the polymer doping has reached a steady state before optical measurements took place. We note that the polymer films were thoroughly de-doped at +0.50 V (vs. Ag/Ag+) between each doping step. In each case, the potential was applied from negative to positive direction. The differential absorption spectra of BBL-P thin films as a function of doping potential and solution pH are also shown in Fig. 4d, e, S9d, e, and S10d, e.† Since results obtained in electrolyte solution of pH 1.04 and 2.46 (Fig. S9 and S10†) are essentially snapshots of those collected in extremely low pH region, we will focus our discussion on the spectroelectrochemistry of BBL-P in highly acidic electrolyte solution (pH < 1) where all three redox reactions coexist in equilibrium (eqn 1–3).
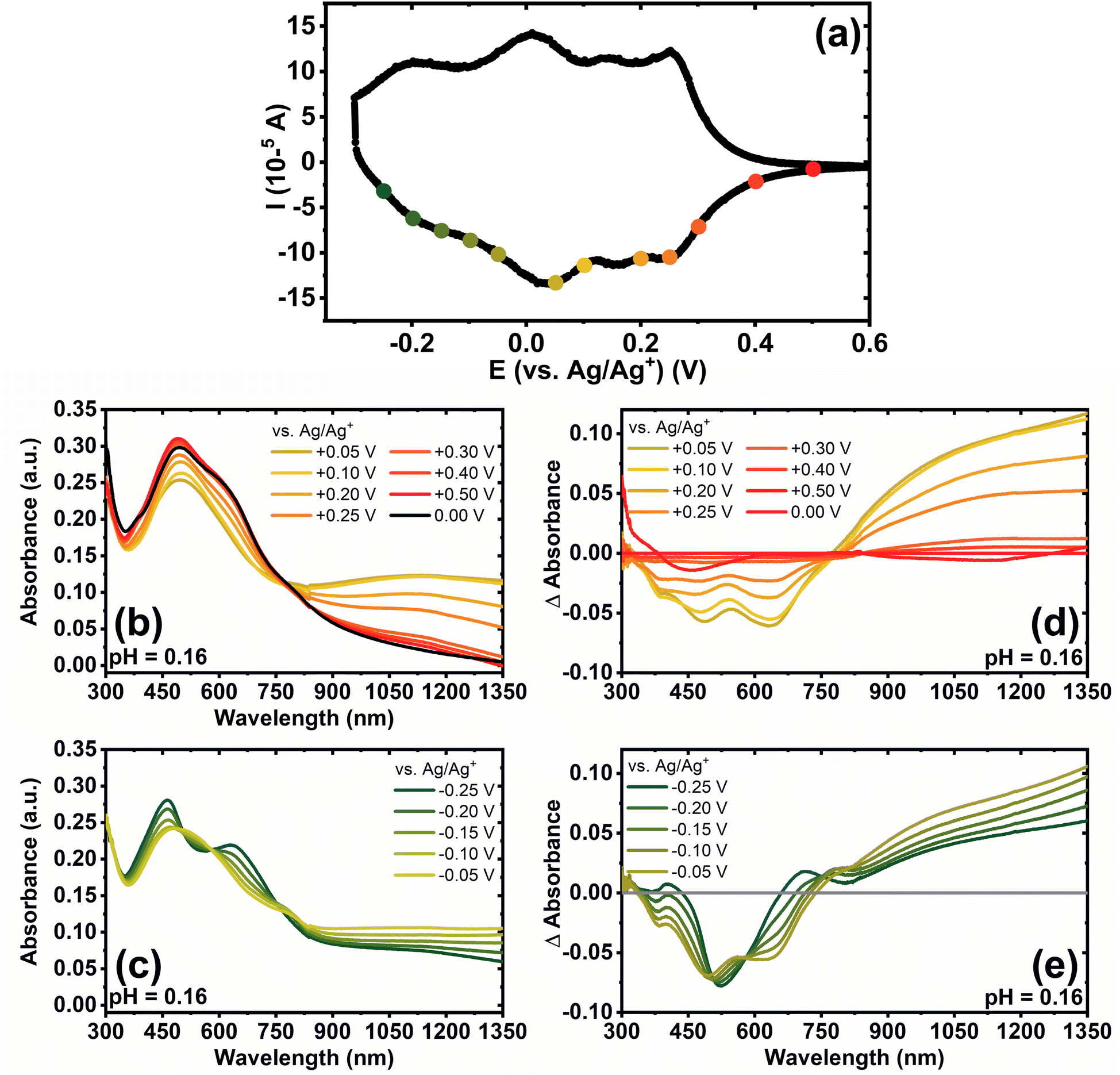 | ||
| Fig. 4 (a) Cyclic voltammogram of BBL-P thin films in aqueous KCl electrolyte of pH 0.29 collected at a scan rate of 25 mV s−1; (b and c) UV-Vis-NIR optical absorption spectra under different potentials of BBL-P thin films in electrolyte of pH = 0.16; (d and e) differential UV-Vis-NIR optical absorption spectra under different potentials of BBL-P thin films. The baseline spectrum was taken at +0.50 V to de-dope the films prior to each doping step. Note that spectrum labeled 0 V corresponds to absorption of the polymer films when submerged in the electrolyte without any applied potential. | ||
The spectral responses of BBL-P in highly acidic electrolyte (pH = 0.16) can be divided into two potential ranges as illustrated in Fig. 4b and c. As the applied potential approached the first redox peak at around +0.25 V (vs. Ag/Ag+) (Fig. 4a) from +0.50 V, the π–π* transition of the ground state (λmax = 496 nm) decreased in intensity accompanied by a new broad absorption band in the near infrared (near-IR) region centered at 1115 nm (Fig. 4b and d), which indicates the formation of polarons. As the applied potential was swept towards the second redox event (E = +0.05 V, vs. Ag/Ag+), a combination of progressive increase of the near-IR absorption band (λ = 1115 nm) and gradual bleaching of the visible absorption band (λ = 496 nm) was observed (Fig. 4b and d), which means that the density of polarons increased with increasing doping level. Within this potential range from +0.50 V to +0.05 V (vs. Ag/Ag+), only one isosbestic point at around 770 nm (Fig. 4b and d) can be observed, indicating the direct conversion of neutral to the singly charged state of BBL-P. Furthermore, the density of polarons was found to be maximized at the second redox event (E = +0.05 V, vs. Ag/Ag+) as evidenced by the highest intensity of the near-IR band (Fig. 4d and S11a†). The differential absorption spectra also suggests that the density of polarons at the first redox event is about 2–2.5 times lower than that of the second redox event (Fig. S11a†).
As the applied potential approached the third redox event (E < 0 V, vs. Ag/Ag+), the π–π* transition of the ground state (λmax = 496 nm) was significantly bleached giving rise to two sharp absorption bands centered at 461 nm and 640 nm (Fig. S11b†). The intensity of the near-IR absorption band (λ = 1115 nm) was also found to decrease from its maximum (Fig. S11a†), which suggests the transition of singly charged polarons to multiply charged species. Indeed, three isosbestic points at 505 nm, 580 nm, and 755 nm (Fig. 4c and e) can be seen indicating the conversion of singly charged to multiply charged states of BBL-P. Thus, the results show that the visible absorption bands at 461 nm and 640 nm can be assigned to the absorption of multiply charged BBL-P whereas the near-IR absorption band at 1115 nm is associated with the singly charged BBL-P. The extremely broad absorption features of BBL-P seen at applied potentials of −0.15 V to −0.05 V (vs. Ag/Ag+) (Fig. 4c) can be explained by the coexistence of both singly and multiply charged states, leading to an overlapping of several absorption features; thus, distorting the optical absorption spectra. As a result, both singly charged as well as multiply charged species can be charge carriers in the third redox event of BBL-P (E1/2 = −0.17 V, vs. Ag/Ag+).
Coulometry of BBL-P films as a function of pH
Next, we quantified the doping level by calculating the number of electrons per repeat unit (eru) via coulometry assay, which would also enable us to also verify the spectroelectrochemistry results. To avoid the effects of oxygen reduction reaction (ORR) and overestimation of charge integration, we used the de-doping current to calculate the electron density in the reduced BBL-P films after the optical absorption measurements.50 The number of electrons per repeat unit was calculated by normalizing the total charge generated in the reduced BBL-P films obtained by integration of the de-doping current to the number of monomers present on the electrode by following a previously reported method.51 The pH-dependence of the doping level is shown in Fig. 5a.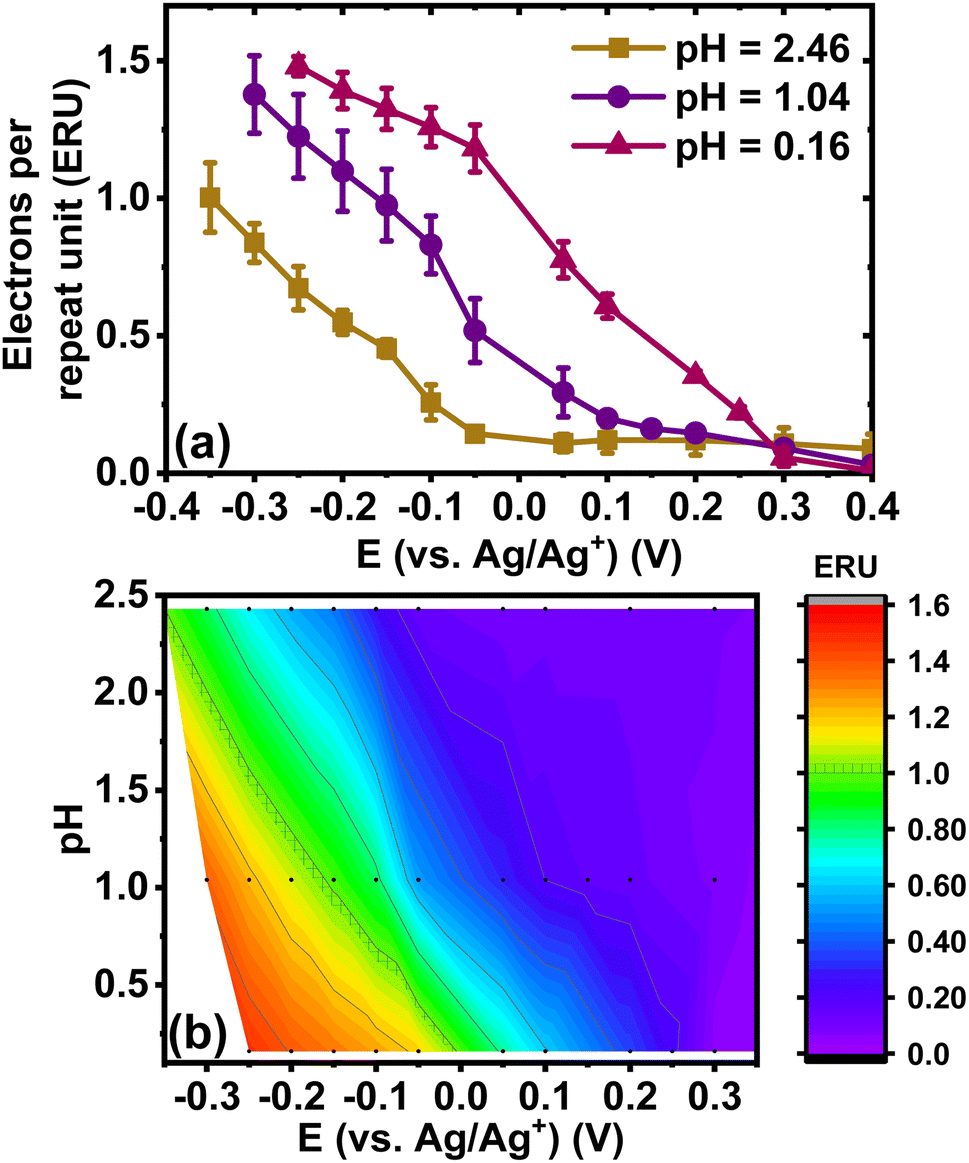 | ||
| Fig. 5 (a) Evolution of doping level (number of electrons per repeat unit) as a function of doping potential. (b) Dependence of doping level on electrolyte pH and doping potentials. | ||
In highly acidic electrolyte (pH = 0.16), the first reduction event (E1/2 = +0.25 V, vs. Ag/Ag+) resulted in a relatively low electron density with an average eru of 0.22 ± 0.02. This means that about one electron is produced per four to five repeat units of reduced unprotonated BBL-P. Further decrease of doping potential led to gradual increase of the electron density (Fig. 5a) showing an average doping level of 0.78 ± 0.07 eru upon the second reduction event; thus, suggesting that approximately three to four electrons are injected per four repeat units of reduced protonated BBL-P at the naphthalene imine sites. These findings indeed corroborate the spectroelectrochemistry results indicating that the electron density of the second redox process is about 3-fold higher than that of the first redox reaction. At an applied potential of −0.15 V (vs. Ag/Ag+) which corresponds to the third redox process, the doping level reached 1.33 ± 0.07 eru, which is equivalent to about five electrons are injected per four repeat units. These results indicate that multiply charged species must exist, which agrees with the optical absorption spectroscopy results.
It is important to note that the electron density as a function of pH is in excellent agreement with the observed CV and Pourbaix diagrams presented earlier. For example, a horizontal line representing a doping level of 0.75 eru was found to intersect the pH = 1.04 line at −0.086 V and the pH = 2.46 line at −0.27 V, both of which are the formal potentials of the second reduction event of BBL-P in the respective electrolyte solutions. Using the calculated electron density at three different pH values, we can also interpolate and generate a heat map to illustrate the number of electrons per repeat unit as a function of doping potentials and electrolyte pH as shown in Fig. 5b. The doping level of the parent BBL ladder polymer in neutral aqueous electrolyte has been previously shown to reach 0.75–1.00 eru at more negative doping potentials between −0.5 V and −0.9 V (vs. Ag/Ag+) depending on the polymer molecular weights and the electrolyte.50,52 In our case, by tuning the electrolyte's pH value, we can achieve comparable doping level (i.e. 0.75–1.00 eru) at significantly lower doping potentials suggesting that potential BBL-P OECT devices operating in acidic environment could have reduced threshold voltage and lower power consumption.
Probing the nature of charge carriers in electrochemically n-doped BBL-P
The unique optical signatures of neutral, singly charged, and multiply charged species of BBL-P (Fig. 6a) from the above spectroelectrochemistry results enable us to track their formation upon electrochemical n-doping. In particular, Fig. 6a is the reconstruction of the spectroelectrochemistry results (Fig. 4b and c) in energy space (E = 1240/λ, where E is the energy (eV), and λ is the wavelength (nm)). The spectrum labeled 0.7–1.0 eru corresponded to the optical absorption spectrum collected with an applied potential of +0.05 V (vs. Ag/Ag+) in aqueous electrolyte of pH = 0.16 (Fig. 4b). The spectra labeled 1.3 eru and 1.5 eru corresponded respectively to the optical absorption spectra collected with an applied potential of −0.15 V (vs. Ag/Ag+) and −0.25 V (vs. Ag/Ag+) in electrolyte of pH = 0.16 (Fig. 4c). Based on the optical transition observed in Fig. 6a, we quantitatively constructed the evolution of the electronic band structure as a function of doping level (Fig. 6b). | ||
| Fig. 6 (a) Optical signatures of polarons (P), polaron pairs (PP), and singlet bipolarons (BP); (b) electronic band structure of different polaronic species including polaron, bipolaron, and polaron pairs. | ||
Neutral BBL-P has a medium bandgap of 2.5 eV (Fig. S12a†) which is in good agreement with previously reported DFT results.28 As the polymer chains are reduced to an intermediate doping level (0.7–1.0 eru), which corresponds to the second redox event, a new absorption band arose centering at 1.1 eV (Fig. 6a and S12a†) which can be ascribed to the optical signature of polarons. Based on the observed optical transitions at 2.5 eV and 1.1 eV (Fig. S12a†), we proposed two possible electronic band diagrams of polarons (Fig. S12b†). In the first scenario, a bandgap (BG) transition corresponding to 2.5 eV is accompanied by a 1.1 eV transition from the valence band (VB) to the bonding state of the charged species (P1). This picture would mean that a finite gap of 0.3 eV (2.5–2 × 1.1 = 0.3) would remain (Fig. S12b†). In the second scenario, a BG transition at 2.5 eV is followed by a 1.1 eV transition from the bonding state to the anti-bonding state of the charged species (P2); thereby, leaving the P1 transition to be 0.7 eV (0.5 × (2.5 − 1.1) = 0.7). Most doped conjugated polymers are found to exhibit P2 transition with higher energy than P1 transition;30,53,54 thereby, we attributed the electronic band diagram presented in scenario 2 (Fig. S12b†) to that of polarons in doped BBL-P.
As the polymer is further reduced to a very high doping level (∼1.5 eru), the electronic band is widened from 2.5 eV to 2.7 eV (Fig. 6a and S13a†), which agrees with the formation of multiply charged species.54 In most doped conjugated polymers, multiply charged species can be categorized into either singlet bipolarons (BP) or triplet bipolarons which is also known as polaron pairs (PP). A recent study has suggested that strongly coupled polarons with lower energy P1 transition or red-shifted optical absorption could be more favorably formed in electrochemically doped poly(3-hexylthiophene) (P3HT) depending on the experimental conditions (e.g. potential bias, counterion size, counterion mobility, etc.).30 On the other hand, if such a transition has higher energy, it would be the optical signature of bipolarons agreeing with the traditional picture of doped conjugated polymers.30,53–57 When we assigned the 1.9 eV optical transition (Fig. S13a†) to the BP1 transition, the electronic band structure would be invalid as shown in scenario 1 of Fig. S13b† (1.9 × 2 > 2.7). This result suggests that bipolarons are unlikely to be the multiply charged species when BBL-P is doped to a high doping level of 1.5 eru. Therefore, the absorption peak at 1.9 eV (Fig. S13a†) is assigned to the transition from the VB to the anti-bonding state of the charged species; thus, implying that the gap between the VB and the bonding state of the charged species is 0.4 eV (0.5 × (2.7 − 1.9) = 0.4). This result means that the P1 transition seen in the multiply charged species has lower energy than that of polarons (Fig. S12b†); therefore, we assign strongly coupled polaron pairs (triplet bipolarons) rather than singlet bipolarons to be the charge carriers in heavily reduced BBL-P. This finding is in agreement with the reported DFT results for BBL polymer58 where triplet bipolarons are formed in highly doped samples.
As mentioned earlier in the spectroelectrochemistry results, the optical absorption spectra corresponding to the third redox event of BBL-P (1.3 eru) were severely distorted, suggesting the coexistence of different polaronic species. We thus performed linear superpositions of the optical signatures of polaron (P) and polaron pairs (PP) to quantify the distribution of these polaronic species in the reduced BBL-P films. Despite varying PP:P compositions, the superimposed spectra could only match the main electronic excitations in the visible (4–1.7 eV) and near-IR (0.9–1.5 eV) regions and failed to capture the electronic transition at 1.6 eV (Fig. S8b†). This result suggests that there must be some electronic interactions between polarons and polaron pairs to give rise to a new charged species with an electronic transition of 1.6 eV (Fig. S14a†). Indeed, two isosbestic points at 1.5 eV and 1.7 eV (Fig. S14a†) correspond to the transformation of polaron pairs and polarons into a new charged species. In other words, the third redox process of BBL-P should consist of a ternary mixture of polaron, polaron pairs, and a new multiply charged species.
We propose that this new multiply charged species are singlet bipolarons arising from the hybridization between the bonding states of polarons and polaron pairs (Fig. S14b†). The optimal energy level distribution of polarons and polaron pairs enables the formation of two new mid-gap levels at E1 = 0.2 eV and E2 = 0.9 eV (Fig. S14b†) whereby E1 + E2 = P1, polaron + P1, polaron pairs = 1.1 eV by virtue of conservation of energy. Thus, the 1.6 eV transition can be assigned to the transition from the new hybridized E1 state to the non-bonding state of polarons (Fig. 6b and S14b†). These findings provide the first experimental evidence of the coexistence of polarons, polaron pairs, and bipolarons in an n-type π-conjugated polymer and clearly mapped out the transition between each polaronic species in energy space. We note that previously reported DFT calculations have predicted the coexistence of polarons and bipolarons on the same polymer chain of BBL;58 however, the calculation did not provide a mechanism as to how polarons can turn into bipolarons.
In summary, we have experimentally provided a complete picture of the formation and transition of different polaronic species in an n-doped π-conjugated polymer. At a low to intermediate doping level of 0.1–1.0 eru, polarons are the main charge carrier, and they show two mid-gap transitions of 1.1 eV and 0.7 eV, which corresponds to the electronic transition from the valence band to the polaron nonbonding state and the polaron bonding state, respectively. At a very high doping level of 1.5 eru, strongly coupled polaron pairs, rather than bipolarons, are the dominant charge carriers, and they show a wider bandgap accompanied by two mid-gap transitions of 0.4 eV and 1.9 eV. At an intermediate-high doping level of ∼1.3 eru, polarons and polaron pairs coexist in equal amount, and the hybridization between their bonding states gives rise to singlet bipolarons; thus, a ternary combinations of polarons, polaron pairs, and bipolarons are in equilibrium.
In operando Raman spectroscopy of BBL-P films
We then used in operando Raman spectroscopy to probe the molecular structure and electronic structure evolution of BBL-P upon electrochemical doping. We will similarly focus our discussion on results obtained in solution of pH < 1 as all three redox states of BBL-P coexist in equilibrium under this condition. The Raman spectra of BBL-P in highly acidic electrolyte (pH = 0.19) are shown in Fig. 7, and the close-up spectra of relevant vibrational modes are shown in Fig. 7b–g. The Raman spectra of BBL-P in electrolyte of higher pH values (pH ∼2 and 3) are shown in Fig. S15.† The Raman intensity decreased over the whole measurement ranges indicating the formation of polaronic species upon electrochemical reduction.52,59 The most intense Raman vibrational modes of undoped BBL-P are located at w1 = 1713 cm−1, w2 = 1630 cm−1, w3 = 1601 cm−1, w4 = 1538 cm−1, w5 = 1385 cm−1, w6 = 1319 cm−1, and w7 = 1157 cm−1. The w1 Raman mode can be assigned to the symmetric CO vibrations which gradually decreased and was completely bleached at E < +0.05 V (vs. Ag/Ag+) (Fig. 7b). The disappearance of the w1 vibrational modes implies a very high doping level beyond 1.0 eru was obtained at more negative potential,52 which corroborates well with our coulometry results.
 | ||
| Fig. 7 (a) In operando Raman spectra of BBL-P films with 532 nm excitation in the range of 1000 –1800 cm−1 region. (b–g) Zoomed in Raman spectra of BBL-P films in the regions of (b) 1125–1220 cm−1, (c) 1250–1350 cm−1, (d) 1300–1500 cm−1, (e) 1450–1590 cm−1, (f) 1565–1680 cm−1, and (g) 1670–1800 cm−1. Raman measurements were collected from BBL-P films coated on gold covered glass substrates as the working electrode with 0.1 M KCl(aq) (pH = 0.19) as the electrolyte, Pt mesh as the counter electrode, Ag/AgCl pellet as reference electrode positioned away from the illuminated area. | ||
The w2 Raman mode (1630 cm−1) can be assigned to the CN stretch arising from both the naphthalene ring and the phenazine unit60 whereas the w3 mode (1601 cm−1) originates from the CC/C–C vibrations. Upon electrochemical doping, the w3 peak intensity decreased and gradually shifted to lower frequency (Fig. 7c and S16a†). In particular, the w3 Raman band was found at 1589 cm−1 (12 cm−1 shift) upon the second redox reaction (E = +0.05 V, vs. Ag/Ag+). The substantial frequency decrease suggests that the polarons that are formed cause the structure to adopt a more quinoidal character.58,61,62 As the polymer chains are further reduced (E = −0.15 to −0.20 V, vs. Ag/Ag+), the w3 vibrational mode increased in intensity and shifted back to higher frequency centering at 1620 cm−1 (Fig. 7c and S16a†). This result implies that the strongly coupled polaron pairs and bipolarons that are formed upon the third redox event results in a pro-benzoidal structure. As a result, these multiply charged species would be much less delocalized compared to polarons.
Moreover, the w2 Raman band (1630 cm−1) was found to progressively shift to higher wavenumber as the applied potential was swept from positive to negative (Fig. S16b†). In particular, this vibrational mode centered at 1637 cm−1 for E = +0.05 V (vs. Ag/Ag+) and at 1663–1664 cm−1 for E = −0.15 to −0.20 V (vs. Ag/Ag+). The significant red-shift is indicative of the protonation of the imine bond to yield –CN+– on both the phenazine rings and the naphthalene rings.63 The higher Raman intensity and more red-shifted Raman band seen at −0.15 to −0.20 V compared to that at +0.05 V suggests that the concentration of –CN+– is higher in this reduced form of BBL-P. We note that at low doping level (E = +0.25 V, vs. Ag/Ag+), the –CN– vibrational mode exhibited marginal shift which means that very few number of imine sites are protonated at this potential.
The w4 mode (1538 cm−1) corresponds to the aromatic C–C and C–N skeletal vibrations. The slight intensity decrease seen at E = +0.25 V (Fig. 7d) suggests that there are marginal conformational changes in the polymer backbone, which makes sense given the low doping level (∼0.2 eru). However, the w4 Raman band was completely bleached at more negative potentials (E < +0.05 V) suggesting significant structural changes upon the formation of charged species.59 This finding indeed corroborates the high doping level (>1.0 eru) explained by coulometry earlier.
The w5 mode (1385 cm−1) can be assigned to the –C–N– single bond stretching.28 As the doping level increased, the Raman intensity was found to gradually decrease, and the peak position shifted to lower wavenumber (Fig. 7e and S16c†). In particular, the peak shifting can be divided into three regions where the first region (E > 0.3 V, vs. Ag/Ag+) showed minimal change in frequency, the second region (0 V < E < 0.3 V, vs. Ag/Ag+) featured a 4 cm−1 shift, and the third region (E < 0 V, vs. Ag/Ag+) had a substantial shift of 23 cm−1 (Fig. S16c†). The significant peak shift of this –C–N– vibrational mode seen in the second and third region is indicative of formation of charged species or –C–Ṅ+– as previously reported for polyaniline and its derivatives.61,64,65 Moreover, the substantial peak shift corroborates the formation of strongly coupled polaron pairs and bipolarons upon the third redox reaction, which can be explained by an increase of conducting (charged) environment around the –C–N– stretch,66 whereas polarons are the main charge carrier in the second redox reaction.
The w6 Raman mode (1319 cm−1) in undoped BBL-P can be assigned to the –C–N– stretching of the phenazine ring. This vibrational mode was shifted to much lower energy (1285 cm−1) (Fig. 7f), which can be ascribed to the formation of polaronic species. Lastly, the w7 Raman mode (1157 cm−1), which corresponds to in-plane C–H bending, was substantially upshifted to 1179 cm−1 upon the third reduction reaction (Fig. 7g). This large increase in vibrational energy of this mode can be explained by the –C–H– vibrational of the radical cation segments.
In summary, our in operando Raman spectroscopy results provide important insights on the molecular and electronic structures of BBL-P upon electrochemical doping, all of which are found to be in excellent agreement with the spectroelectrochemistry and coulometry analyses. The first reduction reaction of BBL-P results in minimal structural changes mainly due to the low and non-uniform distribution of polarons across the polymer backbone (1 electron per 4–5 repeat units). The second reduction reaction of BBL-P leads to the formation of polarons and induces significant changes in the molecular structures, including more quinoidal characteristics and an intermediate concentration of protonated imine sites. The third reduction reaction of BBL-P, involving the formation of polarons, polaron pairs, and bipolarons, results in greater structural changes, including more benzoidal characteristics and a high concentration of protonated imine sites.
Proposed schemes for BBL-P electrochemical reactions
Taking all the above data together, we now provide a comprehensive scheme to explain the multiple electrochemical reduction reactions of BBL-P in acidic aqueous electrolytes.At pH > 3, the BBL-P film remains unprotonated and facilely undergoes a redox reaction to form the first reduced form (Scheme 2). At this stage, the doping level is rather low, featuring about 1 electron per 4–5 repeat units as suggested by spectroelectrochemistry and coulometry results. As a result, minimal changes in the molecular structure are observed as evidenced by the in operando Raman spectroscopy analysis.
 | ||
| Scheme 2 A two-proton/one-electron electrochemical doping reaction (wave I) of BBL-P consisting of 1H+/1e− at the carbonyl oxygen sites followed by 1H+ either at the naphthalene imine or the phenazine imine sites. | ||
At 1 < pH < 3, the BBL-P is partially protonated, specifically at the naphthalene imine bond and then undergoes a redox reaction at a more negative potential to form the second reduced form. The density of polarons in this structure is shown to be around 3–4 electrons per 4 repeat units via spectroelectrochemistry and coulometry. In operando Raman spectroscopy reveals that this reduced form adopts a more quinoidal character than the undoped unprotonated BBL-P. The molecular structure of this reduced form is thus shown in Scheme 3.
 | ||
| Scheme 3 A two-proton/one-electron electrochemical doping reaction (wave II) of BBL-P comprising of 1H+/1e− transferred at each naphthalene imine site followed by 1H+ at the phenazine imine sites. | ||
At pH < 1, the imine bonds of the phenazine rings are protonated, and the protonated polymer undergoes a redox reaction to form the third reduced form at significantly more negative potential. Multiply charged BBL-P with a high doping level of approximately 5 electrons per 4 repeat units are found via spectroelectrochemistry and coulometry. The polymer structure of this reduced state preferentially adopts a more benzoidal character that features a large concentration of –CN+– sites on both the phenazine rings and the naphthalene rings. The molecular structure of this reduced state of BBL-P is thus illustrated in Scheme 4.
 | ||
| Scheme 4 A two-proton/one-electron electrochemical doping reaction (wave III) of BBL-P consisting of a 1H+/1e− transferred at each phenazine imine sites accompanied by 1H+ at the naphthalene imine sites. | ||
It is important to reiterate that all three final products are formed as a result of two-proton per one-electron transferred redox reactions, which is to be contrasted with commonly seen one-proton/one-electron PCET. The extra proton insertion to the polymer backbone implies that all reduced products of BBL-P should have higher redox capacity and volumetric capacitance than the parent BBL.67,68 Our present results have thus established important fundamental frameworks regarding the electrochemical reactivity and resulting optoelectronic and structural evolution of BBL-P, all of which could be broadly applicable to device applications.
Redox capacity of BBL-P films as a function of pH and applied potential
We have also explored the charge storage capability of BBL-P in acidic environment which have implications for applications in organic electrochemical transistors (OECTs), all-organic proton batteries, and proton supercapacitors. The redox capacity (ρ) is defined as the change in volumetric charge density per infinitesimal change of equilibrium potential relative to a reference electrode:67,68where ρ is the redox capacity (F cm−3), Q is the charge stored in the electrode (C), V is the volume of the electrode (cm3), and Eeq is the equilibrium potential.
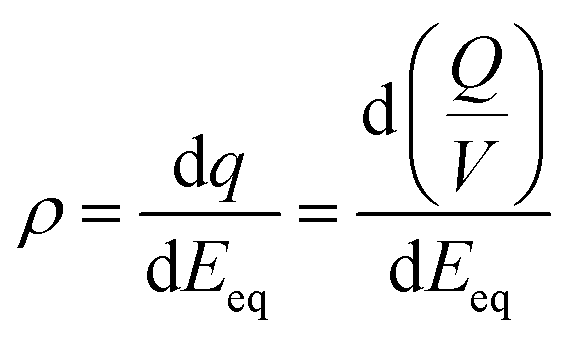
Using the total charge obtained from coulometry analysis, we computed the redox capacity of BBL-P with respect to the doping potential in electrolyte of pH < 1 as shown in Fig. 8a, and the heatmap showing the evolution of redox capacity as a function of both doping potential and electrolyte pH is illustrated in Fig. 8b. The redox capacity of BBL-P in highly acidic electrolyte exhibited three different peaks at around +0.25 V, +0.05 V, and −0.15 V (vs. Ag/Ag+) which coincided with the formal potential of the first, second, and third redox event, respectively. This result makes sense since electrons are injected into the polymer films at these potentials. Moreover, the maximum ρ value of BBL-P in acidic electrolyte (ρ = 1272 F cm−3) is significantly higher than that of the parent BBL in neutral aqueous environment (540–1000 F cm−3)50,69 and neutral non-aqueous environment (∼150 F cm−3).70 We note that the molecular weight of BBL-P used in this study is considered to be relatively low with an intrinsic viscosity around 1.1 dL g−1; thus, further enhanced redox capacity can be expected if higher molecular weight sample is synthesized.50 Our present results demonstrate that the unique molecular structure of BBL-P, which features multiredox sites with varying pKa values, enables a remarkably high redox capacity upon protonation and reduction.
 | ||
| Fig. 8 (a) Evolution of redox capacity (ρ) and doping level (eru) of BBL-P thin films as a function of doping potential in electrolyte of pH < 1; (b) dependence of redox capacity of BBL-P thin films as a function of electrolyte pH and doping potentials. | ||
Conclusions
We have investigated the proton-coupled electron transfer (PCET) process involved in electrochemical n-doping of a novel multiredox π-conjugated polymer, phenazine-substituted poly(benzimidazobenzophenanthroline) (BBL-P), in aqueous electrolyte. From the measured Pourbaix diagram of each of the three redox processes in BBL-P, we have discovered that the PCET response is super-Nernstian, exhibiting two protons per one electron transferred. Although super-Nernstian PCET has previously been observed in some hydrated transition metal oxides9–12 and metalloproteins,4,13,14 our present results represent the first observation of super-Nernstian PCET behavior in a purely organic material/polymer.We have also combined pH-dependent spectroelectrochemistry, in operando Raman spectroscopy, and coulometry to probe the nature of charge carriers in the electrochemically n-doped BBL-P. We found that polarons, delocalized over a pro-quinoidal polymer backbone, are the charge carriers at low to intermediate doping levels (0.1–1.0 eru). At a higher doping level (1.3 eru), a ternary mixture of polarons, polaron pairs, and bipolarons were found to co-exist in equilibrium. At the highest doping level (1.5 eru), polaron pairs were found to be the dominant charge carriers.
Finally, our pH-dependent coulometry analysis of BBL-P showed that it has a very high redox capacity (>1200 F cm−3) in aqueous acidic (pH < 1) electrolyte, which suggests good potential for applications in energy storage and other electrochemical devices.
Data availability
The data that support the findings of this study are available within the paper and its ESI.† Additional data may be requested from the authors.Author contributions
D. K. T. conceived the idea, investigated, conducted the experiments (CV, spectroelectrochemistry, coulometry, in operando Raman spectroscopy), and performed formal analysis and visualization of the experimental data and DFT results. S. M. W. synthesized the polymer and performed Density Functional Theory (DFT) calculations. E. M. K. S. assisted with the synthesis of the polymer. S. A. J. supervised all aspects of the study. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This research was supported by the Department of Energy (DOE) Basic Energy Sciences (DE-SC0023229). Our initial synthesis and characterization of BBL-P and support of S. M. W. were supported by the National Science Foundation (DMR- 2003518). We thank Dr Jiajie Guo and Dr Shinya E. Chen for fruitful discussion. Part of this work was conducted using equipment in the Biomedical Diagnostic Foundry for Translational Research sponsored by the M. J. Murdock Charitable Trust. The DFT calculations were done through the use of advanced computational, storage, and networking infrastructure provided by the Hyak supercomputer system at the University of Washington.References
- M. H. V. Huynh and T. J. Meyer, Proton-Coupled Electron Transfer, Chem. Rev., 2007, 107, 5004–5064 CrossRef CAS PubMed.
- S. Hammes-Schiffer, Exploring proton-coupled electron transfer at multiple scales, Nat. Comput. Sci., 2023, 3, 291–300 CrossRef PubMed.
- A. Migliore, N. F. Polizzi, M. J. Therien and D. N. Beratan, Biochemistry and Theory of Proton-Coupled Electron Transfer, Chem. Rev., 2014, 114, 3381–3465 CrossRef CAS PubMed.
- D. R. Weinberg, C. J. Gagliardi, J. F. Hull, C. F. Murphy, C. A. Kent, B. C. Westlake, A. Paul, D. H. Ess, D. G. McCafferty and T. J. Meyer, Proton-Coupled Electron Transfer, Chem. Rev., 2012, 112, 4016–4093 CrossRef CAS PubMed.
- S. Hammes-Schiffer and A. V. Soudackov, Proton-Coupled Electron Transfer in Solution, Proteins, and Electrochemistry, J. Phys. Chem. B, 2008, 112, 14108–14123 CrossRef CAS PubMed.
- S. Y. Reece and D. G. Nocera, Proton-Coupled Electron Transfer in Biology: Results from Synergistic Studies in Natural and Model Systems, Annu. Rev. Biochem., 2009, 78, 673–699 CrossRef CAS PubMed.
- R. Tyburski, T. Liu, S. D. Glover and L. Hammarström, Proton-Coupled Electron Transfer Guidelines, Fair and Square, J. Am. Chem. Soc., 2021, 143, 560–576 CrossRef CAS PubMed.
- J. M. Mayer, Bonds over Electrons: Proton Coupled Electron Transfer at Solid–Solution Interfaces, J. Am. Chem. Soc., 2023, 145, 7050–7064 CrossRef CAS PubMed.
- M. Dincă, Y. Surendranath and D. G. Nocera, Nickel-borate oxygen-evolving catalyst that functions under benign conditions, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 10337–10341 CrossRef PubMed.
- L. D. Burke, J. K. Mulcahy and D. P. Whelan, Preparation of an oxidized iridium electrode and the variation of its potential with pH, J. Electroanal. Chem. Interfacial Electrochem., 1984, 163, 117–128 CrossRef CAS.
- A. A. Gambardella, N. S. Bjorge, V. K. Alspaugh and R. W. Murray, Voltammetry of Diffusing 2 nm Iridium Oxide Nanoparticles, J. Phys. Chem. C, 2011, 115, 21659–21665 CrossRef CAS.
- K. A. Stoerzinger, R. R. Rao, X. R. Wang, W. T. Hong, C. M. Rouleau and Y. Shao-Horn, The Role of Ru Redox in pH-Dependent Oxygen Evolution on Rutile Ruthenium Dioxide Surfaces, Chem, 2017, 2, 668–675 CAS.
- J. C. Koone, M. Simmang, D. L. Saenger, L. M. Hunsicker-Wang and B. F. Shaw, Charge Regulation in a Rieske Proton Pump Pinpoints Zero, One, and Two Proton-Coupled Electron Transfer, J. Am. Chem. Soc., 2023, 145, 16488–16497 CrossRef CAS PubMed.
- A. R. Reddi, C. J. Reedy, S. Mui and B. R. Gibney, Thermodynamic Investigation into the Mechanisms of Proton-Coupled Electron Transfer Events in Heme Protein Maquettes, Biochemistry, 2007, 46, 291–305 CrossRef CAS PubMed.
- T. Ma, A. D. Easley, R. M. Thakur, K. T. Mohanty, C. Wang and J. L. Lutkenhaus, Nonconjugated Redox-Active Polymers: Electron Transfer Mechanisms, Energy Storage, and Chemical Versatility, Annu. Rev. Chem. Biomol. Eng., 2023, 14, 187–216 CrossRef CAS PubMed.
- Y. Liang, Z. Chen, Y. Jing, Y. Rong, A. Facchetti and Y. Yao, Heavily n-Dopable π-Conjugated Redox Polymers with Ultrafast Energy Storage Capability, J. Am. Chem. Soc., 2015, 137, 4956–4959 CrossRef CAS PubMed.
- S. T. M. Tan, T. J. Quill, M. Moser, G. LeCroy, X. Chen, Y. Wu, C. J. Takacs, A. Salleo and A. Giovannitti, Redox-Active Polymers Designed for the Circular Economy of Energy Storage Devices, ACS Energy Lett., 2021, 6, 3450–3457 CrossRef CAS.
- A. A. Szumska, I. P. Maria, L. Q. Flagg, A. Savva, J. Surgailis, B. D. Paulsen, D. Moia, X. Chen, S. Griggs, J. T. Mefford, R. B. Rashid, A. Marks, S. Inal, D. S. Ginger, A. Giovannitti and J. Nelson, Reversible Electrochemical Charging of n-Type Conjugated Polymer Electrodes in Aqueous Electrolytes, J. Am. Chem. Soc., 2021, 143, 14795–14805 CrossRef CAS PubMed.
- D. Trefz, A. Ruff, R. Tkachov, M. Wieland, M. Goll, A. Kiriy and S. Ludwigs, Electrochemical Investigations of the N-Type Semiconducting Polymer P(NDI2OD-T2) and Its Monomer: New Insights in the Reduction Behavior, J. Phys. Chem. C, 2015, 119, 22760–22771 CrossRef CAS.
- A. Wang, R. Tan, C. Breakwell, X. Wei, Z. Fan, C. Ye, R. Malpass-Evans, T. Liu, M. A. Zwijnenburg, K. E. Jelfs, N. B. McKeown, J. Chen and Q. Song, Solution-Processable Redox-Active Polymers of Intrinsic Microporosity for Electrochemical Energy Storage, J. Am. Chem. Soc., 2022, 144, 17198–17208 CrossRef CAS PubMed.
- A. Aithal, J. J. Samuel, A. Bandyopadhyay, V. K. Karrothu, C. Gangadharappa, S. Patil, A. Narayan and N. P. B. Aetukuri, Extended Conjugation Acceptors Increase Specific Energy Densities in π-Conjugated Redox Polymers, J. Phys. Chem. C, 2023, 127, 5238–5245 CrossRef CAS.
- J. J. Samuel, V. K. Karrothu, R. K. Canjeevaram Balasubramanyam, A. A. Mohapatra, C. Gangadharappa, V. R. Kankanallu, S. Patil and N. P. B. Aetukuri, Ionic Charge Storage in Diketopyrrolopyrrole-Based Redox-Active Conjugated Polymers, J. Phys. Chem. C, 2021, 125, 4449–4457 CrossRef CAS.
- J. Kim, J. H. Kim and K. Ariga, Redox-Active Polymers for Energy Storage Nanoarchitectonics, Joule, 2017, 1, 739–768 CrossRef CAS.
- G. P. Pickup, Conjugated metallopolymers. Redox polymers with interacting metal based redox sites, J. Mater. Chem., 1999, 9, 1641–1653 RSC.
- Z. Jin, Q. Cheng, A. M. Evans, J. Gray, R. Zhang, S. T. Bao, F. Wei, L. Venkataraman, Y. Yang and C. Nuckolls, π-Conjugated redox-active two-dimensional polymers as organic cathode materials, Chem. Sci., 2022, 13, 3533–3538 RSC.
- K. Wilbourn and R. W. Murray, The electrochemical doping reactions of the conducting ladder polymer benzimidazobenzophenanthroline (BBL), Macromolecules, 1988, 21, 89–96 CrossRef CAS.
- T.-L. Choi, K.-H. Lee, W.-J. Joo, S. Lee, T.-W. Lee and M. Y. Chae, Synthesis and Nonvolatile Memory Behavior of Redox-Active Conjugated Polymer-Containing Ferrocene, J. Am. Chem. Soc., 2007, 129, 9842–9843 CrossRef CAS PubMed.
- S. M. West, D. K. Tran, J. Guo, S. E. Chen, D. S. Ginger and S. A. Jenekhe, Phenazine-Substituted Poly(benzimidazobenzophenanthrolinedione): Electronic Structure, Thin Film Morphology, Electron Transport, and Mechanical Properties of an n-Type Semiconducting Ladder Polymer, Macromolecules, 2023, 56, 2081–2091 CrossRef CAS.
- P. Cavassin, I. Holzer, D. Tsokkou, O. Bardagot, J. Réhault and N. Banerji, Electrochemical Doping in Ordered and Disordered Domains of Organic Mixed Ionic–Electronic Conductors, Adv. Mater., 2023, 35, 2300308 CrossRef CAS PubMed.
- E. C. Wu, C. Z. Salamat, O. L. Ruiz, T. Qu, A. Kim, S. H. Tolbert and B. J. Schwartz, Counterion Control and the Spectral Signatures of Polarons, Coupled Polarons, and Bipolarons in Doped P3HT Films, Adv. Funct. Mater., 2023, 33, 2213652 CrossRef CAS.
- A. Babel and S. A. Jenekhe, High Electron Mobility in Ladder Polymer Field-Effect Transistors, J. Am. Chem. Soc., 2003, 125, 13656–13657 CrossRef CAS PubMed.
- D. K. Tran, S. M. West, J. Guo, S. E. Chen, D. S. Ginger and S. A. Jenekhe, Chain Length Dependence of Electron Transport in an n-Type Conjugated Polymer with a Rigid-Rod Chain Topology, J. Am. Chem. Soc., 2024, 146, 1435–1446 CrossRef CAS PubMed.
- R. E. Warburton, A. V. Soudackov and S. Hammes-Schiffer, Theoretical Modeling of Electrochemical Proton-Coupled Electron Transfer, Chem. Rev., 2022, 122, 10599–10650 CrossRef CAS PubMed.
- N. Elgrishi, K. J. Rountree, B. D. McCarthy, E. S. Rountree, T. T. Eisenhart and J. L. Dempsey, A Practical Beginner's Guide to Cyclic Voltammetry, J. Chem. Educ., 2018, 95, 197–206 CrossRef CAS.
- W.-H. Li, X.-Y. Li and N.-T. Yu, Surface-enhanced hyper-Raman scattering and surface-enhanced Raman scattering studies of electroreduction of phenazine on silver electrode, Chem. Phys. Lett., 2000, 327, 153–161 CrossRef CAS.
- Y. Mugnier, L. Roullier and E. Laviron, Reduction mechanism of phenazine in tetrahydrofuran. Influence of added proton donors, Electrochim. Acta, 1991, 36, 803–809 CrossRef CAS.
- Y. Liang, Y. Jing, S. Gheytani, K.-Y. Lee, P. Liu, A. Facchetti and Y. Yao, Universal quinone electrodes for long cycle life aqueous rechargeable batteries, Nat. Mater., 2017, 16, 841–848 CrossRef CAS PubMed.
- M. G. Ullmann, L. Noodleman and D. A. Case, Density functional calculation of pKa values and redox potentials in the bovine Rieske iron-sulfur protein, J. Biol. Inorg. Chem., 2002, 7, 632–639 CrossRef PubMed.
- C. Choi, D. S. Ashby, D. M. Butts, R. H. DeBlock, Q. Wei, J. Lau and B. Dunn, Achieving high energy density and high power density with pseudocapacitive materials, Nat. Rev. Mater., 2020, 5, 5–19 CrossRef.
- V. Augustyn, J. Come, M. A. Lowe, J. W. Kim, P.-L. Taberna, S. H. Tolbert, H. D. Abruña, P. Simon and B. Dunn, High-rate electrochemical energy storage through Li+ intercalation pseudocapacitance, Nat. Mater., 2013, 12, 518–522 CrossRef CAS.
- R. J. Vázquez, G. Quek, S. R. McCuskey, L. Llanes, B. Kundukad, X. Wang and G. C. Bazan, Increasing the molecular weight of conjugated polyelectrolytes improves the electrochemical stability of their pseudocapacitor gels, J. Mater. Chem. A, 2022, 10, 21642–21649 RSC.
- T. Xu, Z. Li, D. Wang, M. Zhang, L. Ai, Z. Chen, J. Zhang, X. Zhang and L. Shen, A Fast Proton-Induced Pseudocapacitive Supercapacitor with High Energy and Power Density, Adv. Funct. Mater., 2022, 32, 2107720 CrossRef CAS.
- X. Wu, H. Zhang, C. He, C. Wu and K.-J. Huang, High-power-energy proton supercapacitor based on interface-adapted durable polyaniline and hexagonal tungsten oxide, J. Colloid Interface Sci., 2021, 601, 727–733 CrossRef CAS PubMed.
- T. Xu, D. Wang, Z. Li, Z. Chen, J. Zhang, T. Hu, X. Zhang and L. Shen, Electrochemical Proton Storage: From Fundamental Understanding to Materials to Devices, Nano-Micro Lett., 2022, 14, 126 CrossRef CAS PubMed.
- Y. Xu, X. Wu and X. Ji, The Renaissance of Proton Batteries, Small Struct., 2021, 2, 2000113 CrossRef CAS.
- R. Emanuelsson, M. Sterby, M. Strømme and M. Sjödin, An All-Organic Proton Battery, J. Am. Chem. Soc., 2017, 139, 4828–4834 CrossRef CAS.
- Z. Zhu, W. Wang, Y. Yin, Y. Meng, Z. Liu, T. Jiang, Q. Peng, J. Sun and W. Chen, An Ultrafast and Ultra-Low-Temperature Hydrogen Gas–Proton Battery, J. Am. Chem. Soc., 2021, 143, 20302–20308 CrossRef CAS PubMed.
- L. Yan, J. Huang, Z. Guo, X. Dong, Z. Wang and Y. Wang, Solid-State Proton Battery Operated at Ultralow Temperature, ACS Energy Lett., 2020, 5, 685–691 CrossRef CAS.
- S. Y. Hong, M. Kertesz, Y. S. Lee and O. K. Kim, Geometrical and electronic structures of a benzimidazobenzophenanthroline-type ladder polymer (BBL), Macromolecules, 1992, 25, 5424–5429 CrossRef CAS.
- J. Guo, L. Q. Flagg, D. K. Tran, S. E. Chen, R. Li, N. B. Kolhe, R. Giridharagopal, S. A. Jenekhe, L. J. Richter and D. S. Ginger, Hydration of a Side-Chain-Free n-Type Semiconducting Ladder Polymer Driven by Electrochemical Doping, J. Am. Chem. Soc., 2023, 145, 1866–1876 CrossRef CAS PubMed.
- M. Quinto, S. A. Jenekhe and A. J. Bard, Polymer Films on Electrodes. 30. Electrochemistry and Scanning Electrochemical Microscopy Characterization of Benzimidazolebenzophenanthroline-Type Ladder (BBL) and Semiladder (BBB) Polymer Films, Chem. Mater., 2001, 13, 2824–2832 CrossRef CAS.
- K. Xu, T.-P. Ruoko, M. Shokrani, D. Scheunemann, H. Abdalla, H. Sun, C.-Y. Yang, Y. Puttisong, N. B. Kolhe, J. S. M. Figueroa, J. O. Pedersen, T. Ederth, W. M. Chen, M. Berggren, S. A. Jenekhe, D. Fazzi, M. Kemerink and S. Fabiano, On the Origin of Seebeck Coefficient Inversion in Highly Doped Conducting Polymers, Adv. Funct. Mater., 2022, 32, 2112276 CrossRef CAS.
- J. L. Bredas and G. B. Street, Polarons, bipolarons, and solitons in conducting polymers, Acc. Chem. Res., 1985, 18, 309–315 CrossRef CAS.
- J. L. Brédas, J. C. Scott, K. Yakushi and G. B. Street, Polarons and bipolarons in polypyrrole: evolution of the band structure and optical spectrum upon doing, Phys. Rev. B: Condens. Matter Mater. Phys., 1984, 30, 1023–1025 CrossRef.
- J. E. Cochran, M. J. N. Junk, A. M. Glaudell, P. L. Miller, J. S. Cowart, M. F. Toney, C. J. Hawker, B. F. Chmelka and M. L. Chabinyc, Molecular Interactions and Ordering in Electrically Doped Polymers: Blends of PBTTT and F4TCNQ, Macromolecules, 2014, 47, 6836–6846 CrossRef CAS.
- M. G. Voss, J. R. Challa, D. T. Scholes, P. Y. Yee, E. C. Wu, X. Liu, S. J. Park, O. León Ruiz, S. Subramaniyan, M. Chen, S. A. Jenekhe, X. Wang, S. H. Tolbert and B. J. Schwartz, Driving Force and Optical Signatures of Bipolaron Formation in Chemically Doped Conjugated Polymers, Adv. Mater., 2021, 33, 2000228 CrossRef CAS PubMed.
- E. Dmitrieva, Y. Harima and L. Dunsch, Influence of Phenazine Structure on Polaron Formation in Polyaniline: In Situ Electron Spin Resonance−Ultraviolet/Visible−Near-Infrared Spectroelectrochemical Study, J. Phys. Chem. B, 2009, 113, 16131–16141 CrossRef CAS PubMed.
- S. Ghosh, V. Gueskine, M. Berggren and I. V. Zozoulenko, Electronic Structures and Optical Absorption of N-Type Conducting Polymers at Different Doping Levels, J. Phys. Chem. C, 2019, 123, 15467–15476 CrossRef CAS.
- E. Tan, J. Kim, K. Stewart, C. Pitsalidis, S. Kwon, N. Siemons, J. Kim, Y. Jiang, J. M. Frost, D. Pearce, J. E. Tyrrell, J. Nelson, R. M. Owens, Y.-H. Kim and J.-S. Kim, The Role of Long-Alkyl-Group Spacers in Glycolated Copolymers for High-Performance Organic Electrochemical Transistors, Adv. Mater., 2022, 34, 2202574 CrossRef CAS PubMed.
- A. Kellenberger, E. Dmitrieva and L. Dunsch, The stabilization of charged states at phenazine-like units in polyaniline under p-doping: an in situATR-FTIR spectroelectrochemical study, Phys. Chem. Chem. Phys., 2011, 13, 3411–3420 RSC.
- G. Ćirić-Marjanović, M. Trchová and J. Stejskal, The chemical oxidative polymerization of aniline in water: Raman spectroscopy, J. Raman Spectrosc., 2008, 39, 1375–1387 CrossRef.
- J. Nightingale, J. Wade, D. Moia, J. Nelson and J.-S. Kim, Impact of Molecular Order on Polaron Formation in Conjugated Polymers, J. Phys. Chem. C, 2018, 122, 29129–29140 CrossRef CAS.
- S. Alex, P. Turcotte, R. Fournier and D. Vocelle, Study of the protonation of simple Schiff bases in solvents of various polarity by means of Raman spectroscopy, Can. J. Chem., 1991, 69, 239–245 CrossRef CAS.
- G. Louarn, M. Lapkowski, S. Quillard, A. Pron, J. P. Buisson and S. Lefrant, Vibrational Properties of PolyanilineIsotope Effects, J. Phys. Chem., 1996, 100, 6998–7006 CrossRef CAS.
- I. Harada, Y. Furukawa and F. Ueda, Vibrational spectra and structure of polyaniline and related compounds, Synth. Met., 1989, 29, 303–312 CrossRef.
- M. C. Bernard and A. Hugot-Le Goff, Quantitative characterization of polyaniline films using Raman spectroscopy: I: polaron lattice and bipolaron, Electrochim. Acta, 2006, 52, 595–603 CrossRef CAS.
- C. E. D. Chidsey and R. W. Murray, Redox capacity and direct current electron conductivity in electroactive materials, J. Phys. Chem., 1986, 90, 1479–1484 CrossRef CAS.
- L. A. Estrada, D. Y. Liu, D. H. Salazar, A. L. Dyer and J. R. Reynolds, Poly[Bis-EDOT-Isoindigo]: An Electroactive Polymer Applied to Electrochemical Supercapacitors, Macromolecules, 2012, 45, 8211–8220 CrossRef CAS.
- H.-Y. Wu, C.-Y. Yang, Q. Li, N. B. Kolhe, X. Strakosas, M.-A. Stoeckel, Z. Wu, W. Jin, M. Savvakis, R. Kroon, D. Tu, H. Y. Woo, M. Berggren, S. A. Jenekhe and S. Fabiano, Influence of Molecular Weight on the Organic Electrochemical Transistor Performance of Ladder-Type Conjugated Polymers, Adv. Mater., 2022, 34, 2106235 CrossRef CAS PubMed.
- K. Wilbourn and R. W. Murray, The d.c. redox versus electronic conductivity of the ladder polymer poly(benzimidazobenzophenanthroline), J. Phys. Chem., 1988, 92, 3642–3648 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc00785a |
| This journal is © The Royal Society of Chemistry 2024 |