Potassium exchanged zirconium hydrogen phosphate Zr(O3POK)2: a heterogeneous basic catalyst for Knoevenagel reaction without solvent
David Fildesa, Vincent Caignaertb, Didier Villemin*a and Paul-Alain Jaffrès*a
aLCMT, UMR, CNRS 6507, Ecole Nationale Supérieure d’Ingénieur de
Caen, Université de Caen, ISMRA, 6Bd du Maréchal Juin, F-14050, Caen, France.. E-mail: jaffres@ismra.fr
bCRISMAT, UMR CNRS 6508, Ecole Nationale Supérieure d’Ingénieur de
Caen, Université de Caen, ISMRA, 6Bd du Maréchal Juin, F-14050, Caen, France
First published on 31st January 2001
Abstract
A simple work-up to prepare crystalline potassium exchanged zirconium hydrogen phosphate Zr(O3POK)2 is proposed. In addition the synthesis of amorphous potassium exchanged zirconium hydrogen phosphate Zr(O3POK)2, obtained from an amorphous sample of Zr(O3POH)2·H2O is reported. All these exchanged materials Zr(O3POK)2 are powerful catalysts for the Knoevenagel reaction. Furthermore the absence of solvent increases the rate of the reaction. Products can be separated from the catalyst by sublimation thus avoiding the use of solvent in the purification step. The catalyst, which is thus easily recovered, shows no appreciable loss of activity when recycled three times.
Green ContextHeterogeneous basic catalysts are of importance in the fine chemicals area, and several have been investigated in reactions such as the Knoevenagel reaction, an important reaction with pronounced solvent dependency. This paper deals with a simplified preparation of one such catalyst, and shows that it is active in the condensation of aromatic aldehydes with C-acids. Of particular interest is the ability to run the reactions without solvent, and to isolate the products by sublimation, so also avoiding the need for solvents in the purification step.DJM |
Introduction
The use of heterogeneous catalysts allows a simplification of the purification step to a simple filtration, separating the catalyst from the reaction media. Heterogeneous basic catalysis is an area of growing interest which has been recently reviewed.1 The structure of inorganic catalysts may be amorphous (silica, alumina) or organised (Al-MCM, zeolite, clay, etc.).2 Even in the latter class of inorganic basic catalysts, their structures are frequently unknown on the atomic scale. This increases the difficulty of characterising the accurate structure of the catalytic centre. Metal–phosphate materials M(O3POH)n are a class of compounds that has been widely studied as proton conductors,3 ionic exchangers,4 catalysts5 or as supports for ionic chromatography.6 The protons of these materials may be exchanged to yield potentially basic materials. Furthermore the structure of these exchanged metal–phosphate materials may be determined at the atomic level. The potassium exchanged zirconium hydrogen phosphate Zr(O3POK)2 has already been used as a basic catalyst for Michael addition,7 Henry reaction,8 cyanosilylation of carbonyl compounds9 and more recently for the desilylation of phenol silyl ethers.10 Zr(O3POK)2, is a layered material which possesses ionic oxygen pointing towards the interlayer space. This compound may be obtained either from zirconium hydrogen phosphate Zr(O3POH)2·H2O via an exchange with potassium hydroxide7 or directly from a solid-phase synthesis at high temperature.11The Knoevenagel condensation is a useful reaction for the generation of double bonds from a carbonyl compound and an active methylene compound. Many homogeneous catalysts have been used to promote this reaction (triethylamine, pyridine–TiCl4, secondary amines etc…).12 More recently the use of heterogeneous basic catalysts have been employed to facilitate the separation of the catalyst from the reaction media. Furthermore in many cases the catalyst may be reused. Metal oxides supported on silica gel, barium hydroxide, zeolites, montmorillonite, magnesium and aluminium oxides, hydrotalcite,13 Cs-MCM-41,14 modified silicagel,15 and more recently Ga/Al-containing layered double hydroxides16 are just a few examples of effective materials which may be used for the Knoevenagel condensation. In some cases, the reaction can be achieved under dry conditions thus decreasing both the cost of the synthesis and the amount of waste flow.17
Here, a simplification of the literature procedure to synthesise crystalline Zr(O3POK)2 is reported as well as the synthesis of amorphous samples of Zr(O3POK)2. The basic properties of these materials are used to catalyse Knoevenagel condensations.
Results and discussion
The synthesis of potassium exchanged zirconium phosphate Zr(O3POK)2 according to the literature [from Zr(O3- POH)2·H2O] is quite laborious as it needs several steps. Nevertheless this method is more convenient for organic chemists, compared to the solid-phase synthesis,11 since it does not need special equipment (high temperature oven). The first step, as reported in the literature, is to increase the specific surface area of the parent material Zr(O3POH)2·H2O. Exfoliation with n-butylamine followed by a reprecipitation leads to an increase in the specific surface of the parent material from 0.5 to 17 m2 g−1. In order to reduce the number of steps to produce Zr(O3POK)2 we have utilized crystalline α-zirconium hydrogen phosphate directly in the exchange process. α-Zirconium hydrogen phosphate Zr(O3POH)2·H2O 1a was obtained according to a modification of the literature procedure18 (the BET specific surface area of Zr(O3POH)·H2O thus obtained was 7.8 m2 g−1). In order to produce a large amount of Zr(O3POK)22a the concentration of the KOH and KCl solutions in ultra-pure water was dramatically increased (1 M instead of 0.1 M). Thus 8 g of Zr(O3POK)22a were produced with a reasonable volume of solution. The X-ray powder diffraction pattern of Zr(O3- POK)2 thus synthesised is compared (Fig. 1) to the structure obtained by Dörffel and Liebertz11 from a single-crystal X-ray diffraction study. The only notable differences concern the cell parameters which are slightly larger in our study (average 0.18%; see Experimental section). As expected from the crystal structure, a singlet is observed for the 31P MAS NMR spectrum of Zr(O3POK)2 (Fig. 2) indicating the equivalence of all the phosphorus atoms in the structure and thus demonstrating the efficiency of the exchange. The chemical shift of Zr(O3POK)22a is displaced towards low field (δiso = −11.4 ppm) compared to the parent material Zr(O3POH)2·H2O (δiso = −18.7 ppm). Previous work describing the high-resolution 31P MAS NMR in some antimony phosphates reported that the 31P chemical shift (δiso) was closely dependent on the P–O bond length.19 In compounds which possess a non-coordinated oxygen, the shorter the P–O bond is the larger the shielding effect is on the phosphorus. It is of note that in the present example, that the opposite behavior is observed. Indeed, the P–O bond length (the bond pointing to the interlayer space) in compounds 1a and 2a are 1.542 Å25 and 1.508 Å,11 respectively. This behaviour could arise from the three P–O bonds connected to zirconium that are longer in compound 2a (1.548 Å) than in compound 1a (average: 1.515 Å). The excellent thermal stability of Zr(O3- POK)22a, which is stable up to 1200 °C as reported earlier,7 was confirmed by TDA analysis which revealed no transition upto 1000 °C except a transition at 150 °C due to the loss of residual water [Fig. 5(c)]. FTIR analysis [Fig. 6(a)] clearly indicates two P–O stretching vibrations: one at 1174 cm−1 arising from the shortest P–O bond (1.508 Å) and one at 976 cm−1 which may be attributed to the three P–O bonds with a length of 1.548 Å. It is worth noting that compound 2a absorbs water from the atmosphere leading to an increase in the broad band at 3450 cm−1 thus decreasing the resolution of the two bands at 974 and 1174 cm−1. The two bands at 552 and 614 cm−1 disappear in the amorphous sample 2c supporting the assumption that these two bands correspond to the vibrations of the network that can be observed only in a well organised structure. | ||
Fig. 1 Calculated (×), observed (—) and difference X-ray profiles
for Zr(O3POK)2. Reflection marks are shown for the
P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) space group. space group. | ||
 | ||
| Fig. 2 31P MAS NMR spectra (12 kHz, NS = 4; d1 = 60 s): (a) Zr(O3POH)2·H2O 1a: (b) Zr(O3POK)22a. | ||
The amorphous samples of Zr(O3POK)2, 2b and 2c, were obtained by an exchange process of Zr(O3POH)2·H2O 1b and 1c in the presence of KCl and KOH aqueous solutions. Compounds 1b and 1c were synthesised by the reaction of ZrOCl2·8H2O with H3PO4 in water without HF and were obtained, owing to the different conditions of reaction (1b: 14 h at reflux; 1c, 4 h at room temperature), with different specific surface areas (BET), 9 and 226 m2 g−1, respectively. The specific surface area (BET) of the potassium exchanged material 2b is 7 m2 g−1 while that of compound 2c is dramatically reduced (55.9 m2 g−1) compared to the specific surface area of the parent material 1c (226 m2 g−1). The FTIR spectra of these exchanged amorphous samples [Fig. 6(c)] clearly indicates the bands corresponding to the P–O stretching (950–1180 cm−1). Figs. 3 and 4 show the 31P MAS NMR of the parent materials Zr(O3POH)2·H2O 1b and 1c and materials 2b and 2c resulting from the exchange of the hydrogen atoms by potassium. In agreement with previous work,20 the amorphous samples of Zr(O3POH)2·H2O 1b and 1c show several signals while the crystalline sample simply shows a singlet (Fig. 2). Both the relative intensities of the signals and the specific surface area depend on the advancement of the crystallisation. The chemical shifts of the exchanged compounds 2b and 2c are displaced towards lower field as found for the crystalline sample 2a (Fig. 2) indicating the efficiency of the exchange.
 | ||
| Fig. 3 31P MAS NMR spectra (12 kHz): (a) Zr(O3POH)2·H2O 1b; (b) Zr(O3POK)22b (peaks marked with * are rotation bands). | ||
 | ||
| Fig. 4 31P MAS NMR spectra (12 kHz): (a) Zr(O3POH)2·H2O 1c; (b) Zr(O3POK)22c (peaks marked with * are rotation bands). | ||
 | ||
| Fig. 5 DTA analyses under argon (heating rate: 10 °C min−1): (a) compound 2c; (b) compound 2b; (c) compound 2a. | ||
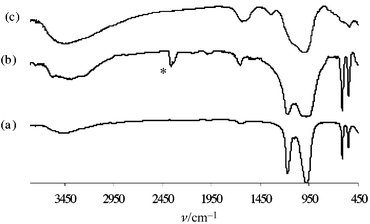 | ||
| Fig. 6 FTIR spectra of Zr(O3POK)2 in KBr: (a) anhydrous crystalline sample 2a; (b) wet crystalline sample 2a (sample left in an open flask for 1 month); (c) anhydrous amorphous sample 2c (peak marked as * is an artefact due to the presence of CO2). | ||
Thermal analysis of these two amorphous exchanged materials 2b and 2c (DTA) reveals an endothermic transition from 80 to 250 °C which is due to the loss of residual water [Fig. 5(a) and 5(b)]. Two (740 and 795 °C) and three (545, 701 and 722 °C) exothermic transitions, respectively are observed for compounds 2c [Fig. 5(a)] and 2b [Fig. 5(b)]. Partial hydrogen exchange in compound 2b could explain its thermal behaviour. Indeed, the first transition (545 °C) could be attributed to the condensation of phosphate groups into pyrophosphates as was previously reported for the non-exchanged materials Zr(O3POH)2 (transition observed at 550 °C20). The two transitions observed for compounds 2b (701 and 722 °C) and 2c (740 and 795 °C), that have almost the same relative intensity, could arise from the same transformation. These transitions were previously reported for a copper salt intercalated in α-zirconium phosphate.21 The different position of these transitions could arise from a partial exchange in compound 2b. Furthermore the absence of a transition around 550 °C for compound 2c indicates a fully exchanged material.
Zr(O3POK)2 was first tested as a basic catalyst for the Knoevenagel condensation of piperonal and Meldrum’s acid. According to the kinetic data obtained using the amorphous catalyst 2b (Table 1, entries 1 and 3), THF was the best solvent while the use of ethanol (Table 1, entry 5) inhibited the reaction. The superiority of THF over CH2Cl2 may be explained by the fact that Knovenagel condensation produces one mol of water per mol of product. Owing to the non-miscibility of water and dichlorormethane and taking into account the polar character of the catalyst, water produced by this condensation could be readily localised on the catalyst when dichloromethane is used as a solvent. Thus the efficiency of the catalyst could be affected. Nevertheless, when the reaction is carried out without solvent the reaction time is even shorter (Table 1, entries 7 and 9). Noteworthy is that the reaction times and yields are identical whatever the structure (amorphous or crystalline) of the catalyst used for the reaction (Table 1, entries 2 and 4). This is the first example of the use of amorphous potassium exchanged zirconium phosphate as a basic catalyst. In absence of solvent a total conversion is reached after only 14 minutes. Interestingly, no difference (yield, reaction times) was observed by the use of microwave heating or classical heating (isomantle). Nevertheless for the expediancy of the procedure, microwave heating (commercial microwave oven Whirlpool) was chosen. The powder X-ray diffraction of the catalyst after the condensation of piperonal with Meldrum’s acid, and before the elimination of the product, did not reveal any modifications of the d001 peak (9.02 Å), indicating that neither the substrate nor the product are present in the interlayer space. This observation indicates that the catalytic process is only located on the surface of the catalyst. Nevertheless the kinetics of the Knoevenagel condensation of piperonal with Meldrum’s acid (experiments carried out in THF, Table 1 entries 2–4) did not show any significant difference between catalysts 2a, 2b and 2c despite their different specific surface areas.
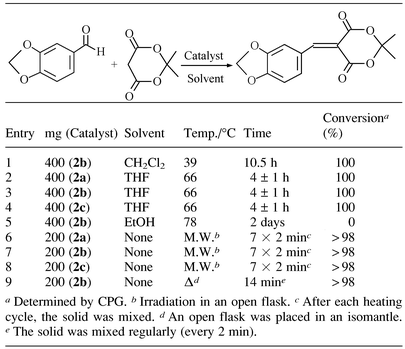
Taking into account the fact that reactions carried out without solvent contribute to a reduction in costs and waste flow it is of note that all the following reactions were achieved without solvent. The mixing step of the catalyst with substrates can affect the reproducibility of the results. The first method, which is completely solvent-free, is to mechanically mix for the catalyst with the substrates for several minutes. The second method is less laborious and consists of dissolving the substrates in a minimum of solvent followed by the addition of this solution to the catalyst. Finally the solvent is removed in vacuo leading to the substrates being absorbed on the catalyst. Although the results of these two methods were identical, we chose the second method due to the rapidity of the work up. Selected data corresponding to the condensation of piperonal with different active methylene compounds are reported in Table 2. The mechanism of the Knoevenagel condensation (under basic conditions) is that one proton from the acidic methylene compound is removed by the base to generate a nucleophile that will react with the aldehyde. In view of the results of Table 2, it appears that the catalyst 2a is efficient for methylene compounds which possess a pKa (determined in water) in the range 4–12. Surprisingly, the use of tetronic acid as the methylenic acid compound (Table 2, entry 6 ; pKa = 3.76) does not yield the condensation product. Despite the coloration of the catalyst almost all the piperonal is recovered; this coloration might be due to the auto-condensation of tetronic acid. Despite the lack of reactivity of tetronic acid it is worth mentioning that other methods are able to catalyse the aldol type condensation of tetronic acid with aldehyde in a 1∶122 or 2∶123 ratio. On the other hand, catalysts 2a are not sufficiently basic to be efficient when methylenic compounds possessing a pKa > 13 are used. To illustrate the reusability of the catalyst, catalyst 2a was engaged three times in the condensation of piperonal with Meldrum’s acid (Table 3). After each reaction the product was separated from the catalyst by sublimation. After three cycles the catalyst was still efficient as reported in Table 3.


Conclusion
The present article deals with the simplification of the synthesis of crystalline potassium exchanged zirconium hydrogen phosphate 2a. Furthermore the synthesis of the amorphous equivalent of compound 2a is described. The basic properties of these materials, which are readily available in large amounts, were used to catalyse the Knoevenagel condensation. Interestingly, both amorphous and crystalline potassium exchanged zirconium hydrogen phosphate are able to catalyse the Knoevenagel reaction. These catalysts are efficient for methylenic compounds possessing pKa values in the range of 4–12. It is noteworthy the efficiency of these catalysts is increased by the absence of solvent. The yields reported and the reaction times are identical to or better than those previously reported. Furthermore the catalyst is easily recovered and can be reused without significant loss of its efficiency.Experimental
Catalyst preparation
a = 5.188(9) [lit.,11a = b = 5.176(1)], c
= 9.022(2) [lit.,11c = 9.012(8)],
number of reflections: 162; GOF = 1.7, Rp = 0.067;
RWp = 0.093; RI = 0.08.Knoevenagel condensations
In a typical experiment 200 mg of compound 2a were added to a solution of piperonal (75 mg; 5 mmol) and Meldrum’s acid (72 mg ; 5 mmol) in dichloromethane (2 ml) placed in a 100 ml round bottomed flask. The solvent was removed under reduced pressure. The round bottomed flask was placed in a microwave oven (Whirlpool, M430, Jet 900) and irradiated (450 W) for a total of 14 min and in 2 min intervals. After cooling, the crude product was purified by sublimation (10 mTorr, isomantle at a temperature between 180 and 220 °C) to give 5-(3,4-methylenedioxyphenyl)methylene-2,2-dimethyl[1,3]dioxane-4,6-dione (0.14 g; 100%). mp 179 °C (lit.,29 178.5–179.5 °C). The results of other experiments are summarised in Table 2.Acknowledgements
We thank Valérie Montouillout and Christian Fernandez (Laboratoire de Catalyse et Spectroscopie, L.C.S. UMR CNRS 6506) for assistance in the solid-state NMR experiments.References
- J. M. Fraile, J. I. Garcia and J. A. Mayoral, Catal. Today, 2000, 57, 3 CrossRef CAS.
- A. Corma, Chem. Rev., 1997, 97, 2373 CrossRef CAS.
- G. Alberti, M. Casciola, S. Cavalaglio and R. Vivani, Solid State Ionics, 1999, 125, 91 Search PubMed.
- A. Clearfield, Chem. Rev., 1988, 88, 125 CrossRef CAS.
- A. Clearfield and D.S. Thakur, Appl. Catal., 1986, 26, 1 CrossRef CAS.
- L. Maya, J. Inorg. Nucl. Chem., 1981, 43, 400 Search PubMed.
- U. Costantino, F. Marmottini, M. Curini and O. Rosati, Catal. Lett., 1993, 22, 333 Search PubMed.
- U. Costantino, M. Curini, F. Marmottini, O. Rosati and E. Pisani, Chem. Lett., 1994, 2215 CAS.
- M. Curini, F. Epifano, M.C. Marcotullio, O. Rosati and M. Rossi, Synlett, 1999, 315 CAS.
- M. Curini, F. Epifano, M.C. Marcotullio, O. Rosati, M. Rossi and A. Tsadjout, Synth. Commun., 2000, 30, 3181 CAS.
- M. Dörffel and J. Liebertz, Z. Kristallogr., 1990, 193, 155 Search PubMed.
- L. F. Tietze and U. Beifuss, in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamon Press, Oxford, 1991, vol. 2, p. 341. Search PubMed.
- A. Corma, S. Iborra, J. Primo and F. Rey, Appl. Catal., 1994, 114, 215 CrossRef CAS.
- K. R. Kloestra and H. Van Bekkum, J. Chem. Soc., Chem. Commun., 1995, 1005 RSC.
- P. M. Price, J. H. Clark and D. J. Macquarrie, J. Chem. Soc., Dalton Trans., 2000, 101 RSC.
- I. Rousselot, C. Taviot-Guého and J. P. Besse, Int. J. Inorg. Mater., 1999, 165 CrossRef CAS.
- D. Villemin, Chem. Ind., 1983, 478 Search PubMed.
- G. Alberti and E. Torracca, J. Inorg. Nucl. Chem., 1968, 30, 317 Search PubMed.
- F. Taulelle, C. Sanchez, J. Livage, A. Lachgard and Y. Piffard, J. Phys. Chem. Solids, 1988, 49, 299 CrossRef CAS.
- H. Benhamza, P. Barboux, A. Bouhaouss, F.-A. Josien and J. Livage, J. Mater. Chem., 1991, 1, 681 RSC.
- C. Ferragia, A. La Ginestra, M. A. Massucci, P. Cafarelli and R. Di Rocco, J. Therm. Anal., 1994, 41, 1469 Search PubMed.
- H. Zimmer, W. W. Hillstrom, J. C. Schmidt, P. D. Seemuth and R. Vögeli, J. Org. Chem., 1978, 43, 1541 CrossRef CAS; D. Villemin and B. Labiad, Synth. Commun., 1990, 20, 3207 CAS.
- L. Wolff and C. Schwabe, Justus Liebigs Ann. Chem., 1896, 291, 231 Search PubMed; N. Olthoff, R. Huttenrauch and H. Brauniger, Z. Chem., 1970, 10, 341 Search PubMed.
- G. Alberti and E. Torracca, J. Inorg. Nucl. Chem., 1968, 30, 317 Search PubMed.
- A. Clearfield and G. D. Smith, Inorg. Chem., 1969, 8, 431 CrossRef CAS.
- N. J. Clayden, J. Chem. Soc., Dalton Trans., 1987, 1877 RSC.
- U. Costantino, F. Marmottini, M. Curini and O. Rosati, Catal. Lett., 1993, 22, 333 Search PubMed.
- A. Clearfield, A. Oskarsson and C. Oskarsson, Ion Exch. Membr., 1972, 1, 91 Search PubMed.
- G. A. Kraus and M. E. Krolski, J. Org. Chem., 1986, 51, 3347 CrossRef CAS.
- H. F. Ebel, CH-Acidität, (Houben-Weyl), Georg Thieme Verlag, Stuttgart, 1970, vol. 13/1, p. 31. Search PubMed.
- B. B. Corson and R. W. Staughton, J. Am. Chem. Soc., 1928, 50, 2829.
- Kauffmann, Chem. Ber., 1919, 1435 Search PubMed.
- D. Villemin and B. Labiad, Synth. Commun., 1990, 3333 CAS.
| This journal is © The Royal Society of Chemistry 2001 |