Which organic crystal structures are predictable by lattice energy minimisation?†
Theresa Beyer, Thomas Lewis and Sarah L. Price*
Centre of Theoretical and Computational Chemistry, University College London, 20 Gordon Street, London, UK WC1H 0AJ. E-mail: s.l.price@ucl.ac.uk
Abstract
A survey of the molecules which have been used in crystal structure prediction studies is presented. The results of these studies have been analysed in terms of whether the experimentally observed crystal structures are found at or near the global minimum in the lattice energy. The results suggest that whilst some crystal structures can be predicted just on the basis of lattice energy searches, there is yet insufficient experience to judge for which molecules this energetic criterion is sufficient, within the limitations of current force-field accuracy. The molecules chosen to test crystal structure prediction methods appear to be biased away from the types that would be expected to be readily predictable and suitable for crystal engineering. The survey highlights the need for more theoretical and experimental collaboration to understand what determines whether a molecule's crystal structure will be so favourable that other polymorphs are unlikely.
 Theresa Beyer | Theresa Beyer obtained her M.Sc. in Chemistry at the University of Dortmund in Germany and was awarded a fellowship of the ‘Studienstiftung des Deutschen Volkes’. She spent six months as a European exchange student at University College London in the group of Professor Sarah L. Price working on her Diplomarbeit and continued her research in this scientific field for her Ph.D. which she completed in May 2001. This work was done in collaboration with the CCDC and generously supported by the EPSRC and the CCDC. |
 Thomas Lewis | Thomas Lewis is currently reading Chemistry with Mathematics at University College London. He performed the literature project that contributed to this article as part of his third year course. He is currently performing a computational and experimental study on the potential polymorphs of parabanic acid to complete an M.Sci. degree. |
 Sarah L. Price | Sarah (Sally) Price obtained her B.A. (Natural Sciences) and Ph.D. at Clare College Cambridge, the latter under the supervision of Anthony Stone. After postdoctoral work at the University of Chicago with Jeremy Burdett, she returned to the University of Cambridge as a Fellow of Fitzwilliam College for further postdoctoral work with Anthony Stone. In 1985 she was awarded a Royal Society 1983 University Research Fellowship, which she transferred to University College London in 1989. She has been promoted through Lecturer, Reader and in 2000 to Professor in Physical Chemistry. Her research interest in developing model intermolecular potentials has spanned molecules from hydrogen to polypeptides. The applications have been so diverse that her hundred publications span the Journal of Molecular Biology to the Journal of Physics C. Her current work develops the computational prediction of organic crystal structures and properties. |
Introduction
There are many practical reasons for developing methods of computationally predicting the crystal structures of a molecule prior to its synthesis. Even when a crystal structure is known, a method of predicting possible polymorphism would have great practical value to avoid problems in the manufacture and patenting of the crystalline product. However, there are also purely scientific reasons for pursuing such studies, since any reliable method of crystal structure prediction would have to reflect the factors that fundamentally control the crystallisation process. There is still a long way to go‡,§ before we can reliably predict the crystal structures of a molecule that is being studied, for example, because of its pharmaceutical properties rather than its potential in crystal engineering. However, if instead of seeking all the possible polymorphs of a given molecule, we are interested in finding molecules that robustly form only one crystal structure, then the unique thermodynamic stability ought to make that structure relatively easy to predict. Thus crystal structure prediction calculations should provide some understanding of principles for use in the engineering of organic crystal structures.The majority of methods that have been developed for crystal structure prediction¶,||,** are based on searching for the crystal structure that corresponds to the global minimum in the lattice energy for the molecule. This is a crude implementation of the assumption that crystallisation is thermodynamically controlled. Nevertheless, implementing such a lattice energy search requires many other assumptions about the molecular structure, intermolecular forces and the molecular packing groups that need to be considered, as indicated by Fig. 1. This difficult search for the global minimum in the lattice energy is obviously insufficient for predicting polymorphism. However, the successes of this method demonstrate that, for some molecules, the observed crystal structure has a thermodynamic advantage over other possibilities such that this first approximation is sufficient. Can we tell which molecules have such predictable crystal structures?
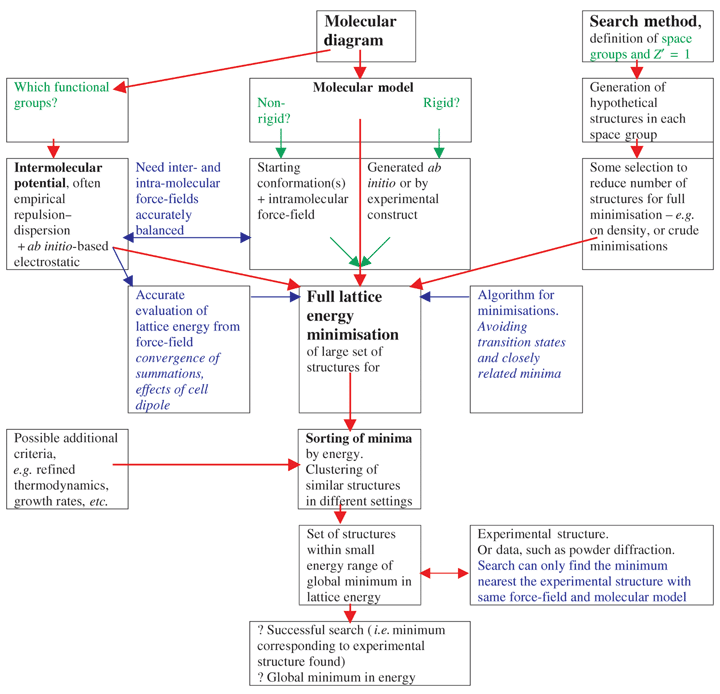 | ||
| Fig. 1 Generalised schematic for the prediction of crystal structures by lattice energy minimisation. Molecule-dependent choices, which require chemical intuition, are shown in green, and some of the technical questions in blue. | ||
We would hope that a study of whichever molecules that had been readily predicted as global minima in the lattice energy, in contrast to those that have many more energetically feasible crystal structures than known polymorphs, might provide insight into what makes a particular crystal structure uniquely favourable. This paper provides a tabulation of molecules that have been used in published crystal structure prediction studies.
Crystal structure prediction studies
There have been many different methods proposed for crystal structure prediction over the past decade or so, since computer power has enabled this huge search problem to be tackled. Table 1 lists the main methods that are based on lattice energy minimisation, noting a few of their features. The program or author name is used to classify loosely the approach within the survey, though it must be emphasised that many of the older approaches have undergone considerable development over the period of the published studies. An even stronger reason as to why the survey cannot be used to judge the efficacy of an approach is that the success of a prediction depends critically upon the accuracy of the force-field, so that it is impossible to evaluate the search method separately from the molecule-type dependent force-field. Indeed, the survey reveals that this is impossible, as there are very few crystal structures that have been studied by more than one approach. Of these examples, benzene and urea are atypical on account of their molecular symmetry and the simplicity of their intermolecular interactions.| Program | Search type | Space group symmetry constrained | Intramolecular flexibility | Electrostatic interactions included | Blind test predictions |
|---|---|---|---|---|---|
| a Some characteristics of the programs used for crystal structure prediction by lattice energy minimisation. In some cases, the hypothetical crystal structures generated are then used as starting points in a lattice energy minimisation using programs such as WMIN, PCK and DMAREL. In other cases, the program includes the minimisation process, though this will often require intermediate clustering of similar structures and rejection of unfavourable structures to reduce the computational requirements. Successful predictions using these programs in the CCDC blind tests are indicated with reference to Chart 1. G denotes a global minimum in the lattice energy search. However, there were no correct predictions of the second polymorph of I, which may be the more stable. N/A indicates programs that were not used in these tests. | |||||
| Chin | Monte Carlo simulated annealing with hydrogen bonding bias | Yes | Yes | Yes | N/A |
| CRYSTALG | Self-consistent basin-to-deformed-basin mapping global optimisation | No | No | Yes | |
| CRYSCA | Random search with steepest descent | Yes | Yes | Yes | |
| ICE9 | Systematic grid search to generate close-packed structures | Yes | No | Molecular multipole moments | N/A |
| MDCP | Constant pressure molecular dynamics to find crude structures | No | No | Yes | N/A |
| MOLPAK | Systematic search for high density structures in common co-ordination types | Yes | No | Only for minimisation | V(G) |
| MPA, extended to | Systematic, or random generation of expanded trial unit cell | No | No | Yes | I(G),V |
| Mpg | Yes | No | Yes | ||
| Perlstein | Aufbau search for low energy 1D and 2D aggregates, primarily for mono-layer predictions | Yes | Yes | Yes | N/A |
| PMC | Random search | Yes | No | Yes | |
| Polymorph Predictor | Monte Carlo simulated annealing with intermediate clustering | Yes | Yes | Yes | I(G), II, IV |
| PROMET | Selecting cohesive dimer, ribbons and layer substructures of partial space group | Yes | No | Optionally in final stages only | I(G) |
| UPACK | Systematic grid or random search, with intermediate clustering | Yes | Yes | Yes | I(G), III, VII(G) |
A more objective test of the current state of crystal structure prediction has been the blind tests††,‡‡ organised by the Cambridge Crystallographic Data Centre. In these international experiments, a large proportion of the scientists working on crystal structure prediction were given the just the information in Chart 1 and later submitted up to three ‘predictions’ of the crystal structures of some of the molecules. Successful predictions in these tests are included in Table 1. Interpreting these results requires recognising that these tests were influenced by published prediction studies. The choice of molecules was limited to general categories (space group, Z′, functional groups and type of conformational flexibility) that were expected to be within the scope of at least some of the crystal structure prediction programs. A referee, with no knowledge of the types of molecules usually studied by the participants, then chose the target molecules (Chart 1) from a short list of recently determined crystal structures provided by crystallographers who were kindly willing to delay publication.
 | ||
| Chart 1 Molecules used in the blind tests of crystal structure prediction. Successful predictions made on the basis of this information are listed in Table 1. | ||
The blind tests undoubtedly demonstrate that lattice energy minimisation is able to predict some crystal structures, but they also demonstrate that the process is far from reliable. The conclusions††,‡‡ from these workshops distinguish between crystal structures that are true and consistent absolute minima in enthalpy, and those that are minima in free energy (possibly including an equilibrium defect concentration), or are metastable and predetermined by nucleation or growth kinetics. In the first case, then incremental developments in search algorithms, inter- and intra-molecular potentials, should make the existing methods more reliable. In the latter cases, then new computational approaches are required to distinguish which of the structures that are near the global minimum in lattice energy will actually be observed. Can we distinguish which molecules are likely to have a crystal structure which falls into the first category? Such molecules would form robust, uniquely favourable crystal structures (i.e. polymorphism is unlikely), provided that the local minima in the enthalpy were sufficiently less thermodynamically stable or kinetically unobtainable.
Method
The survey presented in Table 2 was derived by a search of the Science Citation Index during Autumn 2000 and supplemented by other papers dealing with crystal structure prediction by lattice energy minimisation known to the authors. The molecules have been ordered somewhat arbitrarily. Pharmaceuticals and pigments are listed first, because they were selected for study for their relevance to industry. The other molecules are classified by chemical type, in the order, carboxylic acids and anhydrides, hydrocarbons, alcohols, sugars, and then a miscellaneous collection. The final sections are crystals that might not have been expected in this list. There are molecular materials that are not generally considered organic, but which have been studied by the same methods. The survey finishes with crystals with more than one type of molecule in the unit cell. This is currently only represented by pioneering work on carbohydrate hydrates, and the development to molecular salts and co-crystals would increase the practical use of crystal structure prediction in developing pharmaceuticals and molecular materials. The survey is provided in a downloadable spreadsheet format to allow the reader to re-order, select, or extend at will (see ESI†).| Molecule | Name | No. of known polymorphs | Space group | Z′ (Z) | Program | Year | Reference | Commenta |
|---|---|---|---|---|---|---|---|---|
| a The molecular crystals are sorted into groups (alcohols, carboxylic acids, drugs, hydrocarbons, molecular magnets, pigments, polyalcohols, pyranoses, other and hydrates). Closely related molecules within each group are together, but otherwise the molecules in each group are listed by publication year of first study. The programs are as defined in Table 1, though some significant variants are distinguished. The comments include the following points: G, the experimental structure has been found as the global minimum in the search; L, the experimental structure has been found in the search among the low energy minima, but not as the global minimum; M, the experimental structure has been found in the search, but no further information has been provided about the structures and relative energies of other low energy minima; X, the experimental structure has not been found in the search; a, the prediction has been made in advance of an experimental investigation [NB: in some cases, the prediction was then used to solve the crystal structures which were previously (and are listed as) undetermined]; r, the search has just been restricted to the experimental space group; p, powder diffraction data have been used in the study. | ||||||||
| Pharmaceuticals | ||||||||
 | Aspirin | 1 | P21/c | 1 (4) | PROMET | 1995 | 1 | G |
| Polymorph Predictor | 1999 | 2 | L | |||||
 | Paracetamol | 3 | I: P21/c | 1 (4) | Polymorph Predictor | 1998 | 3 | G(I), L(II) |
| II: Pbca | 1 (8) | MOLPAK/DMAREL | 2001 | 4 | G(I), L(II) | |||
| III: not determined | ||||||||
 | Allopurinol | 1 | P21/c | 1 (4) | MOLPAK/DMAREL | 1997 | 5 | G |
 | Estrone | 3 | P21 | 2 (4) | Polymorph Predictor | 1996 | 6 | G, L |
| P212121 | 1 (4) | r(P212121) | ||||||
| P212121 | 1 (4) | |||||||
 | Progesterone | 2 | α: P212121 | 1 (4) | Polymorph Predictor | 1999 | 7 | G(α), r |
| β: P212121 | 1 (4) | L(β), r | ||||||
 | Prednisolone tert-butylacetate | 2 | P212121 | 1 (4) | Polymorph Predictor | 1998 | 3 | L, r, p |
| +⊕not determined | ||||||||
 | Primidone | 2 | A: P21/c | 1 (4) | Polymorph Predictor | 1999 | 7 | L(A), r, p |
| B: Pbca | 1 (8) | L(B), r | ||||||
| Pigments | ||||||||
 | Pigment red | 1 | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) | 0.5 (1) | Polymorph Predictor | 1993 | 8 | L, p |
 | Quinacridone | 3 | α: not determined | 0.5 (2) | Polymorph Predictor | 1996 | 6 | G(γ), p |
| β: not determined | L(α, β), p, a | |||||||
| γ: P21/c | ||||||||
 | Indigo | 2 | A: P21/c | 0.5 (2) | MOLPAK/DMAREL | 1998 | 9 | G(A) |
| B: P21/c | 0.5 (2) | L(B) | ||||||
 | Yellow pigment ‘0137’ | 1 | Pna21 | 1 (4) | CRYSCA | 1995 | 10 | G, a |
 | Pigment yellow 74 | 1 | P | 1 (2) | CRYSCA | 1999 | 11 | G, r |
| Carboxylic acids and anhydrides | ||||||||
 | Formic acid | 1 | Pna21 (chain) | 1 (4) | CRYSTALG | 2000 | 12 | X |
| MOLPAK/DMAREL | 2000 | 13 | L | |||||
 | Tetrolic acid | 2 | α: P
(dimer) | 1 (2) | PROMET | 1997 | 14 | L(α, β), r |
| β: P21 (chain) | 1 (2) | MOLPAK/DMAREL | 2000 | 13 | L(α, β) | |||
 | Acetic acid | 2 | I: Pna21 (chain) | 1 (4) | Polymorph Predictor | 1998 | 15 | L(I, II), p |
| II: P21/c (dimer, high pressure) | Polymorph Predictor⊕+⊕UPACK (systematic) | 1998 | 16 | L(I, II) | ||||
 | Benzoic acid | 1 | P21/c (dimer) | 1 (4) | MOLPAK/DMAREL | 2000 | 13 | G |
 | Terephthalic acid | 2 | P | 0.5 (1) | Polymorph Predictor | 1996 | 6 | L (refine to either), X, p |
| P | 0.5 (1) | |||||||
 | Bromoacetic acid | 2 | I: P21/c (dimer) | 1 (4) | Polymorph Predictor | 1998 | 15 | X(I) |
| II: Pccn (dimer) | 1 (4) | |||||||
 | Chloroacetic acid | 2 | α: P21/c (tetramer) | 2 (8) | Polymorph Predictor | 1998 | 15 | L(β) |
| β: P21/c (dimer) | 1 (4) | |||||||
 | Fluoroacetic acid | 1 | P21/c (dimer) | 1 (4) | Polymorph Predictor | 1998 | 15 | L |
 | Maleic anhydride | 1 | P212121 | 1 (4) | CRYSTALG | 2000 | 12 | G |
 | Succinic anhydride | 1 | P212121 | 1 (4) | CRYSTALG | 2000 | 12 | G |
 | Trimellitic anhydride | 1 | P212121 | 1 (4) | Polymorph Predictor | 1998 | 17 | L, p |
| Hydrocarbons | ||||||||
 | Benzene | 5 | I: Pbca | 0.5 (4) | PMC | 1984–1989 | 18–22 | G(I), L(III), L(II), a |
| III: P21/c (high pressure) | 0.5 (2) | MPA | 1994–1995 | 23,24 | Low pressure: G(I), L(III) | |||
| II: not determined | P⊕<⊕1.4 GPa: G(I) | |||||||
| IV: not determined | P⊕>⊕1.4 GPa: G(III) | |||||||
| MDCP | 1995 | 25 | G(I), L(III) | |||||
| CRYSTALG | 1995 | 26 | G(I), L(III) | |||||
| ICE9 | 1996 | 27 | L(I) | |||||
| MPA | 1996 | 28 | M(I), M(III) | |||||
| 1996 | 29 | G(II), a, (0.5 GPa⊕<⊕P⊕<⊕1.0 GPa), (special investigation for form II) | ||||||
| Polymorph Predictor | 1996 | 6 | G(I), L(III) | |||||
| UPACK (systematic) | 1998 | 30 | G(I), L(III), II: a, p | |||||
| CRYSTALG | 1998 | 31 | G(I), L(III) | |||||
 | 4,5,6,7-Tetrahydro-2,8-di-isopropylidene-4,7-methano-2H-indene | 1 | P | 1 (2) | PROMET | 1991 | 32 | L, r |
 | 9,10-Di-isopropylidenetricyclo[4.3.0.12,5]deca-3,7-diene | 1 | P212121 | 1 (4) | PROMET | 1991 | 32 | G |
 | 1,4,7,10-Tetramethyldibenzo[a,e]cyclo-octene | 1 | P | 1 (2) | PROMET | 1991 | 32 | G |
 | 1-Methyl-4-hexylbenzene | 1 | P | 1 (2) | PROMET | 1991 | 32 | L, r |
 | cis,cis-Dodecahydrotriptycene | 1 | P | 1 (2) | PROMET | 1991 | 32 | L, r |
 | 1,3,5-Trineopentylbenzene | 1 | P | 1 (2) | PROMET | 1991 | 32 | X, r |
 | 9-tert-Butyl-9-(9-fluorenyl)fluorene | 1 | P | 1 (2) | PROMET | 1991 | 32 | X, r |
 | [2.0.0](1,3)-Benzeno(1,8)-naphthaleno(1,3)-benzenophane | 1 | P | 1 (2) | PROMET | 1991 | 32 | X, r |
 | 2,2-Dimethyl-3-(2-naphthyl)butane | 1 | P21 | 1 (2) | PROMET | 1991 | 32 | X, r |
 | 1-tert-Butyl-4-n-butylbenzene | 1 | Pc | 1 (2) | PROMET | 1991 | 32 | L, r |
 | 9-(2-Methyl-1-naphthyl)fluorene | 1 | P21/n | 1 (4) | PROMET | 1991 | 32 | L, r |
 | Dicyclohepta[de,ij]naphthalene | 1 | P21/c | 1 (4) | PROMET | 1991 | 32 | G |
 | 3,4:5,6-Dibenzophenanthrene | 2 | P21/c | 1 (4) | PROMET | 1991 | 32 | X (P21/c), r |
| C2/c | 1.5 (12) | |||||||
 | Bi(anthracene-9,10-dimethylene) | 1 | P21/c | 0.5 (2) | PROMET | 1991 | 32 | L, r |
 | 9c-Methyl-9cH-cyclopenta[jk]fluorene | 1 | P212121 | 1 (4) | PROMET | 1991 | 32 | X, r |
 | 1,2-Diphenylbenzene | 1 | P212121 | 1 (4) | PROMET | 1991 | 32 | L, r |
 | 1,2-Diphenylcyclopentene | 1 | P21 | 1 (2) | PROMET | 1991 | 32 | X |
 | 3,11-Dimethylene-3a,4,6,7,10,10b-hexahydro-4,10a:7,10-dimethano-3H-cyclohept[e]indene (prediction prior to synthesis) | 0 | Unknown | Unknown | PROMET | 1991 | 32 | a |
 | Ethene | 1 | P21/n | 0.5 (2) | Polymorph Predictor | 1992 | 33 | M |
 | Hexamethylbenzene | 1 | P | 0.5 (1) | Polymorph Predictor | 1992 | 33 | M |
| CRYSCA | 1994 | 34 | G, r | |||||
 | Bicyclohexylidene | 1 | P | 0.5 (1) | ICE9 | 1996 | 27 | G |
 | Pentacene | 1 | P | 1 (2) | ICE9 | 1996 | 27 | X |
 | Dibenz[a,h]anthracene | 2 | A: P21 | 1 (2) | ICE9 | 1996 | 27 | L(A) |
| B: Pcab | 0.5 (4) | L(B) | ||||||
 | Trindan | 1 | P21/c | 1 (4) | ICE9 | 1996 | 27 | L |
 | Pyrene | 1 | P21/a | 1 (4) | ICE9 | 1996 | 27 | L |
 | Durene | 1 | P21/a | 0.5 (2) | ICE9 | 1996 | 27 | L |
 | Naphthalene | 1 | P21/a | 0.5 (2) | ICE9 | 1996 | 27 | L |
 | Anthracene | 1 | P21/a | 0.5 (2) | ICE9 | 1996 | 27 | L |
 | Tetracene | 1 | P | 1 (2) | ICE9 | 1996 | 27 | X |
 | Phenanthrene | 2 | P21 | 1 (2) | ICE9 | 1996 | 27 | L(P21) |
| P21/c | 1 (4) | |||||||
 | Triphenylene | 1 | P212121 | 1 (4) | ICE9 | 1996 | 27 | L |
 | Perylene | 2 | α: P21/a | 1 (4) | ICE9 | 1996 | 27 | L(α) |
| β: P21/c (not fully determined) | 0.5 (2) | |||||||
 | n-Hexane | 1 | P | 0.5 (1) | ICE9 | 1996 | 27 | G |
| Mpg | 1999 | 35 | M | |||||
 | n-Octane | 1 | P | 0.5 (1) | ICE9 | 1996 | 27 | L |
| Mpg | 1999 | 35 | M | |||||
 | n-Pentane | 1 | Pbcn | 0.5 (4) | Mpg | 1999 | 35 | M |
 | Adamantane | 1 | P![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 21/c 21/c | 0.25 (2) | Mpg | 1999 | 35 | M |
 | Iceane | 1 | P63/m | 1/6 (2) | Mpg | 1999 | 35 | M |
 | Pentaspiro[2.0.0.2.0.2.0.0.2.0]tridecane | 1 | Pbcn | 0.5 (2) | Mpg | 1999 | 35 | M |
 | 4,5-Dimethyl-9,10-dihydrophenanthrene | 1 | P | 1 (2) | Mpg | 1999 | 36 | G |
 | sym-Hexahydropyrene | 1 | P21/c | 0.5 (2) | Mpg | 1999 | 36 | G |
 | 1,2-Dihydrocyclobutabenzene | 1 | P | 1 (2) | Mpg | 1999 | 36 | G |
| Alcohols | ||||||||
 | Methanol | 3 | α: P212121 | 1 (4) | Polymorph Predictor⊕+⊕ab initio-derived ff | 1999 | 37 | G(α) |
| β: disordered | ||||||||
| γ: not determined | ||||||||
 | Ethanol | 2 | Pc | 2 (4) | Polymorph Predictor | 1999 | 37 | L(both) |
| P21/c (high pressure) | 1 (4) | +ab initio-derived ff | ||||||
| UPACK (random) | 2000 | 38 | L(both) | |||||
 | Galactitol | 1 | P21/c | 1 (4) | UPACK (systematic) | 1999 | 39 | L, r |
 | D-glycero-L-galacto-Heptitol | 1 | P21 | 1 (2) | UPACK (systematic) | 1999 | 39 | G, r |
 | D-Altritol | 1 | P21 | 1 (2) | UPACK (systematic) | 1999 | 39 | L, r |
 | D-glycero-L-gulo-Heptitol | 1 | P21212 | 1 (4) | UPACK (systematic) | 1999 | 39 | G, r |
 | meso-glycero-allo-Heptitol | 1 | P212121 | 1 (4) | UPACK (systematic) | 1999 | 39 | L, r |
 | muco-Inositol 1,2,4,5,6-cyclohexanehexol | 1 | P21/c | 1 (4) | UPACK (systematic) | 1999 | 39 | G, r |
 | Ribitol | 1 | P21/c | 1 (4) | UPACK (systematic) | 1999 | 39 | G, r |
 | meso-D-glycero-L-ido-Heptitol | 1 | P | 1 (2) | UPACK (systematic) | 1999 | 39 | L, r |
 | meso-D-glycero-L-altro-Heptitol | 1 | Pbca | 1 (8) | UPACK (systematic) | 1999 | 39 | X, r |
 | DL-glycero-DL-galacto-Heptitol | 1 | Cc | 1 (4) | UPACK (systematic) | 1999 | 39 | G, r |
 | D-Iditol | 1 | P21 | 1 (2) | UPACK (systematic) | 1999 | 39 | G, r |
 | D-Glucitol | 1 | P212121 | 1 (4) | UPACK (systematic) | 1999 | 39 | G, r |
 | D-Mannitol | 2 | β: P212121 | 1 (4) | UPACK (systematic) | 1999 | 39 | G(β), r |
| κ: P212121 | 1 (4) | L(κ), r | ||||||
 | Allitol | 1 | P21/c | 0.5 (2) | UPACK (systematic) | 1999 | 39 | G, r |
 | DL-Arabinitol | 1 | Pna21 | 1 (4) | UPACK (systematic) | 1999 | 39 | G, r |
 | DL-Mannitol | 1 | Pna21 | 1 (4) | UPACK (systematic) | 1999 | 39 | G, r |
| epi-Inositol | 1 | P21/c | 1 (4) | UPACK (systematic) | 1999 | 39 | G, r |
 | D-glycero-L-allo-Heptitol | 1 | P21212 | 1 (4) | UPACK (systematic) | 1999 | 39 | G, r |
| L-chiro-Inositol | 1 | P21 | 1 (2) | UPACK (systematic) | 1999 | 39 | L, r |
| DL-Iditol | 1 | P21/c | 1 (4) | UPACK (systematic) | 1999 | 39 | X, r |
 | D-glycero-D-manno-Heptitol | 1 | P212121 | 1 (4) | UPACK (systematic) | 1999 | 39 | G, r |
 | Xylitol | 1 | P212121 | 1 (4) | UPACK (systematic) | 1999 | 39 | G, r |
| neo-Inositol | 1 | P | 0.5 (1) | UPACK (systematic) | 1999 | 39 | G, r |
 | Glycol | 1 | P212121 | 1 (4) | UPACK (systematic)⊕+⊕ab initio-derived ff | 2000 | 40 | L |
| +⊕extended ab initio ff and lattice vibrations | 2001 | 41,42 | G(free energy) | |||||
 | Glycerol | 1 | P212121 | 1 (4) | UPACK (systematic)⊕+⊕ab initio-derived ff | 2000 | 40 | G |
| +⊕extended ab initio ff and lattice vibrations | 2001 | 41,42 | L(2nd free energy) | |||||
| Pyranoses | ||||||||
 | α-D-Galactose | 1 | P212121 | 1 (4) | UPACK (systematic) | 1995 | 43 | G, r |
| UPACK (systematic) | 1999 | 39 | L | |||||
 | α-D-Glucose | 1 | P212121 | 1 (4) | UPACK (systematic) | 1995 | 43 | L, r |
| UPACK (systematic) | 1999 | 39 | L | |||||
 | α-D-Talose | 1 | P212121 | 1 (4) | UPACK (systematic) | 1995 | 43 | G, r |
| UPACK (systematic) | 1999 | 39 | L | |||||
 | β-D-Allose | 1 | P212121 | 1 (4) | UPACK (systematic) | 1995 | 43 | L, r |
| UPACK (systematic) | 1999 | 39 | L | |||||
 | β-D-Galactose | 1 | P212121 | 1 (4) | UPACK (systematic) | 1995 | 43 | L, r |
| UPACK (systematic) | 1999 | 39 | L | |||||
 | β-D-Glucose | 1 | P212121 | 1 (4) | UPACK (systematic) | 1995 | 43 | L, r |
| UPACK (systematic) | 1999 | 39 | L | |||||
 | Methyl α-D-altropyranoside | 1 | P212121 | 1 (4) | UPACK (systematic) | 1999 | 39 | L |
 | Methyl α-D-glucopyranoside | 1 | P212121 | 1 (4) | UPACK (systematic) | 1999 | 39 | L |
 | Methyl α-D-mannopyranoside | 1 | P212121 | 1 (4) | UPACK (systematic) | 1999 | 39 | G |
 | Methyl β-D-galactopyranoside | 1 | P212121 | 1 (4) | UPACK (systematic) | 1999 | 39 | G |
 | β-L-Arabinose | 1 | P212121 | 1 (4) | UPACK (systematic) | 1999 | 39 | L |
 | α-D-manno-2-Heptulose | 1 | P21 | 1 (2) | UPACK (systematic) | 1999 | 39 | L |
 | α-L-Fucose | 1 | P212121 | 1 (4) | UPACK (systematic) | 1999 | 39 | G |
 | Methyl 7-deoxy-L-glycero-β-D-galacto-heptopyranoside | 1 | P212121 | 1 (4) | UPACK (systematic) | 1999 | 39 | L |
 | β-Glucoheptose | 1 | P212121 | 1 (4) | UPACK (systematic) | 1999 | 39 | G |
 | 2-Deoxy-β-D-arabino-hexapyranose | 1 | P212121 | 1 (4) | UPACK (systematic) | 1999 | 39 | L |
 | 3-O-Methyl-α-D-glucopyranose | 1 | P212121 | 1 (4) | UPACK (systematic) | 1999 | 39 | G |
 | 1-Deoxy-1-methyl-α-D-glucopyranose | 1 | P212121 | 1 (4) | UPACK (systematic) | 1999 | 39 | L |
 | 1,5-Anhydro-D-glucitol | 1 | P21 | 1 (2) | UPACK (systematic) | 1999 | 39 | L |
 | 2-Desoxy-β-D-lyxohexose | 1 | P21 | 1 (2) | UPACK (systematic) | 1999 | 39 | L |
 | β-D-Fructopyranose | 1 | P212121 | 1 (4) | UPACK (systematic) | 1999 | 39 | G |
 | β-L-Lyxopyranose | 1 | P212121 | 1 (4) | UPACK (systematic) | 1999 | 39 | L |
 | Methyl α-L-arabinoside | 1 | P212121 | 1 (4) | UPACK (systematic) | 1999 | 39 | L |
 | Methyl β-L-arabinopyranoside | 1 | P21 | 1 (4) | UPACK (systematic) | 1999 | 39 | L |
 | Methyl β-D-arabinopyranoside | 1 | P212121 | 1 (4) | UPACK (systematic) | 1999 | 39 | G |
 | Methyl β-D-ribopyranoside | 1 | P212121 | 1 (4) | UPACK (systematic) | 1999 | 39 | L |
 | Methyl α-D-lyxopyranoside | 1 | P212121 | 1 (4) | UPACK (systematic) | 1999 | 39 | L |
 | Methyl 6-deoxy-α-D-idopyranoside | 1 | P212121 | 1 (4) | UPACK (systematic) | 1999 | 39 | G |
 | Methyl β-xyloside | 1 | P21 | 1 (2) | UPACK (systematic) | 1999 | 39 | L |
 | α-L-Xylopyranose | 1 | P212121 | 1 (4) | UPACK (systematic) | 1999 | 39 | L |
 | Methyl α-L-fucopyranoside | 1 | P21 | 1 (2) | UPACK (systematic) | 1999 | 39 | L |
 | Methyl α-L-rhamnopyranoside (Methyl 6-deoxy-α-L-mannopyranoside) | 1 | P212121 | 1 (4) | UPACK (systematic) | 1999 | 39 | X |
| α-D-Mannose | 1 | P212121 | 2 (8) | UPACK (random) | 2000 | 38 | X |
| Others | ||||||||
 | Azobis(isobutyronitrile) | 1 | P | 0.5 (1) | Polymorph Predictor | 1992 | 44 | G, r |
 | 4,8-Dimethoxy-3,7-diazatricyclo[4.2.2.22,5]-dodeca-3,7,9,11-tetraene | 1 | P | 0.5 (1) | Polymorph Predictor | 1992 | 44 | G, r |
 | cyclo-L-Alanyl-L-alanyl | 1 | P1 | 1 (1) | Polymorph Predictor | 1992 | 44 | L, r |
| Polymorph Predictor | 1993 | 45 | L | |||||
 | cyclo-Bis(dehydroalanyl)-3,6-dimethylenepiperazine-2,5-dione | 1 | P21 | 1 (2) | Polymorph Predictor | 1992–1993 | 44–46 | L |
 | Isoiridomyrmecin | 1 | P21 | 0.5 (1) | Polymorph Predictor | 1992 | 44 | X |
| Polymorph Predictor | 1992 | 46 | G, r | |||||
| Perlstein | 1999 | 47 | G, r | |||||
 | 2,4,6-Trinitro-N-methyldiphenylamine | 1 | P21/c | 1 (4) | MOLPAK/WMIN | 1993 | 48 | G |
 | Methyl β-D-glucopyranoside tetranitrate | 1 | P212121 | 1 (4) | MOLPAK/WMIN | 1993 | 48 | G |
 | cis-1,3,5,7-Tetranitro-1,3,5,7-tetraazadecalin | 1 | P21 | 1 (2) | MOLPAK/WMIN | 1993 | 48 | G |
 | 2,3-Dimethyl-2,3-dinitrobutane | 1 | P | 1 (2) | MOLPAK/WMIN | 1993 | 48 | L |
 | 1,4-Dinitrocyclo-octatetraene | 1 | Pna21 | 1 (4) | MOLPAK/WMIN | 1993 | 48 | M |
 | 2-Nitro-6,7,8,9-tetrahydronaphtho[2,1-b]furan | 1 | P | 1 (2) | MOLPAK/WMIN | 1993 | 48 | M |
 | 2-Methoxy-N-(4-nitrobenzylidene)-5-pyridylamine | 2 | P21/c | 1 (4) | MOLPAK/WMIN | 1993 | 48 | M(P21) |
| P21 | 1 (2) | |||||||
 | 1,5-Dinitro-3,7-methylene-1,3,5,7-tetraazocane | 1 | P21/c | 1 (4) | MOLPAK/WMIN | 1993 | 48 | M |
 | 1,8-Dinitronaphthalene | 2 | P212121 | 1 (4) | MOLPAK/WMIN | 1993 | 48 | L(P212121) |
| Form II: I2/a | 1 (8) | |||||||
 | 1,1,5,5-Tetranitro[4]peristylane | 1 | P21 | 1 (2) | MOLPAK/WMIN | 1994 | 49 | G, p |
 | Pyrimidine | 1 | Pna21 | 1 (4) | MDCP | 1995 | 25 | X |
| CRYSTALG | 2000 | 12 | X | |||||
 | Imidazole | 1 | P21/c | 1 (4) | CRYSTALG | 2000 | 12 | G |
 | m-Nitroaniline | 1 | Pbc21 | 1 (4) | MPA | 1995 | 50 | G |
 | 1,2-Dimethoxyethane | 1 | C2/c | 0.5 (4) | MDCP | 1995 | 51 | G(C2/c) |
 | 4-Amidinoindanone guanylhydrazone | 2 | A: P21/c | 1 (4) | Polymorph Predictor | 1996 | 52 | L(A) |
| B: P | 1 (2) | G(B), r, p | ||||||
 | 7-Dimethylaminocyclopenta[c]coumarin | 2 | Pbca | 1 (8) | PROMET | 1996 | 53 | G(Pbca) |
| Pna21 | 1 (4) | L(Pna21) | ||||||
 | Urea | 1 | P21m | 0.25 (2) | Polymorph Predictor | 1996 | 6 | G |
| MPA | 1996 | 28 | M | |||||
| Mpg | 1999 | 35 | G | |||||
 | Formamide | 1 | P21/n | 1 (4) | CRYSTALG | 2000 | 12 | G |
 | Alloxan | 1 | P41212 | 0.5 (4) | MOLPAK/DMAREL | 1997 | 54 | G |
 | 2-Pyridone | 1 | P212121 | 1 (4) | PROMET | 1997 | 55 | G |
| MOLPAK/WMIN | +⊕discussion | L | ||||||
 | 6-Azauracil | 1 | P212121 | 1 (4) | MOLPAK/DMAREL | 1997 | 5 | L(∼G) |
 | 5-Azauracil | 1 | Pbca | 1 (8) | MOLPAK/DMAREL | 1999 | 56 | G, a |
 | Uracil | 1 | P21/a | 1 (4) | MOLPAK/DMAREL | 1997 | 5 | G |
 | 2-Amino-5-nitropyrimidine | 3 | I: P21/c | 1 (4) | MOLPAK/DMAREL | 1998 | 57 | G(≈I and II), X |
| II: P21/n | 1 (4) | X(III) | ||||||
| III: Pccn | 1 (4) | |||||||
 | 3,6-Bis(diazo)cyclohexanetetraone | 1 | P21/c | 0.5 (2) | Mpg | 1999 | 35 | M |
 | 3′,5′-Di-O-acetyl-(2′S)-deoxy-2′-fluoro-2′-((4-methoxyphenyl)sulfinyl))uridine | 1 | P1 | 1 (1) | Perlstein | 1999 | 47 | G, r |
 | (−)-1-(4-Dimethylaminophenyl)-2-(2-hydroxypropylamino)cyclobutene-3,4-dione | 1 | P1 | 1 (1) | Perlstein | 1999 | 47 | G, r |
 | Tetrahydrofuran | 1 | C2/c | 0.5 (4) | CRYSCA | 1994 | 34 | G, r |
 | 1,4-Dioxane | 2 | I: P21/c | 0.5 (2) | UPACK (systematic) | 1999 | 37 | G(I) |
| II: P21/c | 0.5 (2) | +⊕ab initio-derived ff | L(II) | |||||
 | 4,4,5,5-Tetramethyl-4,5-dihydro-1H-imidazol-1-oxyl 3-oxide [2-Hydronitronylnitroxide radical (HNN)] | 2 | α: P21/c | 1 (4) | PROMET | 1999 | 58 | G(α), p |
| β: P21/c | 4 (16) | |||||||
 | 3,6-(Cyclotetramethylene)-2,5-diketopiperazine | 1 | P | 0.5 (1) | Chin | 1999 | 59 | G, p |
 | 3,6-(Tetramethyl)-2,5-diketopiperazine | 1 | P | 0.5 (1) | Chin | 1999 | 59 | G, p |
 | 3,6-(4,4-Dimethylcyclohexane)-2,5-diketopiperazine | 1 | P | 0.5 (1) | Chin | 1999 | 59 | G, p |
 | 1,2-Dichlorobenzene | 1 | P21/n | 1 (4) | PROMET | 2001 | 60 | X, a |
 | 1,3-Dichlorobenzene | 1 | P21/c | 2 (8) | PROMET | 2001 | 60 | X, a |
 | 1,4-Dichlorobenzene | 3 | α: P21/a | 0.5 (2) | PROMET | 2001 | 60 | X(α) |
| β: P | 0.5 (1) | L(β) | ||||||
| γ: P21/c | 0.5 (2) | L(γ) | ||||||
 | Hexasilylbenzene | 1 | P | 0.5 (1) | CRYSCA | 1994 | 34 | G, r |
| Inorganics and organometallics treated by the same methods | ||||||||
 | Carbon dioxide | 2 | Pa3 | 1(4) | MDCP | 1995 | 61 | G(Pa3) |
| Cmca (high pressure) | 1(4) | G(Cmca) (at high pressure) | ||||||
 | Chlorine | 1 | Cmca | 0.5 (8) | MPA | 1997 | 62 | L |
 | Hexasulfur | 1 | R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) | 1/6 (1) | CRYSTALG | 1998 | 31 | L |
 | C60 fullerene | Complex with disorder | Pa3-P/H | PMC | 1999 | 63 | G(P/H) | |
 | Pentamethylferrocene, Cp*FeCp | 1 | P | 1 (2) | CRYSCA | 1994 | 11,34,64 | G, a |
 | Decamethylruthenocene, (Cp*)2Ru | 1 | P21/m | 0.5 (2) | CRYSCA | 1994 | 34,64 | G, r |
 | Bis(mesitylene)niobium | 1 | P | 1 (2) | CRYSCA | 1994 | 34,64 | G, r |
 | (µ-η4:η3-1,4-Dimethyl-2,3-dimethylenecyclopentenyl)bis(µ-hydrido)bis(η5-tetramethylcyclopentadienyl)dititanium | 1 | Pnma | 0.5 (4) | CRYSCA | 1994 | 34,64 | G, r |
 | (η5-Pentamethylcyclopentadienyl)(η4-trimethylenemethane)rhodium, Cp*Rh(TMM) | 1 | P21/c | 1 (4) | CRYSCA | 1994 | 34,64 | G, r |
 | (η4-Trimethylenemethane)tricarbonylruthenium, (TMM)Ru(CO)3 | 1 | P | 1 (2) | CRYSCA | 1994 | 34,64 | G |
 | (η4-Cyclobutadiene)tricarbonyliron, (C4H4)Fe(CO)3 | 1 | Pnma | 0.5 (4) | CRYSCA | 1994 | 34,64 | G |
 | Chromium hexacarbonyl | 1 | Pnma | 0.5 (4) | CRYSCA | 1994 | 34 | G |
 | Nickel tetracarbonyl | 1 | Pa | 1/3 (8) | CRYSCA | 1994 | 34,64 | G |
| Hydrates | ||||||||
 | 2,4-Di-O-methyl-α-D-galactopyranose monohydrate | 1 | P212121 | 1 (4) | UPACK (random) | 2000 | 38 | L |
 | Methyl 7-deoxy-D-glycero-β-D-galacto-heptopyranoside monohydrate | 1 | C2 | 1 (4) | UPACK (random) | 2000 | 38 | L |
 | Methyl α-D-galactopyranoside monohydrate | 1 | P212121 | 1 (4) | UPACK (random) | 2000 | 38 | L |
 | α-L-Rhamnose monohydrate | 1 | P21 | 1 (2) | UPACK (random) | 2000 | 38 | L |
 | (+)-Ononitol monohydrate | 1 | P1 | 1 (1) | UPACK (random) | 2000 | 38 | L |
| α-D-Glucose monohydrate | 1 | P21 | 1 (2) | UPACK (random) | 2000 | 38 | L |
 | D-manno-3-Heptulose monohydrate | 1 | P212121 | 1 (4) | UPACK (random) | 2000 | 38 | L |
| meso-D-glycero-L-altro-Heptitol monohydrate | 1 | C2/c | 1 (8) | UPACK (random) | 2000 | 38 | X |
| myo-Inositol dihydrate | 1 | P21/a | 1 (4) | UPACK (random) | 2000 | 38 | X |
We have attempted to classify the success of the prediction as to whether the experimental structure was the global minimum found in the search (G), another local minimum (L) or not found (X). Many early papers are not explicit as to whether the structure was the global or local minimum, in which case it is classified as M. Often there is insufficient information in the papers to have any criterion beyond the author's conclusions as to the success of the search. Hence an apparent success (G or L) could be recorded for a poor force-field which did not provide a good reproduction of the crystal structures. Unfortunately, we also found that too few papers gave sufficient information to make the G/L classification as meaningful as we might like. The energy gap between the global minimum and all other hypothetical structures in a reasonably exhaustive search would have been very useful. A significant energy gap implies a readily predicted robust crystal structure, provided that the experimental structure corresponds to the global minimum. An insignificant gap, with the experimental structures being close to the global minimum in energy, suggests potential polymorphism. (This is, of course, very dependent upon the accuracy of the force-field, as the relative energies have to be interpreted according to their likely inaccuracies. If the experimental structure corresponds to a local minimum that is significantly less stable than the global minimum, there are probably deficiencies in the force-field.)
A few other indicative qualifiers are in the Comment section of Table 2. An ‘a’ indicates a prediction made in advance of the determination of the crystal structure. This occasionally refers to predictions published as challenges to experimentalists, though for entries where the crystal structure is given this usually means a collaborative project. In some of these cases the powder patterns of the hypothetical structures were utilised in solving the structure from X-ray powder data. This is an important application of crystal structure prediction, but the distinction between the application of ab initio crystal structure predictions and lattice energy modelling to assist structure solution (which we are not including) is often unclear. Thus, any study that involved the use of powder data is denoted by ‘p’. All searches performed only in the experimental space group are denoted ‘r’, though the significance of this restriction obviously depends upon the space group. Searches restricted by known cell dimensions are not included.
Results and discussion
The most obvious feature of the survey in Table 2 is that of the almost two hundred molecules (189) that have been used in published crystal structure prediction studies, the majority fall into classes that would generally be considered hard to predict. We appear to have been testing some of the boundaries of crystal structure prediction methods, rather than going for easy targets.Sugars are notoriously hard to crystallise, and the conformational flexibility of the OH groups in pyranoses and alcohols produces considerable variation in the directions of the structure-determining hydrogen bonds. The success of the Utrecht group in predicting almost all of the 60 such crystal structures as minima, with almost 30 as global minima, is impressive, despite the restrictions on space group and conformational freedom that were initially imposed. The careful work that has been involved in improving the force-fields, and including lattice vibration effects and more than one molecule in the asymmetric cell, is appreciable, and has pushed crystal structure prediction on the basis of thermodynamic quantities to the practical limits of current computational chemistry. They have also pioneered work on more than one type of molecule in the asymmetric unit by studying hydrates. This considerable body of work advances crystal structure prediction and provides an understanding of sugar crystal structures, rather than undermining the general expectation that carbohydrates have a multitude of low energy crystal structures.
Similarly, over 40 hydrocarbon crystal structures were investigated, mainly because the intermolecular forces are well parameterised, rather than because these forces were expected to be sufficiently strong and directional to determine the crystal structures. About a quarter of the searches found the experimental structure as a global minimum, including some molecules whose three-dimensional shape is sufficiently irregular and lumpy that the packing of the bumps of one molecule into the hollows of another might be expected to produce only one close packing. However, the crystal structures of other such molecules were not found as minima. In most cases, lattice energy minimisation produced many energetically feasible structures for hydrocarbons, as might have been expected from the weakness of the forces.
The carboxylic acid group is a synthon in crystal engineering,§§,¶¶ but once the dimers or chains have been formed, then the many ways in which the chains or dimers pack are controlled by the rest of the molecule. Indeed, acetic acid is now widely cited as the classic example of a molecule where there is a multitude of low energy structures, both chains and dimers, within a small energy range of the global minimum. Terephthalic acid is the only dicarboxylic acid to have been considered, and although it forms the expected hydrogen bonded chains, nature has already found two ways of packing these chains.
Systems that are known to be polymorphic are obviously not readily predicted. The survey includes 29 molecules with more than one known crystal structure, though many of the polymorphs were either not structurally determined or are outside the capabilities of current search methods. Whilst the true extent of polymorphism is very debatable, 15% of our survey molecules is orders of magnitude higher than the proportion of molecules with two or more well characterised structures deposited in the Cambridge Structural Database. It is worth noting that the number of well characterised polymorphic systems is rapidly increasing. In 1995 only 163 organic (C/H/N/O/F/Cl/S) molecules in the Cambridge Structural Database were found|||| with complete X-ray crystallographic structural determinations of more than one polymorphic form, at room temperature and with comparable refinement accuracy, whereas a similar search*** in 1999 showed that the number of such systems had almost doubled to 321. Crystal structure prediction studies of polymorphic systems are essential for learning more about this controversial phenomenon,†††,‡‡‡ and have also been used to help determine crystal structures of polymorphs from powder data. However, polymorphic systems are almost by definition not robust and are difficult to predict. (A grey area in such a distinction is when the known polymorphs are found to be more stable than all hypothetical structures. Indigo provides one such example, where the hydrogen bonded sheet is the same in both known polymorphs.)
Thus, given this caveat about the sampling, it is very encouraging to note that out of 253 searches for a particular polymorphic form, it was found as the global minimum in 103 cases, and as a local minimum 112 times, with 16 cases not differentiating. The 26 failures to locate the experimental structure is not a reliable guide to the difficulties of crystal structure predictions: these are mainly examples in the large datasets studies of sugars and hydrocarbons, or one missing structure in a polymorphic system. Inevitably, crystal structure prediction studies that the authors chose to work on and publish (Table 2) give a rather different impression of the reliability of the methods from the results of the blind tests where only three submissions were allowed (crudely represented in Table 1). Apart from the factor of failed studies not being published, it must be remembered that many participants in the blind tests report having found the experimental structures as minima which were not in their submitted three choices. In addition, considerably more time and effort can go into force-field development for major projects, whereas blind test challenges were often tackled with considerable concern as to whether the hastily constructed force-field would be adequate.
It is encouraging that approximately half the crystal structures have been found as global minima in crystal structure prediction studies (allowing that some local minima are correct predictions of metastable polymorphs). This is likely to represent an underestimate of crystal structures that are minimum enthalpy structures. Some crystal structures may have been found as local minima because of inadequacies in the force-field rather than because of a plurality of energetically feasible structures. Missing structures (X) probably represent inadequacies in the search procedure, provided that there is a local minimum in the potential energy surface reasonably close to the experimental structure. However, the number of polymorphic systems where one known structure has been found as the global minimum emphasises that there may be other accessible polymorphs. Without knowing whether there are other structures within an energy range of probable polymorphism, it is impossible to deduce how reliably the global minimum structure will be formed. Such analysis will remain difficult whilst we are unsure of the experimental energy differences between known polymorphs, let alone of the allowance that should be made for errors in the computational methods.
The survey does show a much higher proportion of global minima amongst the industrial and miscellaneous molecules than amongst the hydrocarbons, carboxylic acids and pyranoses, indicative that crystal packings of molecules with multiple rigidly disposed (non-hydrocarbon) functional groups are more predictable. The survey obviously lacks the range of studies for any statistically valid conclusions, but it is consistent with steric and electronic complementarity§§§ regulating the predictability of crystal structures. The number of very non-planar molecules is fairly limited, and so only gives an indication that an irregular shape may favour predictability, but certainly does not guarantee it. However, the number of molecules with such shapes that do not show conformational flexibility is also limited.
The results illustrate that “the supramolecular behaviour of a particular functional group depends acutely on the nature and even the location of the other functionalities in the molecule”.§§§ Multiple hydrogen bond donors and acceptors may generally favour highly directional interactions, but this can be overcome by other factors. This is shown by alloxan, whose experimental structure (found as a global minimum) has no conventional length hydrogen bonds. Isomers have very different degrees of robustness in their crystal structures: 5-azauracil has an energetically favoured hydrogen bonded sheet structure, whereas 6-azauracil has a variety of different hydrogen bonding motifs within a very small energy range of its global minimum. The lattice energy of the more predictable structure (5-azauracil) is ca. 12 kJ mol−1 more stable than that of its isomer. This shows that the relative disposition of the functional groups determines which isomer can pack in a crystal structure optimally, satisfying the potentially strong intermolecular interactions, and forming a robust network.
Molecules containing supramolecular synthons designed to give robust, multiple hydrogen bonded motifs are poorly represented in the survey. Notable examples are the diketopiperazine derivatives, which are correctly predicted to form the observed hydrogen bonded tapes. In contrast, the tape of double parallel N–H⋯N hydrogen bonds expected from the 2-aminopyrimidine motif was found in one polymorph of 2-amino-5-nitropyrimidine, but not predicted because it required a torsional rotation of the nitro group to allow a favourable packing of the tapes. The alternative hydrogen bonding network found in the other two polymorphs was predicted. Thus, crystal structure prediction does have considerable potential for aiding the crystal engineer, by testing both whether the engineered motif is sufficiently robust to survive any competitive effects from problems of packing the other functional groups, and whether polymorphism is likely. This potential is yet to be realised, probably because the objectives and modus operandi of ab initio crystal structure prediction and crystal engineering have been perceived as very different.¶¶¶ We hope that this survey will encourage more collaboration between theoreticians and crystal engineers, to realise the potential complementarity. The survey shows that the range of functional groups where adequate force-fields exist is increasing, and the hydrate studies represent progress towards computational predictions for structures with more than one molecular fragment in the asymmetric unit. Hence, crystal structure prediction methods should become more widely applicable to molecular materials in the near future.
Conclusions
This survey of crystal structure prediction papers has been presented to highlight and stimulate thought on the following problem. The crystal structures of many molecules have been predicted by searching for a global minimum in the lattice energy, in some cases without knowledge of the experimental structures. The same methods have failed on similar molecules. In individual cases this has been attributed to problems with the molecular model, intermolecular potential or search method. However, there is also the more fundamental problem that determines the robustness of the prediction, as to whether the molecule has one thermodynamically favoured structure or many structures within a few kJ per mole of the global minimum. Although this survey has not been able to distinguish these cases, the number of crystal structures correctly predicted as global minima in the lattice energy does suggest that many molecules have one sufficiently thermodynamically preferred structure so as to be predictable by lattice energy minimisation. Unfortunately, we cannot yet tell whether a molecule's crystal structure is so readily predictable without performing extensive calculations.When there are several energetically feasible structures, then searching for low energy structures is only the first phase in attempting to predict which structures are likely to be observable polymorphs. We are beginning to see studies that take some sort of account of temperature effects on the thermodynamics, growth rates, mechanical stability, nucleation. etc., either by computational modelling or indirectly, using the known crystal structures in the Cambridge Structural Database as the criterion|||||| for the prediction. However, predicting polymorphism reliably remains a great challenge.
If we ask whether crystal structure predictions can help crystal engineering by suggesting what will favour a molecule having one thermodynamically favoured structure, then the computational methods are reaching a state of development when they should be able to help. Unfortunately, the survey shows that a large proportion of crystal structure prediction studies have been made on molecules that are either known to be polymorphic or are expected to be hard to predict. The survey, reflecting the literature, lacks the detailed information and statistically significant numbers of molecules to support the deduction of any guidelines for supramolecular engineering. However, we hope that by highlighting this problem we may stimulate some thought and collaborative research.
Acknowledgements and caveats
The survey table was originally part of Theresa Beyer's Ph.D. thesis, and we thank the EPSRC and CCDC for financial support of her studies. We would also like to thank many colleagues who have provided corrections and updates as this was prepared for publication. We apologise for the remaining errors and omissions, and must stress that in trying to keep the number of categories and volume of information manageable, many simplifications have been made. The reader should consult the original literature for more definitive information on the prediction methods, crystal structures, force-field dependence and the success of the studies.References to survey
- A. Gavezzotti and G. Filipinni, J. Am. Chem. Soc., 1995, 117, 12
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 299 CAS.
299 CAS. - R. S. Payne, R. C. Rowe, R. J. Roberts, M. H. Charlton and R. Docherty, J. Comput. Chem., 1999, 20, 262 CrossRef CAS.
- P. Verwer and F. J. J. Leusen, in Reviews in Computational Chemistry, ed. K. B. Lipkowitz and D. B. Boyd, John Wiley and Sons, Inc., New York, 1998, p. 327 Search PubMed.
- T. Beyer, G. M. Day and S. L. Price, J. Am. Chem. Soc., 2001, 123, 5086 CrossRef CAS.
- S. L. Price and K. S Wibley, J. Phys. Chem. A, 1997, 101, 2198 CrossRef CAS.
- F. J. J. Leusen, J. Cryst. Growth, 1996, 166, 900 CrossRef CAS.
- R. S. Payne, R. J. Roberts, R. C. Rowe and R. Docherty, Int. J. Pharm., 1999, 177, 231 CrossRef CAS.
- H. R. Karfunkel, B. Rohde, F. J. J. Leusen, R. J. Gdanitz and G. Rihs, J. Comput. Chem., 1993, 14, 1125 CrossRef CAS.
- S. L. Price and T. Beyer, Trans. ACA, 1998, 33, 23 Search PubMed.
- M. U. Schmidt and U. Englert, European Crystallographic Meeting 16, Lund, Sweden, 1995.
- M. U. Schmidt, in Crystal Engineering: From Molecules to Crystals to Materials, ed. D. Braga, F. Grepioni and A. G. Orpen, Kluwer, Dordrecht, 1999, pp. 331–348 Search PubMed.
- J. Pillardy, R. J. Wawak, Y. A. Arnautova, C. Czaplewski and H. A. Scheraga, J. Am. Chem. Soc., 2000, 122, 907 CrossRef CAS.
- T. Beyer and S. L. Price, J. Phys. Chem. B, 2000, 104, 2647 CrossRef CAS.
- A. Gavezzotti, G. Filippini, J. Kroon, B. P. van Eijck and P. Klewinghaus, Chem. Eur. J., 1997, 3, 893 CrossRef CAS.
- R. S. Payne, R. J. Roberts, R. C. Rowe and R. Docherty, J. Comput. Chem., 1998, 19, 1 CrossRef CAS.
- W. T. M. Mooij, B. P. van Eijck, S. L. Price, P. Verwer and J. Kroon, J. Comput. Chem., 1998, 19, 459 CrossRef CAS.
- J. A. Kaduk, J. T. Golab and F. J. J. Leusen, Cryst. Eng., 1998, 1, 277 CAS.
- A. V. Dzyabchenko, J. Struct. Chem., 1984, 25, 416 CrossRef.
- A. V. Dzyabchenko, J. Struct. Chem., 1984, 25, 559 CrossRef.
- A. V. Dzyabchenko and M. V. Bazilevskii, J. Struct. Chem., 1985, 26, 553 CrossRef.
- A. V. Dzyabchenko, J. Struct. Chem., 1987, 28, 862 CrossRef.
- A. V. Dzyabchenko, Sov. Phys. Crystallogr., 1989, 34, 131 Search PubMed.
- T. Shoda, K. Yamahara, K. Okazaki and D. E. Williams, J. Mol. Struct. (THEOCHEM), 1994, 119, 321 CrossRef.
- T. Shoda, K. Yamahara, K. Okazaki and D. E. Williams, J. Mol. Struct. (THEOCHEM), 1995, 333, 267 CrossRef CAS.
- N. Tajima, T. Tanaka, T. Arikawa, T. Sakurai, S. Teramae and T. Hirano, Bull. Chem. Soc. Jpn., 1995, 68, 519 CAS.
- K. D. Gibson and H. A. Scheraga, J. Phys. Chem., 1995, 99, 3765 CrossRef CAS.
- A. M. Chaka, R. Zaniewski, W. Youngs, C. Tessier and G. Klopman, Acta Crystallogr., Sect. B, 1996, 52, 165 CrossRef.
- D. E. Williams, Acta Crystallogr., Sect. A, 1996, 52, 326 CrossRef.
- M. M. Thiéry and C. Rérat, J. Chem. Phys., 1996, 104, 9079 CrossRef CAS.
- B. P. van Eijck, A. L. Spek, W. T. M. Mooij and J. Kroon, Acta Crystallogr., Sect. B, 1998, 54, 291 CrossRef.
- R. J. Wawak, J. Pillardy, A. Liwo, K. D. Gibson and H. A. Scheraga, J. Phys. Chem. A, 1998, 102, 2904 CrossRef CAS.
- A. Gavezzotti, J. Am. Chem. Soc., 1991, 113, 4622 CrossRef CAS.
- R. J. Gdanitz, Chem. Phys. Lett., 1992, 190, 391 CrossRef CAS.
- M. U. Schmidt, Kristallberechnungen Metallorganischer Molekülverbindungen, Verlag Shaker, Technische Universität Aachen, Aachen, Germany, 1995 Search PubMed.
- D. Gao and D. E. Williams, Acta Crystallogr., Sect. A, 1999, 55, 621 CrossRef.
- D. E. Williams, in Crystal Engineering: From Molecules to Crystals to Materials, ed. D. Braga, F. Grepioni and A. G. Orpen, Kluwer, Dordrecht, 1999, pp. 295–310 Search PubMed.
- W. T. M. Mooij, B. P. van Eijck and J. Kroon, J. Phys. Chem. A, 1999, 103, 9883 CrossRef CAS.
- B. P. van Eijck and J. Kroon, Acta Crystallogr., Sect. B, 2000, 56, 535 CrossRef.
- B. P. van Eijck and J. Kroon, J. Comput. Chem., 1999, 20, 799 CrossRef CAS.
- W. T. M. Mooij, B. P. van Eijck and J. Kroon, J. Am. Chem. Soc., 2000, 122, 3500 CrossRef CAS.
- B. P. van Eijck, W. T. M. Mooij and J. Kroon, J. Comput. Chem., 2001, 22, 805 CrossRef CAS.
- B. P. van Eijck, J. Comput. Chem., 2001, 22, 816 CrossRef CAS.
- B. P. van Eijck, W. T. M. Mooij and J. Kroon, Acta Crystallogr., Sect. B, 1995, 51, 99 CrossRef.
- H. R. Karfunkel and R. J. Gdanitz, J. Comput. Chem., 1992, 13, 1171 CrossRef CAS.
- H. R. Karfunkel, F. J. J. Leusen and R. J. Gdanitz, J. Comput.-Aided Mater. Des., 1993, 1, 177 Search PubMed.
- H. R. Karfunkel and F. J. J. Leusen, speedup, 1992, 6, 43 Search PubMed.
- J. Perlstein, in Crystal Engineering: From Molecules to Crystals to Materials, ed. D. Braga, F. Grepioni and A. G. Orpen, Kluwer, Dordrecht, 1999, pp. 23–42 Search PubMed.
- J. R. Holden, Z. Du and H. L. Ammon, J. Comput. Chem., 1993, 14, 422 CrossRef CAS.
- H. L. Ammon, Z. Du, J. R. Holden and L. A. Paquette, Acta Crystallogr., Sect. B, 1994, 50, 216 CrossRef.
- T. Shoda and D. E. Williams, J. Mol. Struct. (THEOCHEM), 1995, 357, 1 CrossRef CAS.
- T. Arikawa, N. Tajima, S. Tsuzuki, K. Tanabe and T. Hirano, J. Mol. Struct. (THEOCHEM), 1995, 339, 115 CrossRef CAS.
- H. R. Karfunkel, Z. J. Wu, A. Burkhard, G. Rihs, D. Sinnreich, H. M. Bürger and J. Stanek, Acta Crystallogr., Sect. B, 1996, 52, 555 CrossRef.
- A. Gavezzotti, Acta Crystallogr., Sect. B, 1996, 52, 201 CrossRef.
- D. S. Coombes, G. K. Nagi and S. L. Price, Chem. Phys. Lett., 1997, 265, 532 CrossRef CAS.
- A. Gavezzotti, Faraday Discuss., 1997, 106, 63 RSC.
- B. P. Potter, R. A. Palmer, R. Withnall, B. Z. Chowdhry and S. L. Price, J. Mol. Struct., 1999, 485–486, 349 CrossRef CAS.
- C. B Aakeröy, M. Nieuwenhuyzen and S. L. Price, J. Am. Chem. Soc., 1998, 120, 8986 CrossRef.
- G. Filippini, A. Gavezzotti and J. J. Novoa, Acta Crystallogr., Sect. B, 1999, 55, 543 CrossRef.
- D. N. Chin, T. R. Palmore and G. M. Whitesides, J. Am. Chem. Soc., 1999, 121, 2115 CrossRef CAS.
- R. Boese, M. T. Kirchener, J. D. Dunitz, G. Filippini and A. Gavezzotti, Helv. Chim. Acta, 2001, 84, 1561 CrossRef CAS.
- T. Hirano, S. Tsuzuki, K. Tanabe and N. Tajima, Chem. Lett., 1995, 1073 CAS.
- D. E. Williams and D. Gao, Inorg. Chem., 1997, 36, 782 CrossRef CAS.
- A. V. Dzyabchenko, V. Agafonov and V. A. Davydov, J. Phys. Chem. A, 1999, 103, 2812 CrossRef CAS.
- M. U. Schmidt and U. Englert, J. Chem. Soc., Dalton Trans., 1996, 2077 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: downloadable version of Table 2. See http://www.rsc.org/suppdata/ce/b1/b108135g/ |
| ‡ A. Gavezzotti, Acc. Chem. Res., 1994, 27, 309. |
| § A. Gavezzotti, Crystallogr. Rev., 1998, 7, 5. |
| ¶ P. Verwer and F. J. J. Leusen, in Reviews in Computational Chemistry, ed. K. B. Lipkowitz and D. B. Boyd, John Wiley and Sons, Inc., New York, 1998, p. 327. |
| || R. Docherty and W. Jones, in Organic Molecular Solids: Properties and Applications, ed. W. Jones, CRC Press, Boca Raton, FL, 1997, ch. 5. |
| ** R. J. Gdanitz, Curr. Opin. Solid State Mater. Sci., 1998, 3, 414. |
| †† J. P. M. Lommerse, W. D. S. Motherwell, H. L. Ammon, J. D. Dunitz, A. Gavezzotti, D. W. M. Hofmann, F. J. J. Leusen, W. T. M. Mooij, S. L. Price, B. Schweizer, M. U. Schmidt, B. P. van Eijck, P. Verwer and D. E. Williams, Acta Crystallogr., Sect. B, 2000, 56, 697. |
| ‡‡ W. D. S. Motherwell, H. L. Ammon, J. D. Dunitz, A. Dzyabchenko, P. Erk, A. Gavezzotti, D. W. M. Hofmann, F. J. J. Leusen, J. P. M. Lommerse, W. T. M. Mooij, S. L. Price, H. A. Scheraga, B. Schweizer, M. U. Schmidt, B. P. van Eijck, P. Verwer and D. E. Williams, Acta Crystallogr., Sect. B, 2001, in preparation. |
| §§ G. R. Desiraju, Crystal Engineering, Elsevier, Amsterdam, 1991. |
| ¶¶ C. B. Aakeröy, Acta Crystallogr., Sect. B, 1997, 53, 569. |
| |||| A. Gavezzotti and G. Filipinni, J. Am. Chem. Soc., 1995, 117, 12299. |
| *** L. Yu, G. A. Stephenson, C. A. Mitchell, C. A. Bunnell, S. V. Snorek, J. J. Bowyer, T. B. Borchardt, J. G. Stowell and S. R. Byrn, J. Am. Chem. Soc., 2000, 122, 585. |
| ††† J. D. Dunitz and J. Bernstein, Acc. Chem. Res., 1995, 28, 193. |
| ‡‡‡ T. L. Threlfall, Analyst, 1995, 120, 2435. |
| §§§ G. R. Desiraju, Science, 1997, 278, 404. |
| ¶¶¶ B. Moulton and M. J. Zaworotko, Chem. Rev., 2001, 101, 1629. |
| |||||| W. D. S. Motherwell, Nova Acta Leopold. NF, 1999, 79, 89. |
| This journal is © The Royal Society of Chemistry 2001 |