Matching of molecular and supramolecular symmetry. An exercise in crystal engineering
Praveen K.
Thallapally
a,
Kakali
Chakraborty
b,
Amy K.
Katz
c,
H. L.
Carrell
c,
Sambasivarao
Kotha
*b and
Gautam R.
Desiraju
*a
aSchool of Chemistry, University of Hyderabad, Hyderabad, 500 046, India. E-mail: desiraju@uohyd.ernet.in
bDepartment of Chemistry, Indian Institute of Technology, Powai, Mumbai, 400 076, India
cThe Institute for Cancer Research, Fox Chase Cancer Center, 7701 Burholme Avenue, Philadelphia, PA 19111, USA
Abstract
With the aim of understanding the transfer of molecular threefold symmetry into supramolecular systems, the crystal structures of 2,4,6-tris(5-chloro-3-pyridyloxy)-1,3,5-triazine, 1,3,5-tris(3-methylphenyl)benzene and 1,3,5-tripyrrolylbenzene are described. In all three cases, C3 molecular symmetry is carried over into hexagonal crystal packing. This is rationalized in terms of the supramolecular or void symmetry and the relevant intermolecular interactions.
One of the goals of crystal engineering is to obtain crystals that have or lack a particular symmetry element. In this context, it is useful to consider relationships between molecular and supramolecular symmetry under three categories: (1) the symmetry of the molecule is lowered in the crystal; (2) the crystal symmetry is higher than the molecular symmetry; (3) the molecular and crystal symmetries are matched. The first of these categories is very common but not so useful in crystal design,1 being merely a restatement of Kitaigorodskii's close-packing principle.2 A common example in the second category is when carboxylic groups with 1-symmetry are hydrogen bonded as dimers with
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) -symmetry. But, such symmetry increase has not been explored systematically. The third category listed above, symmetry matching, is very useful in crystal engineering. However, structural chemists
and crystal engineers are used to thinking of the close-packing principle as being of near-universal applicability, and, therefore, such studies of symmetry carry-over of higher-fold symmetry elements have generally not been pursued.
-symmetry. But, such symmetry increase has not been explored systematically. The third category listed above, symmetry matching, is very useful in crystal engineering. However, structural chemists
and crystal engineers are used to thinking of the close-packing principle as being of near-universal applicability, and, therefore, such studies of symmetry carry-over of higher-fold symmetry elements have generally not been pursued.
Here, we discuss the packing features of three diverse molecules, 1–3, with threefold symmetry that crystallize in space groups with 3-axes. In all of these three C3 molecules – 2,4,6-tris(5-chloro-3-pyridyloxy)-1,3,5-triazine31, 1,3,5-tris(3-methylphenyl)benzene42 and 1,3,5-tripyrrolylbenzene53
– the symmetry of the molecule coincides with a crystal symmetry element.

Triazine 1 when crystallized from 1∶1 hexane–EtOAc adopts the centrosymmetric space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) (no. 148)
(Table 1). The asymmetric unit contains 1/3 molecule. The C3i-symmetric Piedfort Unit7
(C3i-PU) formed by an assembly of two stacked molecules of 1 may be identified (Fig. 1). The two molecules in the C3i-PU are related by a -axis and are held together by weak intermolecular interactions,8
π⋯π, C–H⋯O and C–H⋯N (Table 2). The C3i-PUs are stacked along [0
0 1] in a staggered manner to produce a columnar structure, which in turn is stabilized by short N⋯Cl interactions (3.18 Å, 148.5°). The PU stacks are themselves related by 31-axes being connected with C–H⋯N hydrogen bonds (Fig. 2). This helical arrangement is finally stabilized by C–H⋯Cl interactions. Both the formation of the C3i-PUs via
π⋯π stacking and the interconnection of the PUs via C–H⋯N hydrogen bonds are important in preserving the supramolecular threefold symmetry in 1.
(no. 148)
(Table 1). The asymmetric unit contains 1/3 molecule. The C3i-symmetric Piedfort Unit7
(C3i-PU) formed by an assembly of two stacked molecules of 1 may be identified (Fig. 1). The two molecules in the C3i-PU are related by a -axis and are held together by weak intermolecular interactions,8
π⋯π, C–H⋯O and C–H⋯N (Table 2). The C3i-PUs are stacked along [0
0 1] in a staggered manner to produce a columnar structure, which in turn is stabilized by short N⋯Cl interactions (3.18 Å, 148.5°). The PU stacks are themselves related by 31-axes being connected with C–H⋯N hydrogen bonds (Fig. 2). This helical arrangement is finally stabilized by C–H⋯Cl interactions. Both the formation of the C3i-PUs via
π⋯π stacking and the interconnection of the PUs via C–H⋯N hydrogen bonds are important in preserving the supramolecular threefold symmetry in 1.
 | ||
| Fig. 1 Stacking of triazine 1 molecules to form a C3i-Piedfort Unit. Molecules at different levels are shaded differently. C–H⋯O and C–H⋯N interactions are shown as dashed lines. | ||
 | ||
| Fig. 2 C–H⋯N (pyridine) hydrogen bonds (dashed lines) connect Piedfort Units in a threefold helical arrangement in the crystal structure of 1. Notice the synthon (void) symmetry. Click image or 2.htm to access a 3D representation. | ||
| Parameter | 1 | 2 | 3 |
|---|---|---|---|
| a All data were collected at T⊕=⊕120 K using a MADNES-Messerschmidt and Pflugrath diffractometer with MoKα radiation (λ⊕=⊕0.71073 Å). b Full-matrix, least squares refinement on F2 using SHELXL-97.6 c Click b104431c.txt for full crystallographic data (CCDC 164454–164456). | |||
| Empirical formula | C18H9N6O3Cl3 | C27H24 | C18H15N3 |
| M | 463.66 | 348.46 | 273.33 |
| Crystal system | Hexagonal | Hexagonal | Hexagonal |
| Space group |
R |
P63 |
Rc |
| a/Å | 21.189(3) | 12.3060(17) | 19.420(3) |
| b/Å | 21.189(3) | 12.3060(17) | 19.420(3) |
| c/Å | 7.1030(14) | 7.5710(15) | 6.6760(13) |
| V/Å3 | 2761.8(8) | 992.9(8) | 2180.4(6) |
| Z | 6 | 2 | 6 |
| D c/Mg m−3 | 1.673 | 1.166 | 1.166 |
| F(000) | 1404 | 372 | 864 |
| μ/mm−1 | 0.535 | 0.066 | 0.076 |
| θ Range for data collection/° | 3.08–28.55 | 3.63–27.48 | 3.30–27.48 |
| Index ranges | −27⊕≤⊕h⊕≤⊕26; −23⊕≤⊕k⊕≤⊕25; −9⊕≤⊕l⊕≤⊕8 | 0⊕≤⊕h⊕≤⊕15; −13⊕≤⊕k⊕≤⊕0; 0⊕≤⊕l⊕≤⊕9 | 0⊕≤⊕h⊕≤⊕25; −21⊕≤⊕k⊕≤⊕0; −8⊕≤⊕l⊕≤⊕8 |
| Reflections collected | 4168 | 818 | 1020 |
| Unique reflections | 1456 | 818 | 543 |
| R 1 | 0.0425 | 0.0613 | 0.0547 |
| wR 2 | 0.1060 | 0.1631 | 0.1396 |
The next compound of interest is 2. This hydrocarbon crystallizes in the non-centrosymmetric space group P63 (no. 173) (Table 1). The asymmetric unit contains 1/3 molecule. The 63-related molecules are connected by C–H⋯π interactions (2.94 Å, 3.914 Å, 150.1°) involving the CH3 groups as donors9 (Fig. 3). Further stabilization is provided by C(phenyl)–H⋯π interactions (2.78 Å, 3.791 Å, 154.3°). Additionally, adjacent CH3 groups of threefold-related molecules form a hydrophobic core (methyl pool) that effectively defines the supramolecular void in this packing. All these interactions are combined to generate the hexagonal network structure.
 | ||
| Fig. 3 Stereoview of the crystal structure of hydrocarbon 2. Molecules shaded differently are located on 3-axes and are connected to 63-related molecules by C–H⋯π interactions. Notice the methyl-rich hydrophobic core enclosing a void with C3 symmetry. Click image or 3.htm to access a 3D representation. | ||
When heterocycle 3 is crystallized from chlorobenzene, it crystallizes in the centrosymmetric hexagonal space group Rc
(no. 167)
(Table 1).† The asymmetric unit contains 1/6 molecule. Molecules are stacked at a distance of 3.34 Å and, remarkably, with no offset (Fig. 4). The π–π stacking interactions are very significant. They arise from the electron-deficient nature of the aromatic rings10 and could be major determinants
in the generation of a hexagonal crystal structure, in that there is no offset. The reader should note that the existence of any lateral offset would necessarily break the hexagonal symmetry.
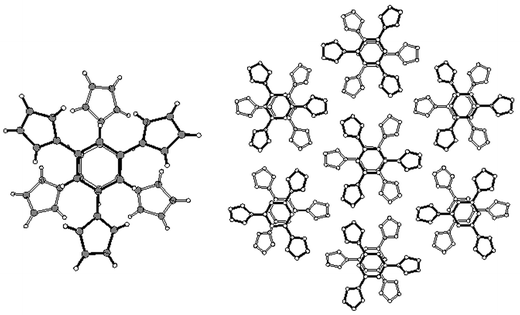 | ||
| Fig. 4 Dimer (left) and complete structure (right) of heterocycle 3. H atoms are removed in the right view for clarity. Notice how the absence of lateral offset in π–π stacking is compatible with supramolecular hexagonal symmetry. | ||
Finally, it should be noted that for none of the compounds studied here is the full molecular symmetry transferred into the crystal. For example, in the tripyrrolyl derivative 3, there is the possibility that the molecule could crystallize with Z′⊕=⊕1/12 with the molecule being planar. In the observed structure, Z′⊕=⊕1/6 and the heterocyclic rings are inclined at an angle of 31° to the plane of the central phenyl ring. This is not surprising. The ubiquity and near-universality of the close-packing principle is such that even partial retention of molecular symmetry in the crystal is noteworthy.
This study shows that matching of molecular and supramolecular symmetry is achieved when the symmetry of the molecule matches the symmetry of the intermolecular voids, in other words the supramolecular synthon11 symmetry. Synthon symmetry follows from the nature, number and relative positioning of intermolecular interactions; by no means need these be connected to the molecular symmetry, at least in a general sense. For example, closely related analogues of compounds 1–3 crystallize in low symmetry, close-packed structures.12
These difficulties do not apply for the most part in substituted tetraphenylmethanes where there is a facile symmetry carry-over between molecules and crystals. In these compounds, S4 molecular symmetry relates to tetragonal crystal symmetry because of the characteristic tetraphenyl embrace,13 which encloses a tetrahedral void. Even the space groups of these compounds may be roughly predicted to be either P![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 21c or I.14 In trigonal molecules, such symmetry carry-over is more difficult because there is no clear classification of the pertinent supramolecular synthons in terms of their symmetry properties. The present work attempts to identify some synthons that connect C3 molecular symmetry and hexagonal crystal packing. The next step would be to identify more molecules that crystallize with these synthons, thus establishing
robustness.
21c or I.14 In trigonal molecules, such symmetry carry-over is more difficult because there is no clear classification of the pertinent supramolecular synthons in terms of their symmetry properties. The present work attempts to identify some synthons that connect C3 molecular symmetry and hexagonal crystal packing. The next step would be to identify more molecules that crystallize with these synthons, thus establishing
robustness.
Acknowledgements
We thank Dr Ashwini Nangia (Hyderabad) for discussions and Professor Joseph Zyss (Cachan) for measuring the β value of compound 3. P. K. T. thanks the CSIR for fellowship support; K. C. and S. K. thank RSIC-Mumbai for recording spectral data; A. K. K. and H. L. C. acknowledge financial support from grant CA-10925 of the National Institute of Health; G. R. D. acknowledges financial support from CSIR project 01(1570)/99/EMR-II.References
- G. R. Desiraju, Crystal Engineering: The Design of Organic Solids, Elsevier, Amsterdam, 1989 Search PubMed.
- A. I. Kitaigorodskii, Molecular Crystals and Molecules, Academic Press, New York, 1973 Search PubMed.
- F. C. Schaefer, J. T. Thurston and J. R. Dudley, J. Am. Chem. Soc., 1951, 73, 2990 CrossRef CAS.
- S. Kotha, K. Chakraborty and E. Brahmachary, Synlett., 1999, 1621 CrossRef CAS.
- H. A. M. Biemans, C. Zhang, P. Smith, H. Kooijman, W. J. J. Smeets, A. L. Spek and E. W. Meijer, J. Org. Chem., 1996, 61, 9012 CrossRef CAS PubMed.
- G. M. Sheldrick, SHELXL-97, Program for Refinement of Crystal Structures, University of Göttingen, Germany, 1997 Search PubMed.
- A. S. Jessiman, D. D. MacNicol, P. R. Mallinson and I. Vallance, J. Chem. Soc., Chem. Commun., 1990, 1619 RSC.
- G. R. Desiraju and T. Steiner, The Weak Hydrogen Bond in Structural Chemistry and Biology, Oxford University Press, Oxford, 1999 Search PubMed.
- V. R. Thalladi, R. Boese, S. Brasselet, I. Ledoux, J. Zyss, R. K. R. Jetti and G. R. Desiraju, Chem. Commun., 1999, 1639 RSC.
- F. Cozzi, M. Cinquini, R. Annunziata, T. Dwyer and J. S. Siegel, J. Am. Chem. Soc., 1992, 114, 5729 CrossRef CAS.
- G. R. Desiraju, Angew Chem., Int. Ed. Engl., 1995, 34, 2311 CrossRef CAS.
- P. K. Thallapally, K. Chakraborty, H. L. Carrell, S. Kotha and G. R. Desiraju, Tetrahedron, 2000, 56, 6721 CrossRef CAS.
- I. Dance and M. Scudder, Chem. Eur. J., 1996, 2, 481 CrossRef CAS PubMed.
- M. A. Lloyd and C. P. Brock, Acta Crystallogr., Sect. B, 1997, 53, 780 CrossRef.
Footnote |
| † The nonlinear hyperpolarisability (β) of 3 measured at 1.064 µm in CHCl3 is (1.9⊕±⊕0.4)⊕×⊕10−30 esu. |
| This journal is © The Royal Society of Chemistry 2001 |