Principles of crystal packing and intermolecular motifs for Ph3XSnXPh3, X⊕=⊕C, Si, Ge, Sn
Basem Alia, Ian Dance*b, Marcia Scudderb and Don Craigb
aDepartment of Chemistry, Al-al Bayt University, Mafraq, Jordan
bSchool of Chemistry, University of New South Wales, Sydney, NSW 2052, Australia. E-mail: I.Dance@unsw.edu.au
Abstract
The crystallisation and crystal structure of Ph3CS3CPh31 are reported and analysed in the context of the known crystal structures of the class of compounds Ph3XSnXPh3, n⊕=⊕1–6, X⊕=⊕C, Si, Ge, Sn. The goal is to understand the crystal packing forces and patterns. A general pattern of intermolecular motifs is evident. Molecules Ph3XSnXPh3 with n⊕>⊕1 normally crystallise with sixfold phenyl embraces (6PEs) of XPh3 at both ends, forming parallel but curved strands of molecules ·Ph3XSnXPh3·6PE·Ph3XSnXPh3·6PE·. For n⊕=⊕3, 4, 5 and 6 these strands are further associated by a mixed sulfur phenyl embrace with composition S2(Ph)4, named 2S4PE. This embrace includes local interactions between phenyl groups and the lone-pair electron density of sulfur, and is calculated to have a stabilising energy within the range 4–8 kcal mol−1, compared with 10.5–13.0 kcal mol−1 for the 6PEs in these molecules. In addition to the 6PEs and 2S4PEs there are some local herringbone arrays of phenyl groups, generating efficient crystal packing. The exception to this pattern, 1, does not form 6PEs or a 2S4PE, but instead forms lateral and end-to-end motifs with less concerted multiple phenyl embraces. This set of compounds demonstrates substitutional polymorphism, that is polymorphism where the molecules are not identical but have internal iso-stereochemical substitution. Recrystallisations of 1 have not yet yielded its expected dimorph.
Introduction
The sixfold phenyl embrace (6PE) is an established intermolecular motif occurring between XPh3 groups in a variety of molecules.1–5 The 6PE is comprised of a set of six phenyl rings engaged in a cycle of six concerted edge-to-face (EF) local interactions between phenyl groups and is commonly centrosymmetric. The 6PE is widespread in crystals containing the Ph4P+ cation,3,6–8 in metal complexes containing PPh3 ligands,2,9–11 and in the structures of diverse molecules like Ph3ER with molecular C3 symmetry.12,13Here, we describe and analyse the crystal packing and supramolecularity of molecules Ph3XSnXPh3 where two XPh3 groups occur at the ends of a simple flexible polysulfane chain Sn. These differ from previous instances of molecules with two or more XPh3 groups where the intramolecular geometry restricts the directions of the XPh3 groups, as, for example, in orthogonal M(PPh3)2 moieties (cis and trans), and trigonal M(PPh3)3.11 Polysulfane chains provide conformationally variable linkages, and use the minimum number of atoms needed for a connecting chain. Unlike most other systems, molecules Ph3XSnXPh3 contain low-volume and flexible connectors between XPh3 groups. Furthermore, molecules Ph3XSnXPh3 are chemically simple, and allow only two types of intermolecular interaction – S⋯Ph3X and S⋯S – in addition to multiple phenyl embraces. This simplicity of atom type is important for the purpose of unravelling and assessing the relative influences of supramolecular motifs.
A search of the Cambridge Structural Database14,15 (CSD) (version 5.20, October 2000) for structures of the type Ph3XSnXPh3 (where X⊕=⊕any non-P atom) revealed 11 examples. These, together with the new crystal structure for Ph3CS3CPh3 reported here, are listed with cell dimensions and space groups in Table 1. Only one crystal (refcode YAVDAA, Ph3CS5CPh3·CHCl316) includes solvent, and the general non-occurrence of solvent in this set of 12 crystals is an indication of efficient crystal packing.
| n | X | Source (REFCODE) | Space group | Cell dimensions | |
|---|---|---|---|---|---|
| a, b, c/Å | (α), (β), (γ)/° | ||||
a This structure has also been determined at −130![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) °C (DTPGES).b There are three entries for this structure in the CSD.c Includes disordered chloroform in the lattice.16 °C (DTPGES).b There are three entries for this structure in the CSD.c Includes disordered chloroform in the lattice.16 | |||||
| 1 | C | PHMESF | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) | 8.83, 9.47, 17.59 | 90.15, 92.58, 106.58 |
| 1 | Si | DEBYAK | P21/n | 17.08, 14.55, 12.25 | 97.27 |
| 1 | Ge | DTPGES02a | P21/c | 11.08, 15.71, 18.81 | 107.29 |
| 1 | Ge | DTPGES01 | P212121 | 9.62, 17.35, 18.41 | — |
| 1 | Sn | TPSNSLb | P212121 | 18.50, 17.62, 9.81 | — |
| 2 | C | PEKZAG | P21/c | 13.94, 12.10, 17.30 | 103.63 |
| 3 | C | 1, this work | P21/c | 13.92, 12.79, 20.68 | 124.39 |
| 3 | Ge | CAFFAQ | P21/n | 11.97, 17.98, 16.74 | 100.37 |
| 4 | Si | WEYHOX | P | 9.44, 9.46, 18.82 | 82.11, 78.95, 83.15 |
| 5 | C | YAVDAAc | P21/n | 8.76, 16.97, 25.23 | 95.40 |
| 6 | C | YAVDEE | P | 9.52, 10.33, 18.54 | 85.78, 80.04, 67.45 |
Table 1 reveals the occurrence of polymorphism – crystal packing isomerism – which is also a valuable phenomenon in understanding crystal supramolecularity. Ph3GeSGePh3 crystallises in two different lattices, as conventional dimorphs. However, the polymorphism of the Ph3XSnXPh3 set of crystals also involves molecules with internal homologous substitution, and is an extension of the conventional view of polymorphism as crystal isomers of the same molecule.17–23 In the Ph3XSnXPh3 set of compounds the atom X is C, Si, Ge or Sn, varying only in size and not stereochemistry, and this variation occurs inside the surface of the molecule and does not directly influence the intermolecular interactions. This substitutional polymorphism is analogous to the ‘iso-electronic’ polymorphism described by Braga and Grepioni for variation of metal,24 and illustrates the need for a wider perspective of polymorphism in inorganic contexts.25 In the current collection of compounds there are four molecules Ph3XSXPh3 with X⊕=⊕C, Si, Ge, Sn, with four different crystal lattices (Table 1): these are substitutional polymorphs. Further, there are two molecules Ph3XS3XPh3 with X⊕=⊕C, Ge, and with different crystal lattices. The question that arises here is whether these subtle internal molecular differences are sufficient to cause distinctly different crystal packing. This bears on a fundamental issue in the fields of crystal supramolecularity, crystal structure prediction, and crystal engineering, namely the relative influences of (1) the energy wells in the potential energy surface for molecular configurations and (2) the energy wells in the potential energy surface for crystal packing.
Previous investigations of intermolecular interactions involving S atoms in molecular crystals have focussed on statistical analyses of S⋯S metrical properties in crystals, looking at the environs of individual surface S functions.26–30 In order to investigate the fundamental issues mentioned in the previous paragraph we analyse the full crystal packing for the compounds to be described, and we calculate some relevant intermolecular energies.
Experimental
Crystallisation and structure analysis of Ph3CS3CPh3, 1
The compound Ph3CS3CPh31 crystallised from a mixture of Ph3CSH, Et3N and MePh3P+Br− (1 mmol each) in 20 ml warm CH3CN, exposed to air. Oxidation to Ph3CS3CPh3 and S–C scission are presumed to have generated Ph3CS3CPh3, which was identified by X-ray diffraction (see Table 2).| Parameter | 1 |
|---|---|
| a Click b103381f.txt for full crystallographic data (CCDC 162199). | |
| Empirical formula | C38H30S3 |
| M | 582.8 |
| Crystal system | Monoclinic |
| Space group | P21/c |
| a/Å | 13.919(10) |
| b/Å | 12.792(4) |
| c/Å | 20.684(15) |
| β/° | 124.39(3) |
| V/Å3 | 3039(3) |
| T/K | 294 |
| Z | 4 |
| Dc/g cm−3 | 1.27 |
| μ(Mo-Kα)/mm−1 | 0.258 |
| 2θmax/° | 50 |
| Unique reflections | 5338 |
| Observed reflections | 3246 |
| Rmerge | 0.014 |
| R | 0.040 |
| Rw | 0.047 |
In view of the substitutional dimorphism with Ph3GeS3GePh3 (CAFFAQ, Table 1), Ph3CS3CPh3 was recrystallised in various ways to investigate the possibility of forming other crystalline phases. Chlorocarbons were investigated because CHCl3 occurs in the reported crystals of Ph3CS5CPh3.16,31 The following crystallisation methods all yielded only the original crystalline phase (checked by diffraction).
Recrystallisation of Ph3CS3CPh3 from CHCl3–EtOH (9∶1) by slow evaporation at room temperature.
Recrystallisation from CHCl3–EtOH by slow cooling of a boiling solution to room temperature.
Slow diffusion of liquid EtOH into a saturated solution of the compound in CHCl3.
Recrystallisation from CH2Cl2 solution by evaporation or by diffusive addition of liquid EtOH.
Slow or fast cooling of a hot, saturated solution in DMF.
Calculation of intermolecular energies
Intermolecular energies are calculated in the summed atom approximation, eqn. (1). The intermolecular potential for atoms i, j with charges qi, qj separated by dij is given by eqn. (2), comprising the van der Waals and coulombic energies. The parameter daij is the distance corresponding to the maximum van der Waals stabilising energy eaij. The van der Waals atom parameters ea and da, expressed as ea (kcal mol−1) and ra (Å), are: C (aromatic), 0.080, 1.95; X, 0.054, 2.0; S, 0.20, 2.1; and H, 0.02, 1.62. The combination rules are given in eqn. (3) and eqn. (4). The permittivity ε in eqn. (2) is distance-dependent, ε⊕=⊕dij, in our calculations. | (1) |
 | (2) |
 | (3) |
 | (4) |
The atom partial charges qi used for the electrostatic component were determined with guidance from density functional calculations of electronic structure and atomic charges, while maintaining overall consistency for partial charges used for benzene and other XPh3 systems. The charges used were: qC(aromatic)⊕=⊕−0.1, qH⊕=⊕0.12, qS⊕=⊕−0.02, and qX in the range 0.01–0.06 to give a neutral molecule.
Results
Ph3XSnXPh3 linked by 6PE into strands
Crystals of the molecules Ph3XSnXPh3 with central sections CS2C, GeS3Ge, SiS4Si, CS5C and CS6C contain molecules connected by a 6PE at each end, as shown in Fig. 1. The dominant feature in the crystal packing of these compounds is these infinite strands of molecules, which can also be regarded as centrosymmetric 6PE motifs linked by polysulfane chains with variable lengths and conformations. All of the 6PEs are well formed. In two cases the 6PE motifs at either end of the molecule are crystallographically equivalent. The variability of the X⋯X distance in these 6PEs is not due to the size of X, but is a manifestation of the relatively soft energy potential for a 6PE. In all cases except CAFFAQ the S–X bonds at the ends of a polysulfane chain are non-parallel, and the XSnX section is curved, such that the strands occur as waves. This curvature is pronounced for Ph3CS6CPh3 YAVDEE, where there is a distinctive wave structure in the crystal packing as shown in Fig. 2. | ||
| Fig. 1 The five instances of infinite strands of molecules Ph3XSnXPh3 linked by 6PE at each end, in the crystals identified by the CSD refcodes marked. X is blue, S is yellow. In this and subsequent figures the 6PE motif is symbolised by a black and white X⋯X candystripe. The 6PE motifs are identified by their X⋯X separations (Å). | ||
 | ||
| Fig. 2 Crystal packing in Ph3CS6CPh3 YAVDEE, showing the parallel waves of curved strands of molecules connected by 6PE at each end. The tetrahedral (trityl) C atom is marked blue, and the S atoms are marked as larger orange spheres; H atoms are not shown. | ||
In Ph3CS3CPh31 the Ph3C ends are not involved in 6PE motifs, and only one of the four Ph3XSXPh3 crystals contains a 6PE motif (at one end of one molecule). These different structures will be presented after the main features of the crystal packing of the compounds in Fig. 1 are described and analysed.
Fig. 2 shows that the strands in Ph3CS6CPh3 YAVDEE are parallel, and this feature recurs in each of the five structures of Fig. 1. In this set of five, Ph3CS5CPh3 YAVDAA is affected by the presence of chloroform disordered in the lattice, and will be described separately. In order to appreciate the similarities and differences in the crystal packing of the four structures YAVDEE (CS6C), WEYHOX (SiS4Si), CAFFAQ (GeS3Ge) and PEKZAG (CS2C), we present in Fig. 3 comparative representations of these four crystal structures viewed along the direction of propagation of the strand.
 | ||
| Fig. 3 Comparative representations of the crystal packing of (a) YAVDEE (P), (b) WEYHOX (P), (c) CAFFAQ (P21/n) and (d) PEKZAG (P21/c), viewed along the strands connected by the 6PE. In each case the X atom is blue, and the 6PEs are marked with black and white candystripes; H atoms are omitted. The unit cell of WEYHOX has been transformed from that published33 to a more comparable cell. | ||
A number of features are observed in Fig. 3. (1) The array of strands is approximately square in PEKZAG and WEYHOX, closer to hexagonal in YAVDEE, and less regular in CAFFAQ. (2) The position of the Sn chain in the projection of the strand is on the edge of the strand in YAVDEE and (to a lesser extent) in WEYHOX, near the centre in CAFFAQ, and nearer the edge in PEKZAG. This is related to the curvature of the strands. (3) There are variations in the inclinations and mutual relationships of the 6PE X⋯X vectors as projected along the strand. In YAVDEE the projections of all 6PE vectors are oriented in the same direction, and the two different 6PE motifs are superimposed in projection, even though this is not required by crystallographic symmetry. This is possible because the S6 chain can extend outside the ends of the 6PE. In WEYHOX the two distinct 6PE vectors are inclined slightly differently, whereas in CAFFAQ the 6PEs are aligned close to the strand propagation direction, and in PEKZAG the 6PE directions form a distinct herringbone pattern. There are some analogies between these projections of 6PE directions and those of the zig-zag infinite chains of 6PEs which occur in many crystals of Ph4P+.6
The next question is about the interactions between the strands: what are these interactions, and how are they responsible for the various strand juxtapositions shown in Fig. 3? We believe that the influential inter-strand interactions are not between the Sn chains protruding from the strands, such as those which appear to be close in projection (Fig. 3). The S⋯S separation for maximum van der Waals attractive energy is ca. 4.2 Å (not the sum of the crystallographic van der Waals radii, which corresponds to weak repulsion1). In the present set of structures the shortest inter-strand S⋯S distance is 4.7 Å (in YAVDAA), which is weakly attractive. Instead of Sn⋯Sn attraction, there is a recurring motif involving four phenyl rings and two S atoms, as described in the next section.
The recurring S2(Ph)4 embrace, 2S4PE
The two-sulfur four-phenyl embrace (2S4PE) is comprised of (1) two motifs that are phenyl edge (or vertex) over phenyl face (EF or VF), and (2) involvement of S lone-pair electron density with H atoms of phenyl groups of the other molecule. Instances of the 2S4PE are illustrated in Fig. 4, and are identified by the X⋯X distance. These pictures show also the probable location of the relevant lone-pair electron density on sulfur, because the H and C atoms of the opposing phenyl groups are close to this density. Usually the α-S atom is involved, although YAVDEE-6.59 uses the α-S and γ-S atoms whereas YAVDEE-7.26 uses the β-S and γ-S atoms. We postulate that S lone-pair electron density provides weak attraction for the H atoms on the other molecule, analogous to a very weak hydrogen bond. The recurrence of this motif with similar characteristics in the various crystal structures is evidence that it is a significant factor in crystal packing and not a consequence of other factors. There are also some intramolecular occurrences of these S⋯Ph interactions.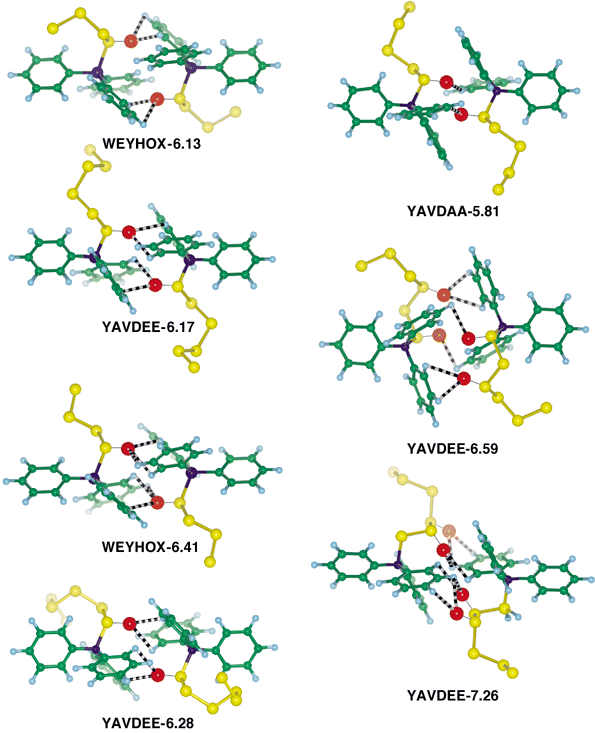 | ||
| Fig. 4 Pictures of the two-sulfur four-phenyl embraces (2S4PEs) occurring in WEYHOX, YAVDAA and YAVDEE. Each embrace is identified with its X⋯X separation (Å). X atoms are blue, S atoms are yellow, and the large red spheres indicate the locations of electron density in relevant lone pairs (LPs) on S in the interaction zone: these assume tetrahedral hybridisation at S, and an LP–S distance of 1.49 Å. Each 2S4PE is centrosymmetric. The shortest LP⋯H interactions in each case are marked as black and white candystripes. Note the approach to collinearity of the distal X–Ph bonds, and therefore the similarity with the 6PE: this embrace is also regarded as a pseudo-6PE (ps-6PE). | ||
Embrace energies
Before locating these 2S4PEs in the crystal lattices, and showing how they connect the 6PE-stranded molecules, we consider their legitimacy and influence in crystal packing by estimation of their energies relative to those of the 6PE. The calculated energies are presented in Table 3. Since the distal sections of the 2S4P-embracing molecules have variable conformations and variable approach to the embrace zone, comparable energies for the 2S4PE and 6PE motifs have been obtained by summing only over the Ph3XS sections of each molecule: note that for the 6PE the full energy (per pair of Ph3XSnXPh3 molecules) is similar to the interaction per pair of Ph3XS sections (Table 3). The conclusion from Table 3 is that the stabilising energy of the 2S4PE is in the range 4–8 kcal mol−1, compared with the 6PE energy in the range 10.5–13 kcal mol−1. The lesser number of phenyl rings and EF local interactions is the main reason for the difference, but nevertheless the 2S4PE is a supramolecular motif energetically comparable with hydrogen bonds.| Crystal | 6PE/kcal mol−1 (X⋯X/ Å) | 2S4PE/kcal mol−1 (X⋯X/ Å) |
|---|---|---|
| Ph3SiS4SiPh3 | −12.8 (5.53) [−13.7] | −7.0 (6.41) |
| (WEYHOX) | −12.9 (5.55) [−15.2] | −8.1 (6.13) |
| Ph3CS5CPh3 | −11.5 (6.02) [−11.9] | −7.3 (5.81) |
| (YAVDAA) | ||
| Ph3CS6CPh3 | −11.4 (5.82) [−12.4] | −5.3 (6.17) |
| (YAVDEE) | −10.6 (6.02) [−15.3] | −4.0 (6.28) |
| −5.7 (7.26) | ||
| −7.2 (6.59) | ||
Nets of embraces
We now describe the locations and roles of these 2S4PEs in the crystal packing. Fig. 5 shows the three-dimensional net of 6PEs and 2S4PEs in WEYHOX. The chains of molecules connected by the 6PE run in the z-direction, and these chains are cross-linked in the x-direction and in the y-direction by the 2S4PE motif. Zig-zag sequences of embraces connect the ends of molecules, and these zig-zag sequences are directly analogous to the ZZI6PE (zig-zag infinite chain of 6PE) one-dimensional motif commonly occurring in crystals containing the Ph4P+ ion.6 This analogy is valid because the 2S4PE is a pseudo-6PE: the pseudo-ZZI6PE chains in WEYHOX are ⋯6PE⋯ps-6PE⋯, compared with ⋯6PE⋯6PE⋯ in the ZZI6PE. In WEYHOX the ps-ZZI6PE chains are orthogonal, comparable with the orthogonal ZZI6PE in [Ph4P]4[Fe4S4(SH)4] (refcode FAGREK).7 | ||
| Fig. 5 Net of 6PEs and 2S4PEs in WEYHOX (Ph3SiS4SiPh3); Ph rings not shown. The 6PE can be identified by the collinearity of S–Si⋯Si–S, and their connection of molecules in chains in the z-direction. The 2S4PEs which cross-link these chains in the y-direction (at z⊕=⊕0.5) are WEYHOX-6.13, and the cross-linking 2S4PEs in the x-direction (at z⊕=⊕0) are WEYHOX-6.41 (Fig. 4). Note the zig-zag chains of alternating 6PE and 2S4PE in the x-direction and in the y-direction, each of which is analogous to the ZZI6PE chains of a 6PE. | ||
The 2S4PEs in WEYHOX cause the twisting of the S–Si⋯Si–S axes of the 6PE at either end of the Ph3SiS4SiPh3 molecule, as shown in Fig. 6. The twisting is in the direction which shortens the 2S4PE.
 | ||
| Fig. 6 Structure of WEYHOX viewed along the chains of molecules. The 6PE motifs are indicated by the candystripe connections within the unit cell projected along z, whereas the 2S4PEs (also indicated by black and white candystripes) cross the x- and y-axes; H atoms omitted. Note that the inclinations of the 6PE are so as to shorten the 2S4PE. | ||
The 3D-net of 6PE and 2S4PE motifs in YAVDEE, for Ph3CS6CPh3, is shown in Fig. 7, comparable with Fig. 5 for WEYHOX. The greater flexibility of the S6 linkage allows additional 2S4PEs not present in WEYHOX, and each end of each molecule participates in one 6PE and two 2S4PEs. Another difference of YAVDEE from WEYHOX is an absence of the twisting of the axes of the 6PE (Fig. 6), also due to the additional embraces which can be formed by the more flexible molecule Ph3CS6CPh3.
 | ||
| Fig. 7 Net of 6PEs and 2S4PEs in YAVDEE (Ph3CS6CPh3); Ph rings not shown. The 6PEs are 6.02 and 5.82 Å: the other four unique connections are 2S4PEs. Each end of each molecule engages one 6PE and two 2S4PEs. | ||
Ph3CS5CPh3 crystallises with CHCl3 which is disordered (YAVDAA), and the structure is poorly determined,16 but the main characteristics of the crystal packing are clear. The chains of molecules linked by the 6PE are more separated, with just one 2S4PE cross-linkage as shown in Fig. 8. The presence of the chloroform, which is not itself engaged in stabilising supramolecular motifs, opens the lattice and disrupts the 2S4PEs that are a feature of the molecules with one less and one more S atom in the connecting chain.
 | ||
| Fig. 8 Curved strands of molecules Ph3CS5CPh3 in YAVDAA, with 6PE embraces (all C⋯C 6.02 Å) between molecules in the z-direction, and one 2S4PE (C⋯C 5.81 Å) linkage in the y-direction. The strands are separated in the x-direction by the disordered chloroform molecule, not shown. | ||
Herringbone arrays of phenyl groups
In Ph3GeS3GePh3 (CAFFAQ) the two S–Ge bonds are almost parallel, the strands of molecules linked by the 6PE are approximately linear (Fig. 1), and these strands are well separated (Fig. 3). What then are the supramolecular motifs that cause this arrangement of the strands? The answer is a herringbone array of phenyl rings from different molecules and different strands. This feature, also recognised in the crystal structures of more rigid molecules of the type (Ph3P)2Ag(O2C2O2)Ag(PPh3)2,32 is explained pictorially in Fig. 9. This shows how eight phenyl groups from molecules involved in four different 6PEs, from four different strands, take part in EF motifs in a herringbone array. Four of these EF motifs are part of the 6PEs, but the remaining six are additional, between 6PE, and are marked with arrows. The eight Ph groups which constitute this herringbone assembly occur in one layer (Fig. 9) and are normal to it. The question arises as to whether these additional (EF)4 motifs dictate the linearity of the Ph3GeS3GePh3 strands, or the reverse. An infinite domain of EF motifs between strands occurs in the crystal WEYHOX. | ||
| Fig. 9 Inter-strand herringbone array of phenyl groups in CAFFAQ, Ph3GeS3GePh3. Only the relevant parts of molecules are shown: the 6PEs are marked as black and white candystripes. There are eight Ph rings in an interaction zone between four strands, engaged in six EF interactions (arrowed) which are not part of the 6PE, and four other EF interactions that are components of the 6PE. The lower view shows how these eight phenyl groups occur in a layer and are normal to it. | ||
The crystal packing of 1
The crystal structure of Ph3CS3CPh31 is the exception to the patterns developed above. There are no 6PE motifs. Chains of molecules occur in the cell, as shown in Fig. 10 which is a projection along the chains, but there are noticeable differences with the corresponding view of the substitutional dimorph, Ph3GeS3GePh3 (Fig. 3, CAFFAQ). The molecules of Ph3CS3CPh3 associate with lateral embraces using phenyl groups at both ends of a molecule, as detailed in Fig. 11. The lateral embrace involving the eight rings coloured green in Fig. 11 alternates in xy layers with lateral embraces of the type shown with red rings. One end of each Ph3CS3CPh3 molecule is associated with the ends of two other molecules, one via (EF)2 embraces for ((Ph)2)2 and the other as an OFF for (Ph)2. All of the embraces in this crystal structure are centrosymmetric.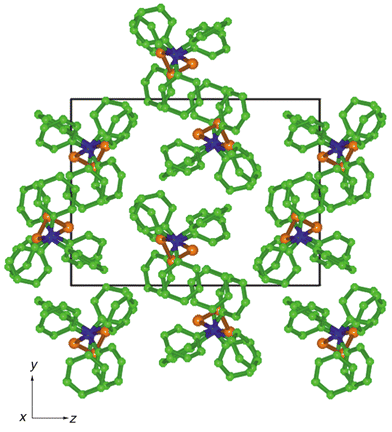 | ||
| Fig. 10 Projection of the crystal structure of Ph3CS3CPh3; H atoms omitted. Note the overlap of some Ph rings in this projection, and the association of molecules in pairs. | ||
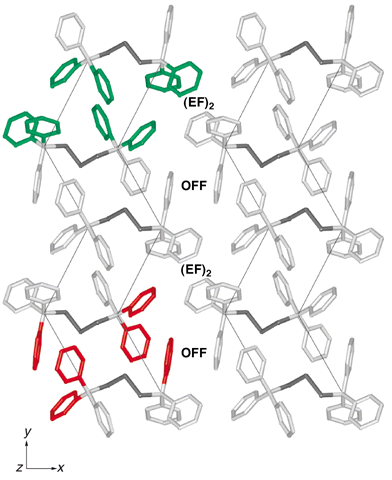 | ||
| Fig. 11 Collection of Ph3CS3CPh3 molecules in crystalline 1, showing the occurrence of (EF)2 and OFF local motifs between the ends of molecules instead of the 6PE, and showing the lateral zig-zag juxtapositions of molecules in the y-direction. Along these lateral zig-zags (thin lines) there is alternation of the assembly of eight phenyl rings marked green at one location and the assembly of six phenyl groups marked red. Only one set of each of these lateral assemblies is coloured: one phenyl ring of each CPh3 group is involved in both types of lateral embrace. Click image or 11.htm to access a 3D representation. | ||
The energies of the lateral embraces in 1 have been calculated to be 16.2 and 15.0 kcal mol−1 stabilisation per pair of Ph3CS3CPh3 molecules. This exceeds the energy of a 6PE between the ends of two such molecules, because the lateral embrace has more atoms in proximity and contributing to the van der Waals attraction. The calculated energies are consistent with the alternative crystal packings of Ph3GeS3GePh3 and Ph3CS3CPh3, and there is no indication that the C/Ge substitution causes the difference.
Finally, we describe part of the crystal packing of Ph3CSCPh3 (PHMESF) which combines features of the packing of Ph3CS3CPh3 and of Ph3GeS3GePh3. Fig. 12 shows that one end of the molecule forms the standard 6PE, while the other end forms a pair of EF interactions, an (EF)2 motif with one molecule and an offset face-to-face, OFF, interaction with another. This occurrence in the one crystal of the different end-to-end motifs observed in the substitutional dimorphs Ph3CS3CPh3 and Ph3GeS3GePh3 is consistent with their approximate energy equivalence.
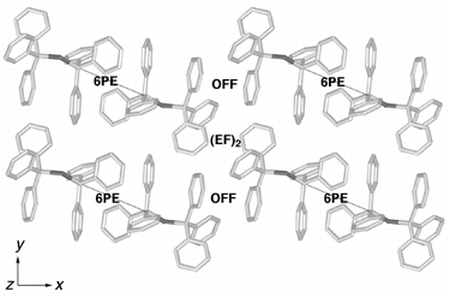 | ||
| Fig. 12 Part of the crystal packing of Ph3CSCPh3 (PHMESF) showing the 6PE (thin lines) at one end of each molecule, and the alternation of (EF)2 and OFF interactions at the other end. All motifs are centrosymmetric. | ||
Discussion
First we summarise. Molecules Ph3XSnXPh3 with n⊕>⊕1 generally crystallise such that the XPh3 functions at either end form the expected 6PE, thereby generating parallel strands. The molecules and the strands are relatively linear for n⊕=⊕2, 3, but longer connectors S4, S5 and S6 are increasingly able to curve, and the strands of molecules become parallel waves. There is one compound and one crystal structure, namely Ph3CS3CPh31, that does not form the 6PE and does not follow this pattern. Molecules that crystallise with the strand-6PE pattern also manifest a general motif between strands. This is an embrace, a centrosymmetric pseudo-6PE, in which two S atoms replace two Ph rings, named the 2S4PE. This 2S4PE is comprised of a pair of EF motifs and a number of Ph⋯S interactions which appear to use the electron density of the S lone pairs. The recurrence of the 2S4PE asserts its validity, and its calculated energy is substantial, although less than that of the 6PE. The more flexible Ph3XSnXPh3 molecules are able to conform the Sn chain to maximise the number of 2S4PEs, and the crystal packing type for this class of compounds is a 3D-net of 6PEs and 2S4PEs. Between the strands there are also regions of herringbone packing of Ph rings, comparable with the distinctive packing in crystalline benzene. The dominant crystal packing pattern for Ph3XSnXPh3 with n⊕>⊕1 is evidently an efficient combination of multiple phenyl embraces. Intermolecular S⋯S motifs do not influence the crystal supramolecularity of these compounds.The high frequency of occurrence of the 6PE and of the 2S4PE in this class of similar compounds with different crystal lattices and space groups attests the significance of these supramolecular motifs. But there is one exception to this pattern, Ph3CS3CPh31. The crystal packing of 1 can be understood, in terms of the occurrence of energetically favourable intermolecular interactions, namely lateral embraces involving Ph groups at both ends of the molecule, and end-to-end (EF)2 and OFF instead of the favourable (EF)6 concert of the 6PE. However, it is not clear why 1 did not conform to the pattern of the other five, or why Ph3GeS3GePh3 did not crystallise like 1. Compound 1 has been crystallised under various conditions in an attempt to find the expected dimorphic structure that fits the pattern, and that occurs in Ph3GeS3GePh3. Further experiments, including alternative crystallisations of Ph3GeS3GePh3 and Ph3SiS3SiPh3,33 are required to understand this anomaly.
The dominant crystal packing motifs are independent of the identity of X and occur for X⊕=⊕C, Si and Ge. These compounds demonstrate that internal substitution – iso-stereochemical – in a molecule need not influence the crystal supramolecularity and packing motifs, even though the crystal lattice and symmetry may change.
There is a need to clarify and possibly extend the terminology used for the phenomena described here, since there are three variables of interest: (1) molecular geometrical structure; (2) crystal lattice dimensions and symmetry (space group); and (3) crystal packing and supramolecular motifs. ‘Isostructural’ normally describes similar geometrical juxtapositions of atoms, in homologous molecules or in crystal lattices. ‘Isomorphous’ normally means similar lattice dimensions and the same lattice symmetry. Both of these descriptors are established and unambiguous. However, it is possible for different compounds to have essentially the same crystal packing motifs and yet not be isomorphous,6 and in the field of crystal supramolecularity a fundamental distinction between crystal lattice properties and crystal packing motifs needs to be made. Different crystal lattice properties need not imply different crystal packing and intermolecular motifs. Polymorphism, like isomorphism, conventionally refers to crystal lattice properties. Polymorphism is frequently claimed to apply only to different crystal lattices of the same (organic) molecule. But where internal substitution involving essentially isostructural molecules is associated with different crystal lattice dimensions, the essential qualities of polymorphism are present, and a category of substitutional polymorphism exists. Variations in crystal packing associated with such substitution, as reported here for Ph3GeS3GePh3 and Ph3CS3CPh3, are legitimate instances of polymorphism. The four compounds Ph3XSXPh3 with n⊕=⊕1 (Table 1) constitute a set of substitutional tetramorphs, and will be analysed in a separate paper. The well studied set of four compounds XPh4, X⊕=⊕C, Si, Sn, Pb,34–36 are isostructural, isomorphous, and have similar crystal packing motifs.
Acknowledgements
This research is funded by the Australian Research Council and the University of New South Wales.References
- I. G. Dance, in Perspectives in Supramolecular Chemistry: The Crystal as a Supramolecular Entity, ed. G. R. Desiraju, Wiley, Chichester, 1996, pp. 137–233 Search PubMed.
- I. G. Dance and M. L. Scudder, J. Chem. Soc., Chem. Commun., 1995, 1039 RSC.
- I. Dance and M. Scudder, Chem. Eur. J., 1996, 2, 481 CrossRef CAS PubMed.
- T. Steiner, Trans. Am. Crystallogr. Assoc., 1998, 33, 165 CAS.
- T. Steiner, New J. Chem., 2000, 24, 137 RSC.
- I. Dance and M. Scudder, J. Chem. Soc., Dalton Trans., 1996, 3755 RSC.
- M. Scudder and I. Dance, J. Chem. Soc., Dalton Trans., 1998, 3167 RSC.
- M. Scudder and I. Dance, J. Chem. Soc., Dalton Trans., 1998, 3155 RSC.
- I. Dance and M. Scudder, New J. Chem., 1998, 22, 481 RSC.
- I. G. Dance and M. L. Scudder, J. Chem. Soc., Dalton Trans., 2000, 1579 RSC.
- I. G. Dance and M. L. Scudder, J. Chem. Soc., Dalton Trans., 2000, 1587 RSC.
- C. Hasselgren, P. A. W. Dean, M. L. Scudder, D. C. Craig and I. G. Dance, J. Chem. Soc., Dalton Trans., 1997, 2019 RSC.
- M. Scudder and I. Dance, J. Chem. Soc., Dalton Trans., 1998, 329 RSC.
- F. H. Allen, J. E. Davies, J. J. Galloy, O. Johnson, O. Kennard, C. F. Macrae and D. G. Watson, Chem. Inf. Comput. Sci., 1991, 31, 204 CrossRef.
- F. H. Allen and O. Kennard, Chem. Des. Automat. News, 1993, 8, 131 Search PubMed.
- M. Kustos, J. Pickardt, J. Albertsen and R. Steudel, Z. Naturforsch., B, 1993, 48, 928 CrossRef CAS.
- W. C. McCrone, in Polymorphism in Physics and Chemistry of the Organic Solid State, ed. D. Fox, M. M. Labes and A. Weissemberg, Interscience, New York, 1965, p. 726 Search PubMed.
- J. Bernstein, in Organic Solid State Chemistry, ed. G. R. Desiraju, Elsevier, Amsterdam, 1987, pp. 471–518 Search PubMed.
- J. D. Dunitz, Pure Appl. Chem., 1991, 63, 177 CrossRef CAS.
- J. Bernstein, J. Phys. D: Appl. Phys., 1993, 26, B66 CrossRef CAS.
- J. D. Dunitz and J. Bernstein, Acc. Chem. Res., 1995, 28, 193 CrossRef CAS.
- J. D. Dunitz, in Perspectives in Supramolecular Chemistry: The Crystal as a Supramolecular Entity, ed. G. R. Desiraju, Wiley, Chichester, 1996, pp. 1–30 Search PubMed.
- J. Bernstein, R. J. Davey and J.-O. Henck, Angew. Chem., Int. Ed., 1999, 38, 3441 CrossRef CAS.
- D. Braga and F. Grepioni, Chem. Soc. Rev., 2000, 29, 229 RSC.
- C. Horn and I. G. Dance, 2001, in preparation.
- R. E. Rosenfield and R. Parthasarathy, J. Am. Chem. Soc., 1974, 96, 1925 CrossRef CAS PubMed.
- R. E. Rosenfield Jr, R. Parthasarathy and J. D. Dunitz, J. Am. Chem. Soc., 1977, 99, 4860 CrossRef.
- T. N. Guru Row and R. Parthasarathy, J. Am. Chem. Soc., 1981, 103, 477 CrossRef.
- F. H. Allen, C. M. Bird, R. S. Rowland and P. R. Raithby, Acta Crystallogr., Sect. B, 1997, 53, 680 CrossRef.
- F. H. Allen, C. M. Bird, R. S. Rowland and P. R. Raithby, Acta Crystallogr., Sect. B, 1997, 53, 696 CrossRef.
- R. Steudel, S. Forster and J. Albertsen, Chem. Ber., 1991, 124, 2357 CrossRef CAS.
- P. A. W. Dean, M. Scudder, D. Craig and I. Dance, CrystEngComm, 2001, 22; http://www.rsc.org/ej/ce/2001/B102399n/index.htm Search PubMed.
- R. Minkwitz, A. Kornath and H. Preut, Z. Anorg. Allg. Chem., 1994, 620, 981 CrossRef CAS.
- N. A. Ahmed, A. I. Kitaigorodskii and K. V. Mirskaya, Acta Crystallogr., Sect. B, 1971, 27, 867 CrossRef CAS.
- A. I. Kitaigorodskii, Molecular Crystals and Molecules, Academic Press, New York, 1973 Search PubMed.
- P. C. Chieh, J. Chem. Soc., Dalton Trans., 1972, 1207 RSC.
| This journal is © The Royal Society of Chemistry 2001 |