DOI:
10.1039/B103307G
(Paper)
CrystEngComm, 2001,
3, 137-140
Unexpected host–guest interactions of a novel V-shaped enediyne and water molecules
Received
12th April 2001
, Accepted 3rd July 2001
Abstract
The coupling of 1-ethynyl-3-hydroxybenzene with half of an equivalent of 1,2-diiodobenzene using Sonogashira conditions provides 1,2-bis(3-hydroxyphenylethynyl)benzene, which crystallises in a ‘screw’-type arrangement. The three-dimensional structure is characterised by hydrogen bonds of the phenolic hydroxyl groups of the enediynes. The resulting tetrahedral cavities are filled with water molecules leading to an unexpected host–guest complex in the crystalline state.
Introduction
Since the discovery of natural enediynes and their biological ability to act as antitumor antibiotics1 through the formation of a cytotoxic benzenoid biradical via a Bergman cyclisation,2 strong efforts have focused on the construction of simple enediyne compounds mimicking the natural specimen.1 As 9- or 10-membered ring enediynes3 are extremely unstable, we are interested in the elaboration of simple acyclic enediynes.4 Here, we describe the preparation and interesting solid state structure of the hitherto unknown enediyne 3. Whereas linear or tetrahedral tectons have been extensively studied due to their role in the formation of 2D or 3D nets,5 V-shaped tectons have received much less attention. The latter building blocks have been shown
to form screw-type chains, mostly, however, in presence of other V-shaped counterparts.6 To the best of our knowledge, enediyne 3 is the first example of a V-shaped tecton to form double screws in the solid state with itself.
Experimental
General synthesis
1-Bromo-3-hydroxybenzene (3-bromophenol) was converted into the trimethylsilylalkynyl-substituted compound 1 using Sonogashira coupling conditions.7 After the removal of the trimethylsilyl group by potassium hydroxide, the resultant 1-ethynyl-3-hydroxybenzene 2 was coupled with half an equivalent of 1,2-diiodobenzene to form the enediyne 3
(Scheme 1).
1-Bromo-3-hydroxybenzene was purchased from ABCR and trimethylsilylacetylene from Aldrich.
Synthesis of 1
1-Bromo-3-hydroxybenzene (2.28 g, 13.2 mmol), copper(I) iodide (515 mg, 2.70 mmol) and tetrakis(triphenylphosphine)palladium(0)
(1.56 g, 1.35 mmol) were suspended, under a nitrogen atmosphere, in anhydrous triethylamine (30 ml) and placed in a sealing tube. Then trimethylsilylacetylene (5.40 g, 55.0 mmol, 7.05 ml) was added and the reaction mixture was allowed to stir at 120![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) °C for a total period of 6 d. The mixture was concentrated, the remaining residue dissolved in methylene chloride (200 ml), washed three times with a 2.5% potassium cyanide solution (100 ml) and three times with water (100 ml). Evaporation of the solvent under reduced pressure and chromatographic purification on silica gel furnished 1.71 g of 3-trimethylsilylethynylphenol 1
(8.98 mmol, 68%) as a yellow oil. TLC (CH2Cl2): Rf⊕=⊕0.3. IR (film,
°C for a total period of 6 d. The mixture was concentrated, the remaining residue dissolved in methylene chloride (200 ml), washed three times with a 2.5% potassium cyanide solution (100 ml) and three times with water (100 ml). Evaporation of the solvent under reduced pressure and chromatographic purification on silica gel furnished 1.71 g of 3-trimethylsilylethynylphenol 1
(8.98 mmol, 68%) as a yellow oil. TLC (CH2Cl2): Rf⊕=⊕0.3. IR (film, ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/char_e0e1.gif) /cm−1): 3408 [s, ν(OH)], 2960 (m), 2158 [s, ν(C
/cm−1): 3408 [s, ν(OH)], 2960 (m), 2158 [s, ν(C![[triple bond, length half m-dash]](https://www.rsc.org/images/entities/char_e007.gif) C)], 1591 [s, δ(OH)], 1579 [s, δ(OH)], 1479 (m), 1445 (m), 1278 (m), 1251 (s), 1156 (s), 953 (s), 845 (s), 783 (m), 760 (m), 686 (m), 646 (m). 1H NMR (200 MHz, CDCl3): δ 0.24 (s, 9H), 6.80 (ddd, 3J⊕=⊕8.1, 4J⊕=⊕2.7, 4J⊕=⊕1.2 Hz, 1H), 6.94 (m, 1H), 7.04 (dt, 3J⊕=⊕8.1, 4J⊕=⊕1.2 Hz,
1H), 7.16 (t, 3J⊕=⊕8.1 Hz, 1H). 13C NMR (50 MHz, CDCl3): δ
−0.09, 94.3, 104.6, 116.0, 118.5, 124.2, 124.6, 129.5, 155.2. Anal. calc. for C11H14OSi (190.3): C 69.42, H 7.41. Found: 69.74, H 7.03%.
C)], 1591 [s, δ(OH)], 1579 [s, δ(OH)], 1479 (m), 1445 (m), 1278 (m), 1251 (s), 1156 (s), 953 (s), 845 (s), 783 (m), 760 (m), 686 (m), 646 (m). 1H NMR (200 MHz, CDCl3): δ 0.24 (s, 9H), 6.80 (ddd, 3J⊕=⊕8.1, 4J⊕=⊕2.7, 4J⊕=⊕1.2 Hz, 1H), 6.94 (m, 1H), 7.04 (dt, 3J⊕=⊕8.1, 4J⊕=⊕1.2 Hz,
1H), 7.16 (t, 3J⊕=⊕8.1 Hz, 1H). 13C NMR (50 MHz, CDCl3): δ
−0.09, 94.3, 104.6, 116.0, 118.5, 124.2, 124.6, 129.5, 155.2. Anal. calc. for C11H14OSi (190.3): C 69.42, H 7.41. Found: 69.74, H 7.03%.
Synthesis of 2
3-Trimethylsilylethynylphenol 1
(223 mg, 1.17 mmol) was dissolved in a mixture of methanol (3 ml) and tetrahydrofuran (3 ml). A solution of potassium hydroxide (500 mg) in water (3 ml) was added and the mixture was stirred at ambient temperature for 1.5 h. The solution was neutralised with 1 M hydrochloric acid, treated with diethyl ether (25 ml), and the aqueous phase was extracted three times with diethyl ether (15 ml). The combined organic layers were dried with magnesium sulfate and the solvent was removed under reduced pressure at ambient temperature. 1-Ethynyl-3-hydroxybenzene82
(137 mg, 1.16 mmol, 99%) was obtained as an orange–brown oil. IR (film, /cm−1):
3377 [s, ν(OH)], 3291 [s, ν(CCH)], 2955 (w), 2108 [w, ν(CC)], 1710 (w), 1592 [s, δ(OH)], 1580 [s, δ(OH)], 1479 (m), 1446 (m), 1310 (m), 1277 (s), 1236 (m), 1148 (m), 1084 (w), 1047 (w), 999 (w), 935 (m), 866 (m), 785 (s). 1H NMR (200 MHz, CDCl3): δ 3.06 (s, 1H), 5.82 (s, 1H), 6.84 (ddd, 3J⊕=⊕7.6, 4J⊕=⊕2.6, 4J⊕=⊕1.2 Hz, 1H), 6.97 (dd, 4J⊕=⊕2.6, 4J⊕=⊕1.2 Hz, 1H), 7.06 (dt, 3J⊕=⊕7.6, 4J⊕=⊕1.2 Hz,
1H), 7.16 (t, 3J⊕=⊕7.6 Hz, 1H). 13C NMR (CDCl3, 50 MHz): δ 67.8, 83.2, 116.3, 118.7, 123.0, 124.4, 129.5, 155.4.
Synthesis of 3
1,2-Diiodobenzene (960 mg, 2.91 mmol, 380 μl) was added under nitrogen to a suspension of copper(I) iodide (63.1 mg, 331 μmol) and bis(triphenylphosphine)palladium(II) chloride (91.9 mg, 131 μmol) in anhydrous triethylamine (10 ml). After the addition of 1-ethynyl-3-hydroxybenzene 2
(830 mg, 7.03 mmol) in triethylamine (10 ml) the reaction mixture was allowed to stir for 5 h at 50°C. The solvent was evaporated in vacuo, the remaining residue was dissolved in methylene chloride (100 ml) and the organic layer was washed three times with a 2.5% potassium cyanide solution (70 ml) and three times with water (70 ml). After drying on magnesium sulfate, the
concentrated residue was purified by column chromatography on silica gel. The fraction causing a fluorescent spot on the TLC was isolated and purified by recrystallisation from chloroform. 1,2-Bis(3-hydroxyphenylethynyl)benzene 3 was obtained (640 mg, 2.06 mmol, 71%) as colourless crystals. TLC (CH2Cl2–ethyl acetate, 9∶1): Rf⊕=⊕0.46. Mp⊕=⊕113–114°C. IR (KBr, /cm−1): ⊕3304 [m, ν(OH)], 1603 [s, δ(OH)], 1578 [s, δ(OH)], 1501 (s), 1445 (s), 1351 (s), 1222 (s), 1164 (m), 1121 (m), 1092 (m), 944 (s), 859 (s), 780 (s), 756 (s), 682 (s). 1H NMR (200 MHz,
acetone-d6): δ 3.34 (s, 2H), 6.98 (ddd, 3J⊕=⊕8.1, 4J⊕=⊕2.6, 4J⊕=⊕1.2 Hz, 2H), 7.15–7.20 (m, 4H), 7.33 (t, 3J⊕=⊕8.1 Hz, 2H), 7.50 (m, 2H), 7.69 (m, 2H). 13C NMR (acetone-d6, 50 MHz): δ 88.3, 94.3, 117.1, 118.7, 123.7, 124.8, 126.3, 129.3, 130.6, 133.0, 158.3. TG/DTG: two endothermic and one exothermic signals were observed. The endothermic signal at 100°C is indicative of the loss of water molecules of crystallisation. Indeed, a TG experiment shows a mass decrease at that temperature. The endothermic signal at 113°C agrees with the measured melting point. The exothermic signal at 275°C indicates the occurrence of the Bergman cycloaromatisation.
The relative high temperature of cyclisation is in good agreement with the long c–d distance (see Scheme 1) of 419 pm. The results of the elemental analysis confirm the presence of a hydrate (3∶H2O⊕=⊕2∶1). Anal. calc. for C22H14O2·1/2H2O (310.4): C 82.74, H 4.73. Found: 82.73, H 4.31%.
Crystal structure determination of 3
X-Ray diffraction intensity collection was carried out with graphite-monochromatised MoKα radiation on a STOE IPDS one-circle image plate diffractometer equipped with an Oxford Cryostream liquid nitrogen cooling device. Crystal data and details of measurement and refinement are summarised in Table 1.
| Parameter |
3
|
|
Full-matrix, least squares refinement on F2.
Click b103307g.txt for full crystallographic data (CCDC 162147).
|
| Empirical formula |
C22H14O2·1/2H2O |
| Crystal dimensions/mm |
0.72⊕×⊕0.34⊕×⊕0.22 |
|
M
|
319.34 |
| Crystal system |
Monoclinic |
| Space group |
C2/c |
|
a/pm |
3720.7(3) |
|
b/pm |
564.04(4) |
|
c/pm |
1642.75(13) |
|
β/° |
104.546(9) |
|
V/nm3 |
3.3370(4) |
|
Z
|
8 |
|
T/K |
173(1) |
|
D
c/Mg m−3 |
1.271 |
|
λ/pm |
71.073 |
|
μ/mm−1 |
0.082 |
|
F(000) |
1336 |
|
θ Range for data collection/° |
2.26–25.98 |
| Limiting indices |
−45⊕≤⊕h⊕≤⊕45; −6⊕≤⊕k⊕≤⊕6; −20⊕≤⊕l⊕≤⊕20 |
| Reflections collected, unique, significant |
17201, 3223, 2833 |
|
R
int, Rσ |
0.0348, 0.0193 |
| Completeness to θ⊕=⊕25.98 |
98.7 |
| Absorption correction |
None (μ⊕×⊕r⊕≤⊕0.05) |
| Max., min. transmission |
0.982, 0.943 |
| Data, restraints, parameters |
3223, 0, 225 |
| Goodness-of-fit on F2 |
1.030 |
| Final R indices [I⊕>⊕2σ(I)] |
R
1⊕=⊕0.044, wR2⊕=⊕0.122 |
|
R indices (all data) |
R
1⊕=⊕0.049, wR2⊕=⊕0.126 |
| Δρmin., Δρmax./10−6 e pm−3 |
−0.31(4), 0.29(4) |
According to the μ⊕×⊕r⊕<⊕0.1 criterion, no absorption correction had to be applied. Crystal structure solution and refinement based on F2 were performed by direct methods and subsequent Fourier syntheses with anisotropic displacement parameters for all non-hydrogen atoms using SHELXS-97 and SHELXL-97-2.9 The co-ordinates of the hydrogen atoms H2 to H23 were placed and refined for idealised geometries with isotropic displacement parameters fixed to the 1.2 times of equivalent isotropic displacement parameters of the bonding carbon atoms. The positions of the hydroxyl and water hydrogen atoms H1, H24 and Hw, respectively, have been located from difference Fourier maps and refined to Uiso values fixed to 1.5 Ueq of the bonded oxygen atoms (O1, O24 and Ow, respectively). Selected bond distances and angles are given in Table 2.
Table 2
Selected bond distances (pm) and angles (°) of 3
|
Symmetry operators: #1 x⊕+⊕1/2, y⊕−⊕1/2, z; #2 −x⊕+⊕1, y⊕−⊕1, −z⊕+⊕1/2; #3 −x⊕+⊕1/2, y⊕+⊕1/2, −z⊕+⊕1/2.
|
|
Phenyl-I
|
Phenyl-III
|
| O1–C3 |
138.23(19) |
C18–C19 |
139.2(2) |
|
| C2–C3 |
138.4(2) |
C18–C23 |
139.85(19) |
|
| C2–C7 |
139.6(2) |
C19–C20 |
138.1(2) |
|
| C3–C4 |
138.8(2) |
C20–C21 |
138.8(2) |
|
| C4–C5 |
138.1(2) |
C21–C22 |
138.2(2) |
|
| C5–C6 |
138.5(2) |
C22–C23 |
138.4(2) |
|
| C6–C7 |
139.8(2) |
C22–O24 |
137.64(18) |
|
| |
|
Phenyl-II
|
Acetylene-bridge
|
| C10–C11 |
139.8(2) |
C7–C8 |
143.47(19) |
|
| C10–C15 |
141.1(2) |
C8–C9 |
119.7(2) |
|
| C11–C12 |
138.4(2) |
C9–C10 |
143.15(19) |
|
| C12–C13 |
138.6(2) |
C15–C16 |
143.09(19) |
|
| C13–C14 |
138.3(2) |
C16–C17 |
119.7(2) |
|
| C14–C15 |
140.44(19) |
C17–C18 |
143.1(2) |
|
| |
|
C8⋯C17 |
418.8(2) |
|
| |
|
Angles
|
| C7–C8–C9 |
176.34(16) |
C15–C16–C17 |
175.81(15) |
|
| C8–C9–C10 |
178.32(17) |
C16–C17–C18 |
177.11(16) |
|
| C4–C10–C15–C21 |
−6.66(12) |
∠(I,II) |
3.09(8) |
|
| ∠(I,III) |
4.05(9) |
∠(II,III) |
7.14(9) |
|
| |
|
H-bonds
a
|
d(D–H)
|
d(H⋯A)
|
d(D⋯A)
|
∠(DHA)
|
O1–H1⋯Ow![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) #1 #1 |
84 |
208 |
276.0(2) |
138.3 |
| O24–H24⋯O1′#2 |
84 |
192 |
272.30(18) |
159.2 |
| Ow–Hw⋯O24#3 |
73(3) |
202(3) |
274.5(2) |
170(3) |
Crystal structure description
Compound 3
(Pearson symbol mC316) crystallises in the monoclinic system with the space group C2/c, with one formula unit in the asymmetric unit. All atoms of 3 are placed on general positions 8f(1). The water molecule of crystallisation is placed on a special position 4e(2). The V-shaped molecule 3 may possess maximum point group symmetry mm2. Owing to (a) the different angles of the alkyne functions (see Table 2), (b) the different orientations of the hydroxyl groups, (c) the torsion angles of the substituents, i.e. C4–C10–C15–C21⊕=⊕−6.66(12)°, and (d) the different coplanar arrangements of the phenyl functions, i.e.
∠(I–II)⊕=⊕3.09(8)°
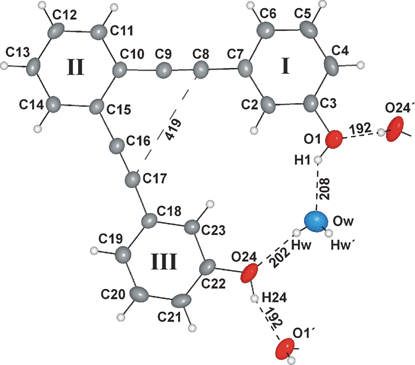
and ∠(II–III)⊕=⊕7.14(9)°, the symmetry of 3 is reduced to 1. Responsible for this break of symmetry may be the hydrogen bonding scheme (Fig. 1) and the spatial arrangement of 3 in the crystal lattice. The 3D arrangement indicates a herring-bone structure (Fig. 2), which is controlled by space-filling motifs and not by CH–π- or π–π-stacking interactions.
 |
| | Fig. 2
The herring-bone arrangement of compound 32·H2O.
| |
The hydroxyl groups act as hydrogen bond donors and acceptors for weak hydrogen bonds10 both to other molecules of 3, i.e.d(O24–H24⋯O1′)⊕=⊕192 pm, and to the included water molecules, i.e.d(O1–H1⋯Ow)⊕=⊕208 pm and d(Ow–Hw⋯O24)⊕=⊕202 pm. Thus, the oxygen atoms of the water molecules are co-ordinated tetrahedrally by hydrogen atoms.
The intermolecular hydrogen bonds O24–H24⋯O1 lead to screw-chains 1∞[3] parallel [010] with an angle of 109.91(1)° for the planes of succeeding molecules. As this arrangement does not achieve a high space-filling, a second screw is inserted [Fig. 3, screw-chains A (blue) and B (grey)]. The resulting tetrahedral cavities are filled by water molecules (3∶H2O⊕=⊕2∶1). The screw-chains A and B are connected via antidromic cooperative hydrogen bond systems formed by the phenolic hydroxyl groups and the water molecules (Fig. 4).11 The hydrogen bond-connected screw-type double chains are quasi-tetragonally
packed in (010).
 |
| | Fig. 3
Pictorial linkage of screw-chain A (blue) and B (grey) to generate the 3D arrangement of 32·H2O. Click image or 3.htm to access a 3D representation.
| |
![The chain-structured antidromic cooperative hydrogen bond system of 32·H2O along [010]. The two different screw-chains A and B (see Fig. 3) are represented by blue and grey dashed lines.](/image/article/2001/CE/b103307g/b103307g-f4.gif) |
| | Fig. 4
The chain-structured antidromic cooperative hydrogen bond system of 32·H2O along [010]. The two different screw-chains A and B (see Fig. 3) are represented by blue and grey dashed lines.
| |
In the sense of crystal engineering the V-shaped molecules of 3 act as a template for a V-shaped macromolecule as shown by the tetrahedral angles between succeeding molecules and the 1D hydrogen bond network.
Acknowledgements
Financial support from the Deutsche Forschungsgemeinschaft, Volkswagen Stiftung and Fonds der Chemischen Industrie is gratefully acknowledged.
References
- K. C. Nicolaou and W.-M. Dai, Angew. Chem., 1991, 103, 1453; K. C. Nicolaou and W.-M. Dai, Angew. Chem., Int. Ed. Engl., 1991, 30, 1387 Search PubMed; M. E. Maier, Synlett, 1995, 13 Search PubMed.
- R. R. Jones and R. G. Bergman, J. Am. Chem. Soc., 1972, 94, 660 Search PubMed.
- K. C. Nicolaou, G. Zuccarello, Y. Ogawa, E. J. Schweiger and T. Kumazawa, J. Am. Chem. Soc., 1988, 110, 4866 Search PubMed; K. C. Nicolaou, G. Zuccarello, C. Riemer, V. A. Estevez and W.-M. Dai, J. Am. Chem. Soc., 1992, 114, 7360 Search PubMed; T. Mita, S. Kawata and M. Hirama, Chem. Lett., 1998, 959 Search PubMed; W.-M. Dai, K. C. Fong, C. W. Lau, L. Zhou, W. Hamaguchi and S. Nishimoto, J. Org. Chem., 1999, 64, 682 Search PubMed.
- M. Schmittel and S. Kiau, Chem. Lett., 1995, 953 Search PubMed.
- O. Ermer and A. Eling, J. Chem. Soc., Perkin Trans. 2, 1994, 925 Search PubMed.
- S. Hanessian, R. Saladino, R. Margarita and M. Simard, Chem. Eur. J., 1999, 5, 2169 Search PubMed.
- K. Sonogashira, Y. Tohda and N. Hagihara, Tetrahedron Lett., 1975, 50, 4467 Search PubMed; A. de Meijere and F. E. Meyer, Angew. Chem., 1994, 106, 2473 Search PubMed; A. de Meijere and F. E. Meyer, Angew. Chem., Int. Ed. Engl., 1994, 33, 2379 Search PubMed; R. Rossi, A. Carpita and F. Bellina, Org. Prep. Proced. Int., 1995, 27, 127 Search PubMed;
R. F. Heck, Palladium Reagents in Organic Synthesis, Academic Press, London, 1995 Search PubMed.
- F. Bohlmann, K.-D. Albrecht and G. Schmidt, Chem. Ber., 1966, 99, 2822 Search PubMed.
-
G. M. Sheldrick, SHELXS-97, A Program for Crystal Solution, University of Göttingen, Germany, 1997 Search PubMed;
G. M. Sheldrick, SHELXL-97-2, A Program for the Refinement of Crystal Structures, University of Göttingen, Germany, 1997 Search PubMed.
- M. C. Etter, Acc. Chem. Res., 1990, 23, 120 Search PubMed.
-
W. Saenger and T. Steiner, Conference Proceedings ‘Water–Biomolecule Interaction’, Vol. 43, ed. M. U. Palma, M. B. Palma-Vittorelli and F. Parak, SIF, Bologna, 1993 Search PubMed; W. Saenger, Nature, 1979, 279, 343 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2001 |
Click here to see how this site uses Cookies. View our privacy policy here.