The ether–1,3-peri aromatic hydrogen interaction
Solhe F.
Alshahateet
,
Roger
Bishop
*,
Donald C.
Craig
and
Marcia L.
Scudder
School of Chemistry, The University of New South Wales, Sydney, 2052, Australia. E-mail: R.Bishop@unsw.edu.au
Abstract
Single crystal X-ray structures of the oxygenated diquinoline derivatives 3 and 4 show that, in both cases, the ether oxygen atom interacts very effectively with two 1,3-peri aromatic hydrogens of an adjacent molecule. A six-membered cycle involving two C–H⋯O weak hydrogen bonds results and in structure 4 this plays a key role in preventing inclusion host behaviour. The occurrence of this motif has been investigated using the Cambridge Structural Database and is compared with the behaviour more commonly observed between ethers and aromatic hydrogens.
Introduction
In earlier work we have reported that while the parent diquinoline derivative 1 packs efficiently by itself in the solid state; its exo,exo-dibromo analogue 2 does not and prefers to crystallise from small polyhalocarbon solvents in the form of clathrate inclusion compounds. The weak intermolecular forces involved in the crystal packing of both these structural types have been analysed and described in detail.1,2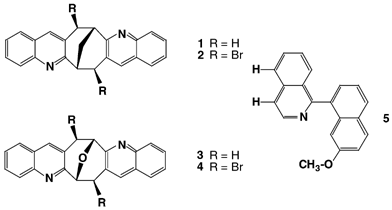
In light of these findings we expected that the oxygenated analogues 3 and 4 would behave rather similarly, perhaps even giving isostructural outcomes, but this proved not to be the case.
Results and discussion
Structures 3 and 4
The oxygenated diquinoline derivative 3 was prepared through Friedländer condensation3 of o-aminobenzaldehyde4 and 9-oxabicyclo[3.3.1]nonane-2,6-dione5,6 in methanol solution containing a small amount of sodium hydroxide. Benzylic bromination using N-bromosuccinimide in CCl4 afforded the required dibromide 4. Crystallisation of 3 from methanol, and 4 from chloroform, gave materials suitable for single crystal X-ray analysis. Numerical details of the solution and refinement of the two structures are presented in Table 1.| Parameter | 3 | 4 |
|---|---|---|
| a Click b103134c.txt for full crystallographic data (CCDC 161963 and 161964). | ||
| Empirical formula | C22H16N2O | C22H14Br2N2O |
| M | 324.4 | 482.2 |
| Crystal system | Monoclinic | Monoclinic |
| Space group | P21/c | P21/c |
| a/Å | 18.433(5) | 16.225(9) |
| b/Å | 6.523(1) | 7.761(2) |
| c/Å | 14.405(4) | 14.691(7) |
| β/° | 113.06(1) | 94.24(3) |
| V/Å3 | 1593.6(6) | 1845(1) |
| Z | 4 | 4 |
| T/°C | 21(1) | 21(1) |
| D c/g cm−3 | 1.35 | 1.74 |
| Radiation, λ/Å | CuKα, 1.5418 | MoKα, 0.7107 |
| μ/mm−1 | 0.624 | 4.363 |
| Scan mode | θ/2θ | θ/2θ |
| 2θmax/° | 120 | 46 |
| No. of intensity measurements | 2362 | 2401 |
| Criterion for observed reflection | I/σ(I)⊕>⊕3 | I/σ(I)⊕>⊕3 |
| No. of independent observed reflections | 1377 | 1727 |
| No. of reflections (m) in final refinement | 1377 | 1727 |
| No. of variables (n) in final refinement | 227 | 244 |
| R⊕=⊕Σm|ΔF|/Σm|Fo| | 0.036 | 0.028 |
| R w⊕=⊕[Σmw|ΔF|2/Σmw|Fo|2]1/2 | 0.046 | 0.034 |
| s⊕=⊕[Σmw|ΔF|2/(m−n)]1/2 | 1.49 | 1.15 |
| Crystal decay (%) | None | None |
| Max., min. transmission coefficient | — | 0.39, 0.62 |
| R for multiple measurements | 0.020 | 0.026 |
| Largest peak in final diffraction map/e Å−3 | 0.34 | 0.85 |
Comparison of the methylene compounds 1 and 2 with the ethers 3 and 4
The crystal structure of 1 results from a combination of size and shape considerations and weak intermolecular attractions which include aryl–aryl face–face,7 aryl–aryl edge–face,7 and dimeric aryl C–H⋯N edge–edge2 synthons (Fig. 1). These interactions are also present in the clathrate inclusion structures formed by 2, but additional host–guest and guest–guest attractive forces are also present. The most significant of these are halogen–halogen interactions.8–10 | ||
| Fig. 1 Centrosymmetric aryl edge–edge C–H⋯N dimer interaction present in the solid diquinoline derivative 1. The C–H⋯N and C–H⋯N distances are 2.56 and 3.55 Å, respectively.2 Weak hydrogen bonds are shown as red and white dashed lines. Colour code: C green, H light blue, N dark blue. | ||
In moving from compounds 1/2 to 3/4 the only molecular change is replacement of a bridging methylene group by an ether oxygen. This minor alteration results in considerable supramolecular change and compounds 3/4 pack quite differently to the earlier ones. Indeed, compound 4 no longer includes the guests trapped by its close analogue 2.2 On a supramolecular level the ether functionality offers new possibilities for intermolecular attractions, the most significant of these being the C–H⋯O weak hydrogen bond.11–13
Analysis of structure 3
Molecules of 3 pack efficiently in the space group P21/c and, as anticipated, both aryl–aryl face–face and edge–face interactions are involved in the crystal lattice. The familiar edge–edge aryl C–H⋯N dimer is also present but, with relatively long C–H⋯N and C–H⋯N distances of 3.13 and 4.07 Å, respectively, it is less effective than usual (Fig. 2). Strikingly, the most noteworthy interaction is that of the ether oxygen atom of one molecule with two 1,3-peri hydrogens of a second molecule of 3. This interaction has C–H⋯O distances of 2.57 and 2.66 Å, respectively. | ||
| Fig. 2 Crystal structure of solid 3 showing the centrosymmetric aryl edge–edge C–H⋯N dimer operating between opposite enantiomers and the intermolecular ether–1,3-peri aromatic hydrogen interaction. Colour code: O red. | ||
This new ether–1,3-peri aromatic hydrogen interaction is illustrated more clearly in Fig. 3 which shows the hypothetical case of naphthalene interacting with a general ether R2O.
 | ||
| Fig. 3 Schematic illustration of the ether–1,3-peri aromatic hydrogen interaction. | ||
In earlier work Murray-Rust and Glusker14 have surveyed cases of hydroxy group hydrogen bonding to ether oxygen atoms and reported a good correlation between the oxygen lone pair and hydrogen bond directions. Similarly, the plane containing the oxygen and lone pair orbitals is ideally placed orthogonal to the aromatic plane. Although the C–H⋯O hydrogen bond is much weaker (ca. 4–8 kJ mol−1)13 similar relationships would be expected here. The optimum geometry for the ether–1,3-peri aromatic hydrogen interaction should have two equal and short C–H⋯O distances, with the aromatic and ether planes orthogonal, and with the ether oxygen atom lying in the aromatic plane.
Analysis of structure 4
Once again, molecules of 4 pack in the space group P21/c and both aryl–aryl face–face and edge–face interactions are prevalent. The edge–edge aryl C–H⋯N dimer, however, is no longer present (Fig. 4). This is a remarkable absence since we have encountered this structural motif in the crystal structures of all other diquinoline compounds that we have investigated. On the other hand, the ether–1,3-peri aromatic hydrogen interaction (C–H⋯O distances of 2.57 and 2.69 Å), once again, is prominent. | ||
| Fig. 4 Part of the crystal structure of 4 showing the highly efficient ether–1,3-peri aromatic hydrogen interaction present. The bromine atoms are coloured brown, and the C–H⋯O weak hydrogen bonds are indicated by red and white dashed lines. | ||
CSD survey of ether–aromatic hydrogen interactions
Examples of ether–aromatic hydrogen interactions were sought in the Cambridge Structural Database (CSD).15 The most common behaviour observed is for an ether oxygen to interact with only one aromatic hydrogen atom. This is illustrated in Fig. 5 where CSD IsoStar16 was used to illustrate the 49 examples of ether oxygens located close to the hydrogen atoms of naphthalene. Note the overwhelming preference for single aromatic hydrogen interactions in this scatter plot and the complete absence of 1,3-peri synthons. | ||
| Fig. 5 Interactions between naphthalene and ether oxygen atoms from the database compiled within IsoStar.16 The ether molecules are drawn as partial molecular structures for simplicity and their oxygens are coloured red. Click image or 5.htm to access a 3D representation. | ||
A specific search for ether–1,3-peri aromatic hydrogen interactions was carried out next. This was restricted to compounds where an ether oxygen interacted with two 1,3-peri aromatic hydrogen atoms in a second molecule (either of the same or of a different type). Functional groups containing an ether oxygen as part of a larger functional group (such as ester, anhydride, carbonate or ketal) were omitted. An upper limit of 3.30 Å was applied to both C–H⋯O distances, and totals of 15 compounds and 19 interactions were located.17
The outcome of this survey indicates that the ether–1,3-peri aromatic hydrogen interaction does occur in a number of other crystal structures, though it is less frequent than we expected. Also, many of the cases recorded were less than ideal in geometry. This interaction is, however, a weak one and will be significantly influenced by other factors operating in a particular crystal. For example, since all the compounds surveyed are aromatic in nature, aryl–aryl face–face and/or edge–face interactions are expected to be prevalent. The weak ether–1,3-peri aromatic hydrogen interactions must co-exist with these and other supramolecular synthons, and it would be foolish to assume that their occurrence and experimental values are unaffected by these other forces.
Fig. 6 shows the ether–1,3-peri aromatic hydrogen H⋯O distances obtained from the CSD survey (plus those for 3 and 4) plotted against each other. Five of the hits (coloured green) involve aromatic molecules which are either π-deficient heterocycles or carry a strong electron-withdrawing substituent, whereas the remaining 14 literature hits are coloured black. One would expect the former group to form stronger interactions with ethers since their hydrogens will be more positive. The plot of C–H⋯O distances supports this view. One such case in particular is worthy of comment. The crystal structure of molecule 5 (refcode COHHAI)18 forms its 1,3-peri interaction through the two isoquinoline hydrogens highlighted in the structural diagram, rather than through those available on the competing electron-rich methoxynaphthalene ring.
 | ||
| Fig. 6 Plot of H1⋯O and H2⋯O distances (Å) for the ether–1,3-peri aromatic hydrogen interactions obtained from the CSD survey. Those examples involving aromatic rings bearing electron-withdrawing substituents or π-deficient heterocycles are marked in green and the remainder in black. The values observed in structures 3 and 4 are shown in red. | ||
The C–H⋯O values for 3 and 4 are shown in red in Fig. 6, and it is immediately clear that these examples are the most effective so far recorded.
Our results show that, in appropriate cases, the ether–1,3-peri aromatic interaction can play a significant role in crystal packing but, despite its apparently compact and convenient nature, only a comparatively small number of examples are currently known. The scatter plot for the ether–1,3-peri aromatic interactions is illustrated in Fig. 7 where the ether oxygen atoms of 3 and 4 are coloured blue.
 | ||
| Fig. 7 Ether–1,3-peri aromatic hydrogen interactions17 plotted in Fig. 6 showing the spatial orientation of the ether oxygen with respect to the aromatic hydrogen atoms. Both the ethers and the aromatic molecule are drawn as partial molecular structures for simplicity. Ether oxygen atoms are coloured red for the CSD references and blue for structures 3 and 4. The near optimum geometry for the latter can be seen. Click image or 7.htm to access a 3D representation. | ||
The highly favourable interactions present in the structures of 3 (2.57, 2.66 Å) and 4 (2.57, 2.69 Å) have the shortest C–H⋯O distances recorded so far for the ether–1,3-peri aromatic hydrogen interaction. They have good interplanar angles of 71.0 and 80.1°, and their oxygen atoms deviate from the aromatic plane by only 1.31 and 0.23 Å, respectively. These near ideal ether–1,3-peri aromatic interactions play a major role in the crystal packing of both 3 and 4. So much so, in the case of compound 4, that the ubiquitous edge–edge aryl C–H⋯N dimer is replaced and this potential host molecule is able to adopt new efficient lattice packing without guest inclusion.
Acknowledgements
We gratefully acknowledge financial support from the Australian Research Council.References
- C. E. Marjo, R. Bishop, D. C. Craig, A. O'Brien and M. L. Scudder, J. Chem. Soc., Chem. Commun., 1994, 2513 RSC.
- C. E. Marjo, M. L. Scudder, D. C. Craig and R. Bishop, J. Chem. Soc., Perkin Trans. 1, 1997, 2513 Search PubMed.
- C.-C. Cheng and S.-J. Yan, Org. React., 1982, 28, 37 CrossRef CAS.
- L. I. Smith and J. W. Opie, Org. Synth., 1955, coll. vol. 3, 56 Search PubMed.
- R. O. Duthaler, K. Wicker, P. Ackermann and C. Ganter, Helv. Chim. Acta, 1972, 55, 1809 CrossRef CAS.
- K. C. Pich, R. Bishop, D. C. Craig, I. G. Dance, A. D. Rae and M. L. Scudder, Struct. Chem., 1993, 4, 41 CrossRef CAS.
- G. R. Desiraju, Crystal Engineering: The Design of Organic Solids, Materials Science Monographs No. 54, Elsevier, Amsterdam, 1989 Search PubMed.
- J. A. R. P. Sarma and G. R. Desiraju, Acc. Chem. Res., 1986, 19, 222 CrossRef.
- V. R. Pedireddi, D. S. Reddy, B. S. Goud, D. C. Craig, A. D. Rae and G. R. Desiraju, J. Chem. Soc., Perkin Trans. 2, 1994, 2353 RSC.
- S. L. Price, A. J. Stone, J. Lucas, R. S. Rowland and A. Thornley, J. Am. Chem. Soc., 1994, 116, 4910 CrossRef CAS.
- G. R. Desiraju and T. R. Steiner, The Weak Hydrogen Bond in Structural Chemistry and Biology, Oxford University Press, Oxford, 1999 Search PubMed.
- R. Taylor and O. Kennard, J. Am. Chem. Soc., 1982, 104, 5063 CrossRef CAS.
- G. R. Desiraju, Acc. Chem. Res., 1991, 24, 290 CrossRef CAS.
- P. Murray-Rust and J. P. Glusker, J. Am. Chem. Soc., 1984, 106, 1018 CrossRef CAS.
- F. H. Allen, J. E. Davies, J. J. Galloy, O. Johnson, O. Kennard, C. F. Macrae, E. M. Mitchell, G. F. Mitchell, J. M. Smith and D. G. Watson, J. Chem. Inf. Comput. Sci., 1991, 31, 187 CrossRef CAS.
- IsoStar: A Library of Information About Nonbonded Interactions; I. J. Bruno, J. C. Cole, J. P. M. Lommerse, R. S. Rowland, R. Taylor and M. L. Verdonk, J. Comput.-Aided Mol. Design, 1997, 11, 525 CrossRef CAS PubMed.
- X-Ray structure refcodes and H1⋯O, H2⋯O distances (in Å): COHHAI (2.65, 2.81); DITBAJ (2.85, 2.91); JODYEG (2.63, 2.93); JUSNOA (2.75, 2.82); KEPKOF (3.00, 3.08); KEPKUL (2.81, 2.97), (2.81, 3.02); LIVTAL (2.68, 2.88); PADBUR (2.77, 2.81); SIVRIY (2.74, 2.78); TERXUJ (2.94, 2.96); VEVBON (2.72, 2.80); WERNOW (2.87, 3.04), (2.89, 3.09), (3.16, 3.24); YAGZOV (3.08, 3.19); ZOHMEO (2.84, 3.10), (2.88, 3.14); ZUXXEV (3.03, 3.04); 3 (2.57, 2.66); 4 (2.57, 2.69).
- G. Chelucci, A. Bacchi, D. Fabbri, A. Saba and F. Ulgheri, Tetrahedron Lett., 1999, 40, 553 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2001 |