Carbon–carbon bond forming reactions of N-bound transition metal α-cyanocarbanions: a mechanistic probe for catalytic Michael reactions of nitriles†‡
Takeshi Naota*, Akio Tannna and Shun-Ichi Murahashi*
Department of Chemistry, Graduate School of Engineering Science, Osaka University, Machikaneyama, Toyonaka, Osaka, 560-8531, Japan.. E-mail: naota@chem.es.osaka-u.ac.jp
First published on 14th December 2000
Abstract
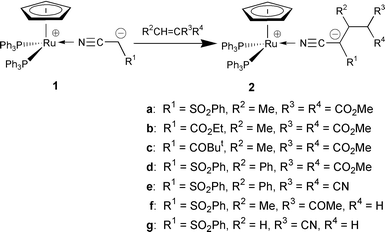
N-Bound α-cyanocarbanion complexes Ru+Cp(NCCH−R1)(PPh3) 2 1 react with electron deficient olefins to afford the conjugate adduct Ru+Cp(NCC−R1CHR2CR3 R4)(PPh3)2 2, kinetic studies of which revealed that complex 1-catalyzed Michael reactions of nitriles proceed via transformation of 1 to 2 and subsequent ligand exchange with nitriles.
Transition metal α-cyanocarbanions have attracted much attention as active species for stoichiometric1 and catalytic2,3 carbon–carbon bond formations of nitriles, and studies on their structure and reactivity4,5 are particularly of importance to develop novel transformations of nitriles bearing high selectivities, atom efficiency and sustainability.6 In 1989 we presented a new methodology for catalytic carbon–carbon bond formation of nitriles initiated by α-C–H activation of nitriles with low-valent transition metal catalysts.2a Capture of the α-cyanocarbanion intermediate with electrophiles provides a family of catalytic C–C bond formations at the α-position of nitriles under neutral conditions.2N-Bound α-cyanocarbanion complexes, mer-Ru+H(NCCH−CO2R)(NCCH 2CO2R)(PPh3)3, derived by α-C–H activation of alkyl cyanoacetates with RuH2(PPh3)4, have proven to be reactive species and active catalysts for the RuH2(PPh3)4-catalyzed aldol and Michael reactions of nitriles.2b,7 Further studies revealed that a variety of N-bound transition metal α-cyanocarbanion complexes act as efficient catalysts for the aldol and Michael reactions of nitriles.8
One of the most important aspects of this chemistry is to clarify and control the C–C bond forming process on the α-cyanocarbanion intermediates. Direct nucleophilic attack of zwitterionic N-bound α-cyanocarbanions to carbon electrophiles has been postulated for the crucial step in these catalytic aldol and Michael reactions.2b,3b,7,8 Isomerization to C-bound α-cyanocarbanions5 and subsequent carbometallation1 are alternative possibilities for this process; however, precise mechanistic information on this step still remains to be explored. During the course of our systematic studies on the structure and reactivity of transition metal α-cyanocarbanions5 we have succeeded in the isolation of intermediate for catalytic Michael reactions of nitriles. We describe here the conjugate addition of N-bound α-cyanocarbanions, Ru+Cp(NCCH−R)(PPh3)2 1 to electron deficient olefins, and mechanism of the catalytic Michael reactions of nitriles.9
When a 25.0 mM solution of N-bound α-cyanocarbanion Ru+Cp(NCCH−SO2Ph)(PPh3) 251a in benzene was allowed to react with dimethyl ethylidenemalonate (1.0 equiv.) at room temperature under argon atmosphere, conjugate addition and subsequent 1,3-hydrogen shift occurred to give the corresponding N-bound α-cyanocarbanion Ru+Cp[NCC−(SO2Ph)CHMeCH(CO2 Me)2](PPh3)22a in 99% isolated yield. Complex 2a was characterized by 1H, 13C{1H}, 31P{1H} NMR, IR, mass spectra and elemental analysis.§ Characteristic downfield shifts of the IR absorption (2153 cm−1) and 13C NMR signal (δ 143.7) of the cyano group indicate that the α-cyanocarbanion is N-bound,¶ and the zwitterionic structure has been unequivocally established by a 1H–13C HMBC experiment.|| Similar treatment of cyanoacetate and ketonitrile analogs 1b (R1 = CO2Et)8d and 1c (R1 = COBut) gave the corresponding adducts 2b, c in 98 and 98% isolated yields. Various electron deficient olefins such as dimethyl benzylidenemalonate, benzylidenemalononitrile, pent-3-en-2-one, and acrylonitrile reacted smoothly with 1a at room temperature to afford 2d–g in 98, 99, 31 and 36% yields, respectively.
 | (1) |
As well as complex 1,8d complex 2 shows comparable catalytic activity for the Michael additions of nitriles. Typically, the reaction of (phenylsulfonyl)acetonitrile 4 (4.00 M) with dimethyl benzylidenemalonate 3 (4.40 M) in the presence of 1a or 2d catalyst (0.120 M) in THF at 25 °C for 24 h gave adduct 5 in 86 and 88% isolated yields, respectively. In order to obtain insight into the mechanism of the complex 1-catalyzed Michael addition of nitriles, kinetic studies on the reaction of 1a with an excess amount of 3 in THF-d8 were carried out by means of 1H NMR (500 MHz) analysis using an internal standard (bibenzyl). The consumption rate of 1a exhibited clean pseudo-first-order dependence on the concentration of 1a (kobs = 6.61 ± 0.04 × 10−4 s−1 at 25.0 °C, [1a]0 = 2.00 × 10−2 M, [3]0 = 2.00 × 10−1 M), and the kobs values showed linear dependence on the initial concentration of 3 ranging from 2.00 to 4.00 × 10−1 M, indicating the second-order rate constant k1 as 2.9 ± 0.1 × 10−3 dm3 mol−1 s−1 at 25.0 °C. Dependences of kobsvs. [3]0 at 25.0–45.0 °C are shown in Fig. 1. The rate data correlate well (R2 = 0.990) with the Arrhenius relationship of ln(k1) vs. 1/T, where the ΔH‡ and ΔS‡ values were determined as 41 ± 3 kJ mol−1 and −148 ± 8 J mol−1 K−1, respectively. The second-order kinetics and the large negative value of ΔS‡ clearly indicate that the conjugate addition of 1a to 3 proceeds via a direct ionic pathway without any contact of olefins with the metal center. Ru+Cp[NCCH(SO2Ph)CHMeC−(CO2 - Me)2](PPh3)2 could not be detected during 1H NMR experiments, indicating that the process involves a fast 1,3-hydrogen shift after the conjugate addition. When complex 2d was allowed to react with nitrile 4 at room temperature in THF-d8, dissociation of Michael adduct 5 and regeneration of complex 1a can be monitored by 1H NMR spectroscopy. The consumption rate of 2d was first-order on the concentration of 2d ([2d]0 = 2.00 × 10−2 M, [4]0 = 2.00 × 10−1 M) and independent on the concentration of 4 ([4]0 = 2.00–3.50 × 10−1 M). The first-order rate constant k2 was determined to be 7.3 ± 0.3 × 10−6 s−1 at 25.0 °C. The observed first-order kinetics on the ligand exchange process is ascribed to rate-determining formation of the 16-electron complex [RuCp(PPh3)2]+ which would be followed by fast rebound process of the carbanion of 4 from outer sphere of the metal.
![Dependence of kobsvs.
[3]0 for the reactions of 1a with 3
in THF-d8 at 25.0 °C (●), 30.0 °C (○) 35.0
°C (■), 40.0 °C (□) and 45.0 °C (▲).](/image/article/2001/CC/b007517p/b007517p-f1.gif) | ||
| Fig. 1 Dependence of kobsvs. [3]0 for the reactions of 1a with 3 in THF-d8 at 25.0 °C (●), 30.0 °C (○) 35.0 °C (■), 40.0 °C (□) and 45.0 °C (▲). | ||
The complex 1a-catalyzed Michael reaction of nitrile 4 with olefin 3 can be rationalized by the mechanism shown in Scheme 1. Direct ionic addition of 1a to 3 and a subsequent 1,3-hydrogen shift affords complex 2d, which undergoes a rate determining process of ligand exchange with 4 to form adduct 5 and regenerate catalyst 1a. In order to verify this mechanism, kinetics on the overall catalytic Michael reaction of 4 with 3 were carried out at 25.0 °C in THF-d8 using the same NMR technique. The initial rate of the formation of 5 was constant (d[5]/dt = kobs = 1.44 ± 0.03 × 10−7 dm3 mol−1 s−1), when the reaction was started at an initial concentration of [1a]0 = 2.00 × 10−2 M, [3]0 = 2.00 × 10−1 M and [4]0 = 2.00 × 10−1 M. Almost the same kobs value of 1.49 ± 0.03 × 10−7 dm3 mol−1 s−1 was obtained when complex 2d was employed as the catalyst ([2d]0 = 2.00 × 10−2 M). These rate constants are first-order on the concentration of catalyst 1a or 2d, and zero-order on the concentration of both nitrile 4 and olefin 3. Thus, the rate law for the catalytic reaction can be expressed as d[5]/dt = k3[Ru] ([Ru] = [1a]0 = [2d]0 = [1a] + [2d]), which leads to the relation k3 = kobs/[Ru]. The calculated k3 value of 7.7 ± 0.9 × 10−6 s−1 (25.0 °C) is well in accord with that of k2; the rate constant of rate-determining step of the proposed catalytic cycle.
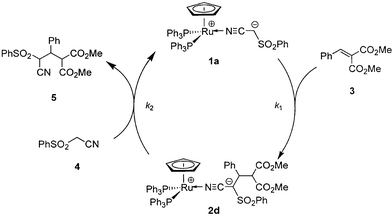 | ||
| Scheme 1 | ||
In summary, we have identified the active species of the catalytic Michael reactions of nitriles, and presented a definitive mechanism for the reactions. This is a rare case because most reported mechanistic investigations for catalytic Michael reactions of nitriles deal mainly with substrate and product analysis while speculating on the crucial step of carbon–carbon bond formation. Efforts are currently underway to investigate more fully the dynamic behavior of transition metal α-cyanocarbanion intermediates in a variety of carbon–carbon bond forming processes of nitriles.
Acknowledgements
This work was supported by Research for the Future program, the Japan Society for the Promotion of Science, and a Grant-in-Aid for Scientific Research, the Ministry of Education, Science, Sports, and Culture, Japan.Notes and references
- P. Knochel, N. Jeong, M. J. Rozema and M. C. P. Yeh, J. Am. Chem. Soc., 1989, 111, 6474 CrossRef CAS; H.-J. Liu and N. H. Al-said, Tetrahedron Lett., 1991, 32, 5473 CrossRef CAS; T. Kauffmann, H. Kieper and H. Pieper, Chem. Ber., 1992, 125, 899 CAS; M. T. Reetz, H. Haning and S. Stanchev, Tetrahedron Lett., 1992, 33, 6963 CrossRef CAS.
- (a) T. Naota, H. Taki, M. Mizuno and S.-I. Murahashi, J. Am. Chem. Soc., 1989, 111, 5954 CrossRef CAS; (b) S.-I. Murahashi, T. Naota, H. Taki, M. Mizuno, H. Takaya, S. Komiya, Y. Mizuho, N. Oyasato, M. Hiraoka, M. Hirano and A. Fukuoka, J. Am. Chem. Soc., 1995, 117, 12436 CrossRef CAS; (c) H. Takaya, T. Naota and S.-I. Murahashi, J. Am. Chem. Soc., 1998, 120, 4244 CrossRef CAS.
- (a) S. Paganelli, A. Schionato and C. Botteghi, Tetrahedron Lett., 1991, 32, 2807 CrossRef CAS; (b) M. Sawamura, H. Hamashima and Y. Ito, J. Am. Chem. Soc., 1992, 114, 8295 CrossRef CAS; (c) Y. Yamamoto, M. Al-Masum and N. Asao, J. Am. Chem. Soc., 1994, 116, 6019 CrossRef CAS; (d) B. M. Trost, P.-Y. Michellys and V. J. Gerusz, Angew. Chem., Int. Ed. Engl., 1997, 36, 1750 CrossRef CAS.
- S. D. Ittel, C. A. Tolman, A. D. English and J. P. Jesson, J. Am. Chem. Soc., 1978, 100, 7577 CrossRef CAS; J. G. Stack, J. J. Doney, R. G. Bergman and C. H. Heathcock, Organometallics, 1990, 9, 453 CrossRef CAS; G. L. Crocco, K. E. Lee and J. A. Gladysz, Organometallics, 1990, 9, 2819 CrossRef CAS; J. S. Ricci and J. A. Ibers, J. Am. Chem. Soc., 1971, 93, 2391 CrossRef.
- T. Naota, A. Tannna and S.-I. Murahashi, J. Am. Chem. Soc., 2000, 122, 2960 CrossRef CAS and references therein..
- S.-I. Murahashi and T. Naota, Bull. Chem. Soc. Jpn., 1996, 69, 1805 CAS.
- Y. Mizuho, N. Kasuga and S. Komiya, Chem. Lett., 1991, 2127 CAS.
- Ru+H(NCCH−CO2R)(dppe)2 : (a) M. Hirano, A. Takenaka, Y. Mizuho, M. Hiraoka and S. Komiya, J. Chem. Soc., Dalton Trans., 1999, 3209 RSC Re+(NCCH−CO2R)(NCCH2 CO2R)(PMe2Ph)4:; (b) M. Hirano, Y. Ito, M. Hirai, A. Fukuoka and S. Komiya, Chem. Lett., 1993, 2057 CAS; (c) M. Hirano, M. Hirai, Y. Ito, T. Tsurumaki, A. Baba, A. Fukuoka and S. Komiya, J. Organomet. Chem., 1998, 569, 3 CrossRef CAS RuCp+(NCCH−- CO2R)(PPh3)2:; (d) S.-I. Murahashi, K. Take, T. Naota and H. Takaya, Synlett, 2000, 1016 CAS.
- A preliminary result has been presented: 74th Annual Meeting of the Chemical Society of Japan, Kyotanabe, March 27–30, 1998, Paper 3A117..
Footnotes |
| † Electronic supplementary information (ESI) available. Experimental section. See http://www.rsc.org/suppdata/cc/b0/b007517p/ |
| ‡ Dedicated to Professor J. F. Normant on the occasion of his 65th birthday. |
§ Characterization data for 2a: mp 110 °C
(decomp.). IR (KBr) 2153 (CN), 1732 (C![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) O), 1480, 1433, 1281
(C–O, SO), 1134 (SO), 1090, 745, 696, 610
cm−1; 1H NMR (500 MHz,
C6D6) δ 1.26 (d, J 7.0 Hz, 3
H, CHCH3), 3.27 (s, 3 H, OCH3), 3.37 (s,
OCH3), 3.63 (dq, J 9.6, 7.0 Hz, 1 H,
CH3CH3), 4.11 [d, J 9.6 Hz, 1 H,
CH4(CO2CH3)2], 4.44
(s, 5 H, C5H5), 6.92–7.02 (m, 21 H, ArH),
7.36–7.44 [m, 12 H, PC6H5 (ortho)],
8.07 [dd, J 8.0, 2.5 Hz, 2 H,
SO2C6H5 (ortho)];
13C{1H} NMR (126 MHz,
C6D6) δ 169.5 (CO), 169.4
(CO), 150.9 [SO2C6H5
(ipso)], 143.7 (CN), 137.9 [PC6H5
(ipso)], 134.0 [SO2C6H5
(ortho)], 133.8 [PC6H5 (ortho)],
129.3 [SO2C6H5 (para)], 129.2
[PC6H5 (para)], 128.5
[PC6H5 (meta)], 126.4
[SO2C6H5 (meta)], 83.6
(C5H5), 59.9
[CH(CO2CH3)2], 58.7
(CCN), 52.0 (OCH3), 51.6 (OCH3), 34.2
(CHCH3), 20.4 (CH3);
31P{1H} NMR (202 MHz,
C6D6) δ 43.2 (s); FAB-MS: m/z
1030 ([M]+). Anal. Calc. for
C56H51NO6P2RuS: C, 65.4; H,
5.00; N, 1.36. Found: C, 65.5; H, 4.82; N, 1.36%. O), 1480, 1433, 1281
(C–O, SO), 1134 (SO), 1090, 745, 696, 610
cm−1; 1H NMR (500 MHz,
C6D6) δ 1.26 (d, J 7.0 Hz, 3
H, CHCH3), 3.27 (s, 3 H, OCH3), 3.37 (s,
OCH3), 3.63 (dq, J 9.6, 7.0 Hz, 1 H,
CH3CH3), 4.11 [d, J 9.6 Hz, 1 H,
CH4(CO2CH3)2], 4.44
(s, 5 H, C5H5), 6.92–7.02 (m, 21 H, ArH),
7.36–7.44 [m, 12 H, PC6H5 (ortho)],
8.07 [dd, J 8.0, 2.5 Hz, 2 H,
SO2C6H5 (ortho)];
13C{1H} NMR (126 MHz,
C6D6) δ 169.5 (CO), 169.4
(CO), 150.9 [SO2C6H5
(ipso)], 143.7 (CN), 137.9 [PC6H5
(ipso)], 134.0 [SO2C6H5
(ortho)], 133.8 [PC6H5 (ortho)],
129.3 [SO2C6H5 (para)], 129.2
[PC6H5 (para)], 128.5
[PC6H5 (meta)], 126.4
[SO2C6H5 (meta)], 83.6
(C5H5), 59.9
[CH(CO2CH3)2], 58.7
(CCN), 52.0 (OCH3), 51.6 (OCH3), 34.2
(CHCH3), 20.4 (CH3);
31P{1H} NMR (202 MHz,
C6D6) δ 43.2 (s); FAB-MS: m/z
1030 ([M]+). Anal. Calc. for
C56H51NO6P2RuS: C, 65.4; H,
5.00; N, 1.36. Found: C, 65.5; H, 4.82; N, 1.36%. |
| ¶ A variety of N-bound complexes RuCp+(NCCH−SO2Ph)(PR3) 2 show IR absorption for the CN triple bond in the range ca. 2150–2170 cm−1, and 13C chemical shift of a nitrile carbon at δ 140–155, while those of the C-bound isomers have been observed at 2190–2200 cm−1 and δ 110–125, respectively.5 |
| || The 1H NMR signal of 2a appearing at δ 4.11 strongly correlates with 13C signals of two carbonyl carbons (δ 169.4 and 169.5), which clearly leads to assignment of the 1H signal as the H4 proton of the zwitterionic α-cyanocarbanion moiety. |
| This journal is © The Royal Society of Chemistry 2001 |