Olefin metathesis in non-degassed solvent using a recyclable, polymer supported alkylideneruthenium†
James Dowden* and Jelena Savović
Wolfson Laboratory of Medicinal Chemistry, Department of Pharmacy and Pharmacology, University of Bath, Claverton Down, Bath, UK BA2 7AY. E-mail: J.Dowden@bath.ac.uk
First published on 11th December 2000
Abstract
Polystyrene-supported ruthenium complex 8 is a robust pro-catalyst for olefin metathesis that can be used in non-degassed solvents and recycled without added stabilisers.
Recent enthusiastic interest in olefin metathesis has been fuelled by the development of well-defined transition metal catalysts.1 These catalysts have found wide application because they provide facile alkene exchange, often late in a synthetic sequence.2 Grubb’s alkylideneruthenium 13 is particularly popular because it is easy to handle. Undesirable features of these systems are that they are not amenable to recycling and that they give rise to high levels of ruthenium contamination. Recent work has sought to address these problems.4–7 Hoveyda and co-workers established 56,7 as a remarkably robust complex that was stable to silica gel chromatography using non-degassed eluent and could therefore be recovered and recycled. Recently, Barrett’s group reported a ‘boomerang’ system 38 for sequestering the catalytic species onto vinyl polystyrene. Indeed 3 could be reused, as long as an alkyl alkene additive was added to intercept the unstable catalytic methylidene carbene (Cl2(PCy3)2Ru
![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) CH2).
8 Complex 2 can be transferred to
vinyl polystyrene in the same way, with corresponding
benefits.9
CH2).
8 Complex 2 can be transferred to
vinyl polystyrene in the same way, with corresponding
benefits.9
We are interested in developing a robust immobilised catalyst for olefin metathesis that can be readily modified to operate in a range of solvent environments. The ability to utilise non-degassed solvent offers practical simplicity and could widen the scope of this reaction. The reported stability of 5 was attractive and we reasoned that a suitably modified version could be attached to a range of polymer supports. In this communication, we describe initial studies leading to a recyclable, polystyrene-supported analogue of complex 5 that promotes olefin metathesis in non-degassed solvents.
We envisaged that the isopropyl portion of the phenol ether could be extended to accommodate some functional handle that would allow attachment to any chosen support (Scheme 1). Thus, opening of racemic δ-hexanolactone 6 with sodium methoxide,11 subsequent Mitsunobu reaction with 2-vinylphenol12 and saponification provided 7. Treating the pendant acid of 7 with aminomethyl polystyrene under standard conditions afforded the polystyrene supported ligand. Direct formation of the alkylideneruthenium7 would be expected to be more efficient but we reasoned that reaction of styrene 7 with 1 would generate 8 more simply and in sufficient quantities to permit our initial study. One disadvantage of this approach is that the accumulation of free phosphine effectively inhibits olefin metathesis.10 This is reflected in the fact that treatment of resin 7 with stoichiometric Grubb’s complex afforded loadings of only 0.12 mmol g−1 (as determined by phosphorus analysis), while treating the resin with five successive portions of 10 mol% of1 essentially doubled the loading to 0.20 mmol g−1. Simple filtration and washing of the resin gave 8 as dark brown beads that could be dried, stored in air and used more than a month later with no apparent decrease in activity. In all cases no colour change, or corresponding decomposition was observed during the loading of the catalyst8 even after extended reaction times.
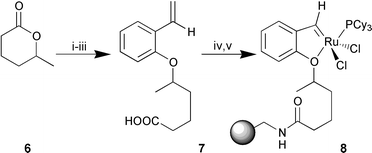 | ||
| Scheme 1 Reagents and conditions: i, sodium methoxide, MeOH, 0 °C to
rt, 6 h, 95%; ii, 2-vinylphenol, diisopropyl azodicarboxylate,
PPh3, THF, 0 °C to rt, 14 h, 64%; iii, 1 M NaOH, dioxane,
rt, 12 h, 88%; iv, polystyrene-NH2, DIC, HOBt,
CH2Cl2–DMF (1∶1), rt, 12 h; v,
Cl2(PCy3)2RuCHPh, DCE, rt, 12
h. | ||
Polymer-supported complex 8 was then tested for activity using representative diene substrates for ring closing metathesis (Table 1). It is notable that the rates of reaction were somewhat slower than those reported using homogenous Grubb’s alkylidene 1, so that reaction of benchmark substrates such as diethyl diallylmalonate (entry 2) was only 42% complete after 90 min. All of these reactions however, provided good to quantitative yield of product within 5 h. Preparative scale transformation (153 mg, 5 mol%, 4 h) of benzyl N,N-diallylcarbamate (entry 1) gave isolated product in 91% yield after chromatography.

The easy manipulation of 8 is noteworthy. In each case, general laboratory grade dichloromethane (DCM) was used without degassing, in an air atmosphere, so that substrates were simply dissolved and added to the resin in a plastic solid phase organic synthesis tube fitted with a glass frit. The tube was then sealed and subjected to agitation by 360° rotation at rt. Filtration and washing with DCM afforded the product and the remaining resin used for further transformations.
Recycling of 8 was tested by treatment of the same batch of resin with equal, successive portions of benzyl N,N-diallylcarbamate, without addition of stabilising alkene (Table 2).8 Indeed 8 proved to be remarkably robust providing good yields over five successive runs at 5 mol% catalyst. In subsequent runs yields declined steadily and the beads turned from dark brown to black.
We have also demonstrated the ability of 8 to perform cross metathesis. Heating a two-fold excess of (Z)-1,4-diacetoxybut-2-ene13 with 4-allylanisole and 8 in DCM under reflux for 9 h furnished the cross-coupled product (33%) along with the anisole homodimer (18%). The recovered resin retained its brown colouration, rather than turning black. Subsequent ring closing metathesis of benzyl N,N-diallylcarbamate using this resin afforded the cyclised product in 85% yield over 90 min, confirming the retention of catalytic activity.
Olefin metathesis using 1 is typically performed in degassed solvent, for example, previous studies of 5 were performed in argon-saturated solvent and atmosphere (only chromatography using non-degassed solvent was reported).7 We believe that this communication is notable because studies involving non-degassed solvent have not been widely reported. We also found, however, that for reactions corresponding to entries 1 and 5 using non-recyclable complex 1 in non-degassed DCM proceeded in good yield and draw attention to recent discussion relating to the oxidative decomposition of alkylidenerutheniums.14 In the case of Barrett’s boomerang system 3 however, it is necessary to add 10 mol% hex-1-ene to prevent decomposition.8 It is therefore interesting that 8 promotes olefin metathesis for up to five cycles, without added stabiliser.
The ability to perform olefin metathesis in polar protic solvents is a desirable goal that has so far been addressed by the development of water-soluble phosphine ligands.14,15 We therefore investigated the effect of a different polymer support on the solubility and catalytic activity of the complex. Thus, amine-functionalised TentaGel (0.3 mmol g−1) was converted into TentaGel-8 and in preliminary experiments we observed ring-closing metathesis in non-degassed MeOH, although there was significant variation between batches of resin. The best reaction proceeded in 31% yield after 6 h of reaction (entry 6) and the worst gave 18% after overnight reaction with double the quantity of resin (100 mg). The lower yield of this transformation presumably reflects the much lower loading offered by this TentaGel resin but does suggest that modification of the supporting polymer is a viable strategy.
In summary, we have reported a novel polymer supported pro-catalyst for olefin metathesis that is robust and easy to use. That 8 is stable to non-degassed solvents and can be recycled without the use of stabilising additives is notable and interesting. An attractive feature of the precursor 7 is that it can be attached to any solid support.
Note added to proof: two notable papers have been published since the submission of this manuscript: (a) S. B. Garber, J. S. Kingsbury, B. L. Gray, A. H. Hoveyda, J. Am. Chem. Soc., 2000, 122, 8168; (b) Q. Yao, Angew. Chem., Int. Ed., 2000, 39, 3896.
Acknowledgements
We are grateful to the University of Bath for a studentship (to J. S.) and to Professor B. V. L. Potter for helpful advice and encouragement.Notes and references
- For reviews see: (a) R. H. Grubbs and S. Chang, Tetrahedron, 1998, 54, 4413; CrossRef; (b) S. K. Armstrong, J. Chem. Soc., Perkin Trans. 1, 1998, 371..
- For examples: (a) R. Roy and S. K. Das, Chem. Commun., 2000, 519 RSC; (b) M. J. Bassindale, A. S. Edwards, P. Hamley, H. Adams and J. P. A. Harrity, Chem. Commun., 2000, 1035 RSC; (c) A. G. M. Barrett, S. D. Baugh, V. C. Gibson, M. R. Giles, E. L. Marshall and P. A. Procopiou, Chem. Commun., 1997, 155 RSC; (d) S. E. Gibson, V. C. Gibson and S. P. Keen, Chem. Commun., 1997, 1107 RSC.
- P. Schwab, R. H. Grubbs and J. W. Ziller, J. Am. Chem. Soc., 1996, 118, 100 CrossRef CAS.
- L. A. Paquette, J. D. Schloss, I. Efremov, F. Fabris, F. Gallou, J. Mendez-Andino and J. Yang, Org. Lett., 2000, 2, 1259 CrossRef CAS.
- H. D. Maynard and R. H. Grubbs, Tetrahedron Lett., 1999, 40, 4137 CrossRef CAS.
- J. P. A. Harrity, D. S. La, D. R. Cefalo, M. S. Visser and A. H. Hoveyda, J. Am. Chem. Soc., 1998, 120, 2343 CrossRef CAS.
- J. S. Kingsbury, J. P. A. Harrity, P. J. Bonitatebus and A. H. Hoveyda, J. Am. Chem. Soc., 1999, 121, 791 CrossRef CAS.
- M. Ahmed, A. G. M. Barrett, D. C. Braddock, S. M. Cramp and P. A. Procopiou, Tetrahedron Lett., 1999, 40, 8657 CrossRef CAS.
- M. Ahmed, T. Arnauld, A. G. M. Barrett, D. C. Braddock and P. A. Procopiou, Synlett, 2000, 1007 CAS.
- E. L. Dias, S. T. Nguyen and R. H. Grubbs, J. Am. Chem. Soc., 1997, 119, 3887 CrossRef CAS.
- A. S. Hernandez, A. Thaler, J. Castells and H. Rapoport, J. Org. Chem., 1996, 61, 314 CrossRef CAS.
- S. L. Tsaur and R. M. Fitch, J. Colloid Interface Sci., 1987, 115, 450 CrossRef CAS.
- D. J. O’Leary, H. E. Blackwell, R. A. Washenfelder and R. H. Grubbs, Tetrahedron Lett., 1998, 39, 7427 CrossRef CAS.
- D. M. Lynn, B. Mohr, R. H. Grubbs, L. M. Henling and M. W. Day, J. Am. Chem. Soc., 2000, 122, 6601 CrossRef CAS.
- T. A. Kirkland, D. M. Lynn and R. H. Grubbs, J. Org. Chem., 1998, 63, 9904 CrossRef CAS.
- Y. S. Shon and T. R. Lee, Tetrahedron Lett., 1997, 38, 1283 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: experimental details. See http://www.rsc.org/suppdata/cc/b0/b007304k/ |
| This journal is © The Royal Society of Chemistry 2001 |